Translational Read-Through Therapy of RPGR Nonsense Mutations

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
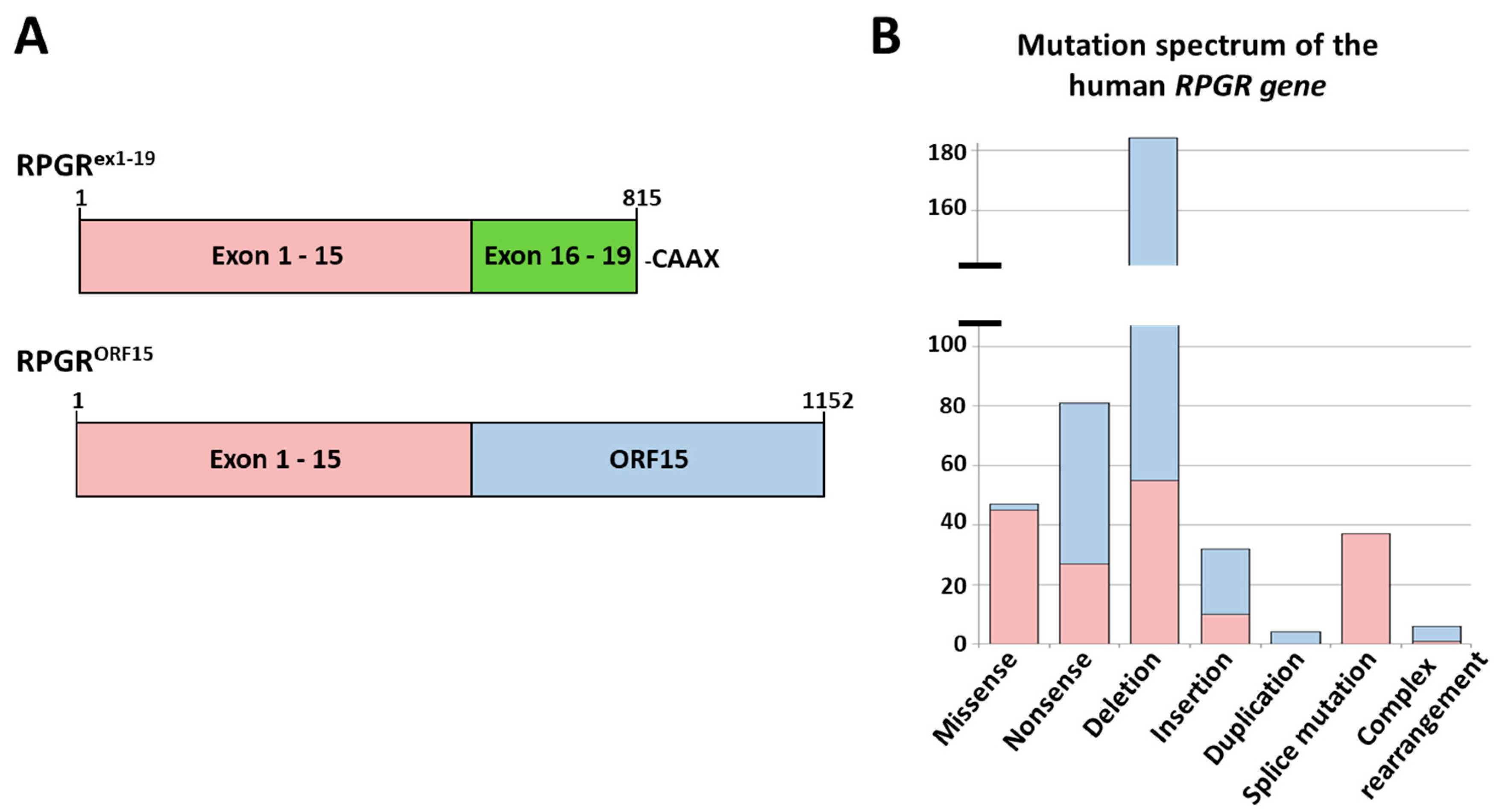
2.1. Analysis of the Mutation Spectrum in the RPGR Gene
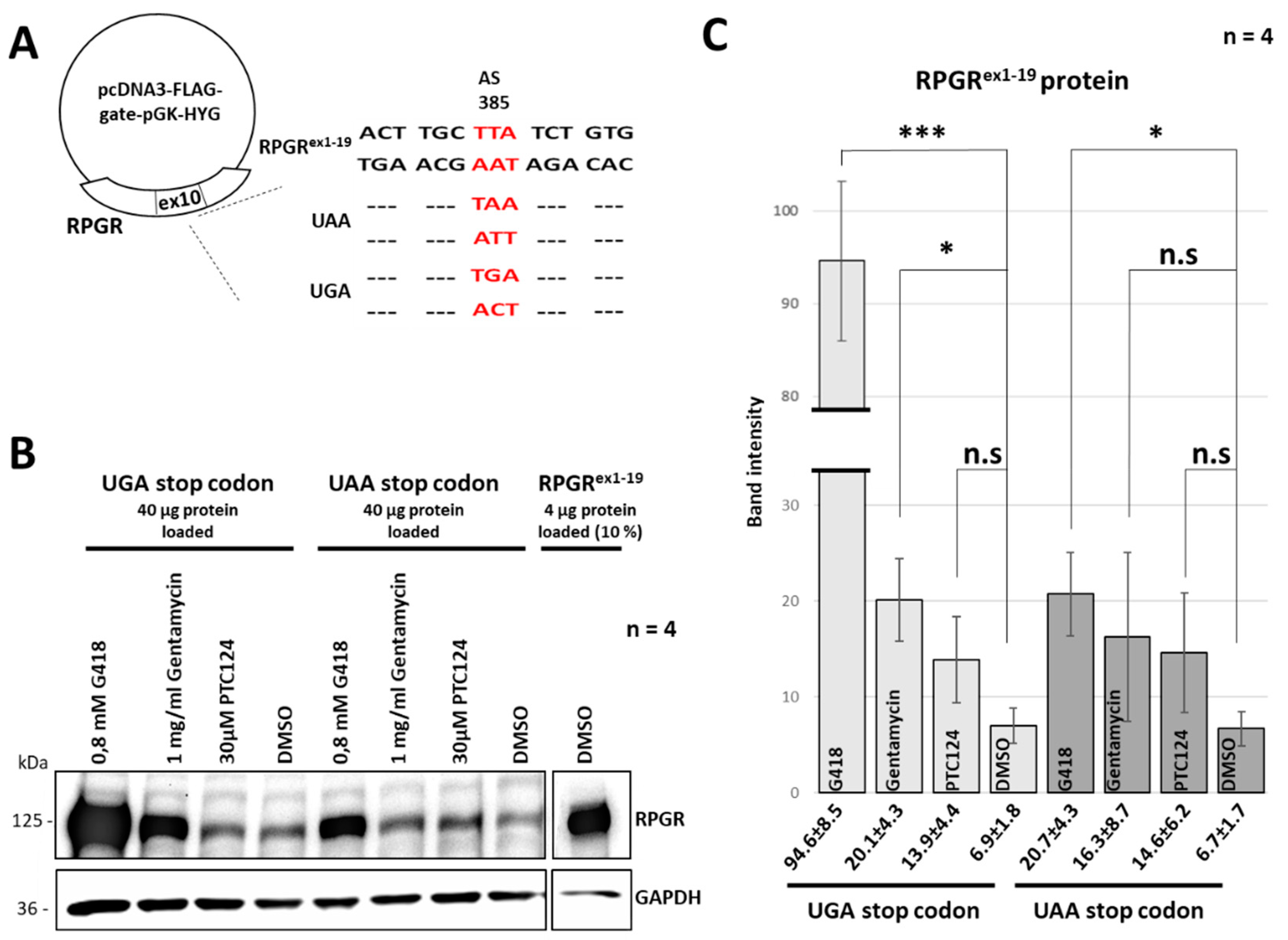
2.2. A New Nonsense Mutation in Exon 10 Leading to a PTC
2.3. The Read-Through Inducing Reagents Partially Restored RPGR Expression
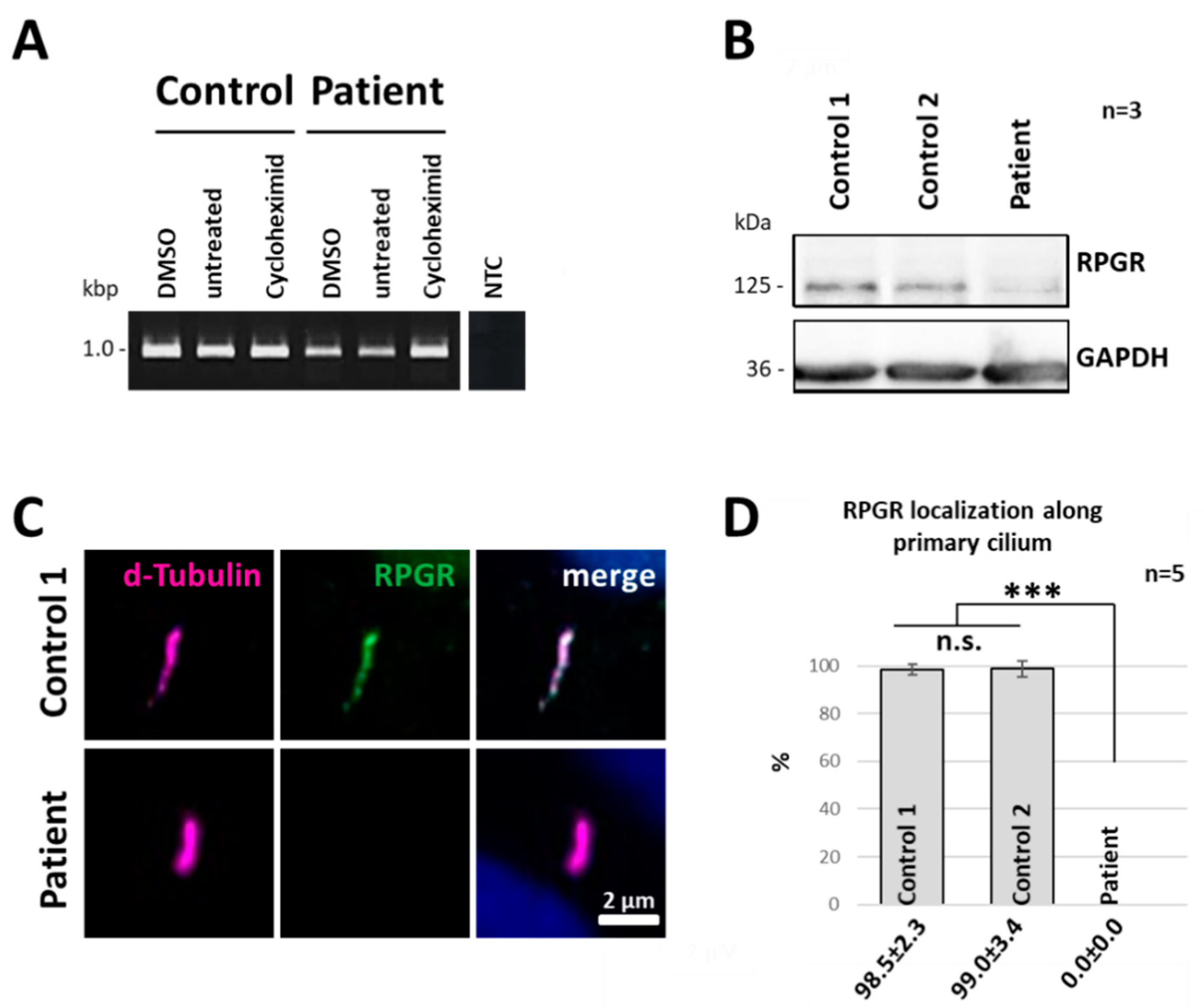
2.4. Nonsense-Mediated mRNA Decay Acts on the Truncated RPGR Transcript
2.5. RPGR Protein Expression Is Reduced in Patient-Derived Cells
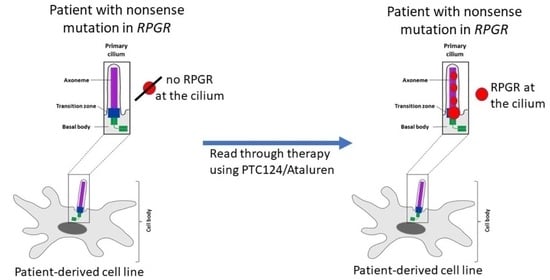
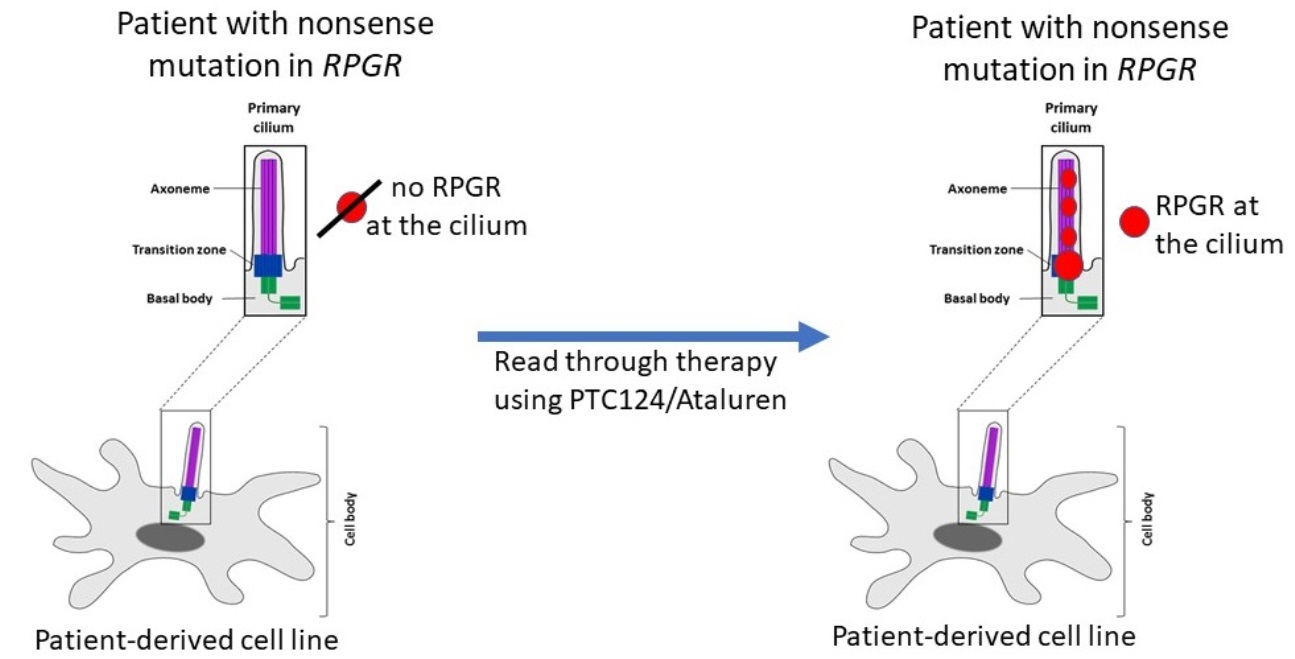
2.6. RPGR Protein Is Not Detectable at the Primary Cilium of Patient-Derived Cells
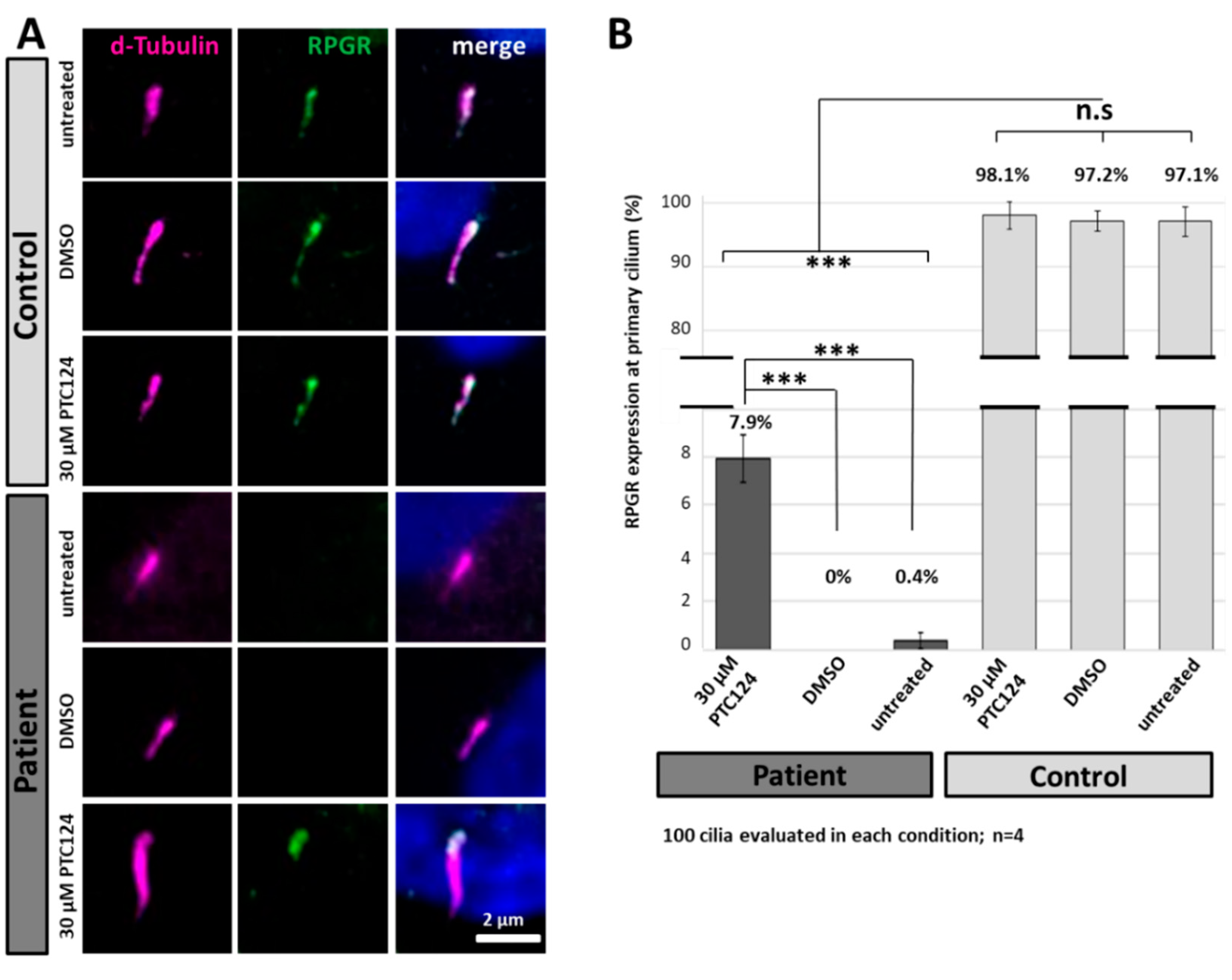
2.7. PTC124 Partially Restores Protein Expression and Localization of RPGR in Patient-Derived Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Patients and Mutation Screening
4.2. Cell Culture of Fibroblast from Skin Biopsies
4.3. Plasmid and Site-Directed Mutagenesis
4.4. Cell Culture of HEK293T Cells and Transfection
4.5. Ciliary Staining and Microscopy
4.6. RNA Isolation and RT-PCR
4.7. Protein Isolation and Western Blotting
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr Genom. 2011, 12, 238–249. [Google Scholar]
- Veltel, S.; Gasper, R.; Eisenacher, E.; Wittinghofer, A. The retinitis pigmentosa 2 gene product is a GTPase-activating protein for Arf-like 3. Nat. Struct Mol. Biol. 2008, 15, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Farber, M.D.; Derlacki, D.J. X-linked retinitis pigmentosa. Profile of clinical findings. Arch. Ophthalmol. 1988, 106, 369–375. [Google Scholar] [CrossRef]
- Branham, K.; Othman, M.; Brumm, M.; Karoukis, A.J.; Atmaca-Sonmez, P.; Yashar, B.M.; Schwartz, S.B.; Stover, N.B.; Trzupek, K.; Wheaton, D.; et al. Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8232–8237. [Google Scholar] [CrossRef]
- Sharon, D.; Sandberg, M.A.; Rabe, V.W.; Stillberger, M.; Dryja, T.P.; Berson, E.L. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am. J. Hum. Genet. 2003, 73, 1131–1146. [Google Scholar] [CrossRef]
- Bader, I.; Brandau, O.; Achatz, H.; Apfelstedt-Sylla, E.; Hergersberg, M.; Lorenz, B.; Wissinger, B.; Wittwer, B.; Rudolph, G.; Meindl, A.; et al. X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1458–1463. [Google Scholar] [CrossRef]
- Schmid, F.; Glaus, E.; Cremers, F.P.; Kloeckener-Gruissem, B.; Berger, W.; Neidhardt, J. Mutation- and tissue-specific alterations of RPGR transcripts. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1628–1635. [Google Scholar] [CrossRef][Green Version]
- Neidhardt, J.; Glaus, E.; Barthelmes, D.; Zeitz, C.; Fleischhauer, J.; Berger, W. Identification and characterization of a novel RPGR isoform in human retina. Hum. Mutat. 2007, 28, 797–807. [Google Scholar] [CrossRef]
- Roepman, R.; Bernoud-Hubac, N.; Schick, D.E.; Maugeri, A.; Berger, W.; Ropers, H.H.; Cremers, F.P.; Ferreira, P.A. The retinitis pigmentosa GTPase regulator (RPGR) interacts with novel transport-like proteins in the outer segments of rod photoreceptors. Hum. Mol. Genet. 2000, 9, 2095–2105. [Google Scholar] [CrossRef]
- Kirschner, R.; Rosenberg, T.; Schultz-Heienbrok, R.; Lenzner, S.; Feil, S.; Roepman, R.; Cremers, F.P.; Ropers, H.H.; Berger, W. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum. Mol. Genet. 1999, 8, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, R.; Glaus, E.; Tiwari, A.; Kloeckener-Gruissem, B.; Berger, W.; Neidhardt, J. Localizing the RPGR protein along the cilium: A new method to determine efficacies to treat RPGR mutations. Gene Ther 2015, 22, 413–420. [Google Scholar] [CrossRef]
- Hong, D.H.; Li, T. Complex expression pattern of RPGR reveals a role for purine-rich exonic splicing enhancers. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3373–3382. [Google Scholar]
- Hong, D.H.; Pawlyk, B.; Sokolov, M.; Strissel, K.J.; Yang, J.; Tulloch, B.; Wright, A.F.; Arshavsky, V.Y.; Li, T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2413–2421. [Google Scholar] [CrossRef] [PubMed]
- Mavlyutov, T.A.; Zhao, H.; Ferreira, P.A. Species-specific subcellular localization of RPGR and RPGRIP isoforms: Implications for the phenotypic variability of congenital retinopathies among species. Hum. Mol. Genet. 2002, 11, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Meindl, A.; Dry, K.; Herrmann, K.; Manson, F.; Ciccodicola, A.; Edgar, A.; Carvalho, M.R.; Achatz, H.; Hellebrand, H.; Lennon, A.; et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat. Genet. 1996, 13, 35–42. [Google Scholar] [CrossRef]
- Renault, L.; Nassar, N.; Wittinghofer, A.; Roth, M.; Vetter, I.R. Crystallization and preliminary X-ray analysis of human RCC1, the regulator of chromosome condensation. Acta Crystallogr. D Biol. Crystallogr. 1999, 55 Pt 1, 272–275. [Google Scholar] [CrossRef]
- Shu, X.; McDowall, E.; Brown, A.F.; Wright, A.F. The human retinitis pigmentosa GTPase regulator gene variant database. Hum. Mutat. 2008, 29, 605–608. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- He, S.; Parapuram, S.K.; Hurd, T.W.; Behnam, B.; Margolis, B.; Swaroop, A.; Khanna, H. Retinitis Pigmentosa GTPase Regulator (RPGR) protein isoforms in mammalian retina: Insights into X-linked Retinitis Pigmentosa and associated ciliopathies. Vision Res. 2008, 48, 366–376. [Google Scholar] [CrossRef]
- Brogna, S.; McLeod, T.; Petric, M. The Meaning of NMD: Translate or Perish. Trends Genet. 2016, 32, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Bhuvanagiri, M.; Schlitter, A.M.; Hentze, M.W.; Kulozik, A.E. NMD: RNA biology meets human genetic medicine. Biochem. J. 2010, 430, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Asiful Islam, M.; Alam, F.; Kamal, M.A.; Gan, S.H.; Wong, K.K.; Sasongko, T.H. Therapeutic Suppression of Nonsense Mutation: An Emerging Target in Multiple Diseases and Thrombotic Disorders. Curr. Pharm. Des. 2017, 23, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Nagel-Wolfrum, K.; Moller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting Nonsense Mutations in Diseases with Translational Read-Through-Inducing Drugs (TRIDs). BioDrugs 2016, 30, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Beltran, W.A.; Cideciyan, A.V.; Lewin, A.S.; Iwabe, S.; Khanna, H.; Sumaroka, A.; Chiodo, V.A.; Fajardo, D.S.; Roman, A.J.; Deng, W.T.; et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2012, 109, 2132–2137. [Google Scholar] [CrossRef]
- Hong, D.H.; Pawlyk, B.S.; Adamian, M.; Sandberg, M.A.; Li, T. A single, abbreviated RPGR-ORF15 variant reconstitutes RPGR function in vivo. Investig. Ophthalmol. Vis. Sci. 2005, 46, 435–441. [Google Scholar] [CrossRef]
- May-Simera, H.; Nagel-Wolfrum, K.; Wolfrum, U. Cilia—The sensory antennae in the eye. Prog. Retin. Eye Res. 2017, 60, 144–180. [Google Scholar] [CrossRef]
- Morais, P.; Adachi, H.; Yu, Y.T. Suppression of Nonsense Mutations by New Emerging Technologies. Int. J. Mol. Sci. 2020, 21, 4394. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Bassuk, A.G.; Zheng, A.; Li, Y.; Tsang, S.H.; Mahajan, V.B. Precision Medicine: Genetic Repair of Retinitis Pigmentosa in Patient-Derived Stem Cells. Sci. Rep. 2016, 6, 19969. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Eleftheriou, C.; Allen, A.E.; Milosavljevic, N.; Pienaar, A.; Bedford, R.; Davis, K.E.; Bishop, P.N.; Lucas, R.J. Restoration of Vision with Ectopic Expression of Human Rod Opsin. Curr. Biol. 2015, 25, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Politano, L.; Nigro, G.; Nigro, V.; Piluso, G.; Papparella, S.; Paciello, O.; Comi, L.I. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003, 22, 15–21. [Google Scholar]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Mendell, J.R. Aminoglycoside-induced mutation suppression (stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Ther. Adv. Neurol. Disord. 2010, 3, 379–389. [Google Scholar] [CrossRef]
- Du, M.; Jones, J.R.; Lanier, J.; Keeling, K.M.; Lindsey, J.R.; Tousson, A.; Bebok, Z.; Whitsett, J.A.; Dey, C.R.; Colledge, W.H.; et al. Aminoglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. J. Mol. Med. (Berl) 2002, 80, 595–604. [Google Scholar] [CrossRef]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investig. 1999, 104, 375–381. [Google Scholar] [CrossRef]
- Linde, L.; Kerem, B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008, 24, 552–563. [Google Scholar] [CrossRef]
- Lubamba, B.; Dhooghe, B.; Noel, S.; Leal, T. Cystic fibrosis: Insight into CFTR pathophysiology and pharmacotherapy. Clin. Biochem. 2012, 45, 1132–1144. [Google Scholar] [CrossRef]
- Goldmann, T.; Overlack, N.; Moller, F.; Belakhov, V.; van Wyk, M.; Baasov, T.; Wolfrum, U.; Nagel-Wolfrum, K. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 2012, 4, 1186–1199. [Google Scholar] [CrossRef]
- Goldmann, T.; Rebibo-Sabbah, A.; Overlack, N.; Nudelman, I.; Belakhov, V.; Baasov, T.; Ben-Yosef, T.; Wolfrum, U.; Nagel-Wolfrum, K. Beneficial read-through of a USH1C nonsense mutation by designed aminoglycoside NB30 in the retina. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6671–6680. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.M.; Nommiste, B.; Amelia, R.L.; Carr, A.F.; Powner, M.B.; Matthew, J.K.S.; Chen, L.L.; Muthiah, M.N.; Webster, A.R.; Moore, A.T.; et al. Rescue of the MERTK phagocytic defect in a human iPSC disease model using translational read-through inducing drugs. Sci. Rep. 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, N.; Carr, A.J.; Lane, A.; Moeller, F.; Chen, L.L.; Aguila, M.; Nommiste, B.; Muthiah, M.N.; Kanuga, N.; Wolfrum, U.; et al. Translational read-through of the RP2 Arg120stop mutation in patient iPSC-derived retinal pigment epithelium cells. Hum. Mol. Genet. 2015, 24, 972–986. [Google Scholar] [CrossRef] [PubMed]
- Guerin, K.; Gregory-Evans, C.Y.; Hodges, M.D.; Moosajee, M.; Mackay, D.S.; Gregory-Evans, K.; Flannery, J.G. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp. Eye Res. 2008, 87, 197–207. [Google Scholar] [CrossRef]
- Samanta, A.; Stingl, K.; Kohl, S.; Ries, J.; Linnert, J.; Nagel-Wolfrum, K. Ataluren for the Treatment of Usher Syndrome 2A Caused by Nonsense Mutations. Int. J. Mol. Sci. 2019, 20, 6274. [Google Scholar] [CrossRef]
- Zhang, Q.; Giacalone, J.C.; Searby, C.; Stone, E.M.; Tucker, B.A.; Sheffield, V.C. Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc. Natl. Acad. Sci. USA 2019, 116, 1353–1360. [Google Scholar] [CrossRef]
- Rao, K.N.; Zhang, W.; Li, L.; Anand, M.; Khanna, H. Prenylated retinal ciliopathy protein RPGR interacts with PDE6delta and regulates ciliary localization of Joubert syndrome-associated protein INPP5E. Hum. Mol. Genet. 2016, 25, 4533–4545. [Google Scholar]
- Roy, B.; Leszyk, J.D.; Mangus, D.A.; Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3038–3043. [Google Scholar] [CrossRef]
- Richardson, R.; Smart, M.; Tracey-White, D.; Webster, A.R.; Moosajee, M. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp. Eye Res. 2017, 155, 24–37. [Google Scholar] [CrossRef]
- Grayson, C.; Chapple, J.P.; Willison, K.R.; Webster, A.R.; Hardcastle, A.J.; Cheetham, M.E. In vitro analysis of aminoglycoside therapy for the Arg120stop nonsense mutation in RP2 patients. J. Med. Genet. 2002, 39, 62–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- El Shamieh, S.; Mejecase, C.; Bertelli, M.; Terray, A.; Michiels, C.; Condroyer, C.; Fouquet, S.; Sadoun, M.; Clerin, E.; Liu, B.; et al. Further Insights into the Ciliary Gene and Protein KIZ and Its Murine Ortholog PLK1S1 Mutated in Rod-Cone Dystrophy. Genes (Basel) 2017, 8, 277. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: Aminoglycosides and ataluren (PTC124). J. Child. Neurol. 2010, 25, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Owczarek-Lipska, M.; Mulahasanovic, L.; Obermaier, C.D.; Hortnagel, K.; Neubauer, B.A.; Korenke, G.C.; Biskup, S.; Neidhardt, J. Novel mutations in the GJC2 gene associated with Pelizaeus-Merzbacher-like disease. Mol. Biol. Rep. 2019, 46, 4507–4516. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vössing, C.; Owczarek-Lipska, M.; Nagel-Wolfrum, K.; Reiff, C.; Jüschke, C.; Neidhardt, J. Translational Read-Through Therapy of RPGR Nonsense Mutations. Int. J. Mol. Sci. 2020, 21, 8418. https://doi.org/10.3390/ijms21228418
Vössing C, Owczarek-Lipska M, Nagel-Wolfrum K, Reiff C, Jüschke C, Neidhardt J. Translational Read-Through Therapy of RPGR Nonsense Mutations. International Journal of Molecular Sciences. 2020; 21(22):8418. https://doi.org/10.3390/ijms21228418
Chicago/Turabian StyleVössing, Christine, Marta Owczarek-Lipska, Kerstin Nagel-Wolfrum, Charlotte Reiff, Christoph Jüschke, and John Neidhardt. 2020. "Translational Read-Through Therapy of RPGR Nonsense Mutations" International Journal of Molecular Sciences 21, no. 22: 8418. https://doi.org/10.3390/ijms21228418
APA StyleVössing, C., Owczarek-Lipska, M., Nagel-Wolfrum, K., Reiff, C., Jüschke, C., & Neidhardt, J. (2020). Translational Read-Through Therapy of RPGR Nonsense Mutations. International Journal of Molecular Sciences, 21(22), 8418. https://doi.org/10.3390/ijms21228418