Molecular Mechanisms of “Antiphospholipid Antibodies” and Their Paradoxical Role in the Pathogenesis of “Seronegative APS”



Abstract
1. Introduction
2. β2-GPI Conformations in the Pathogenesis of APS
3. Post-Translational Modifications and Pathogenesis of APS
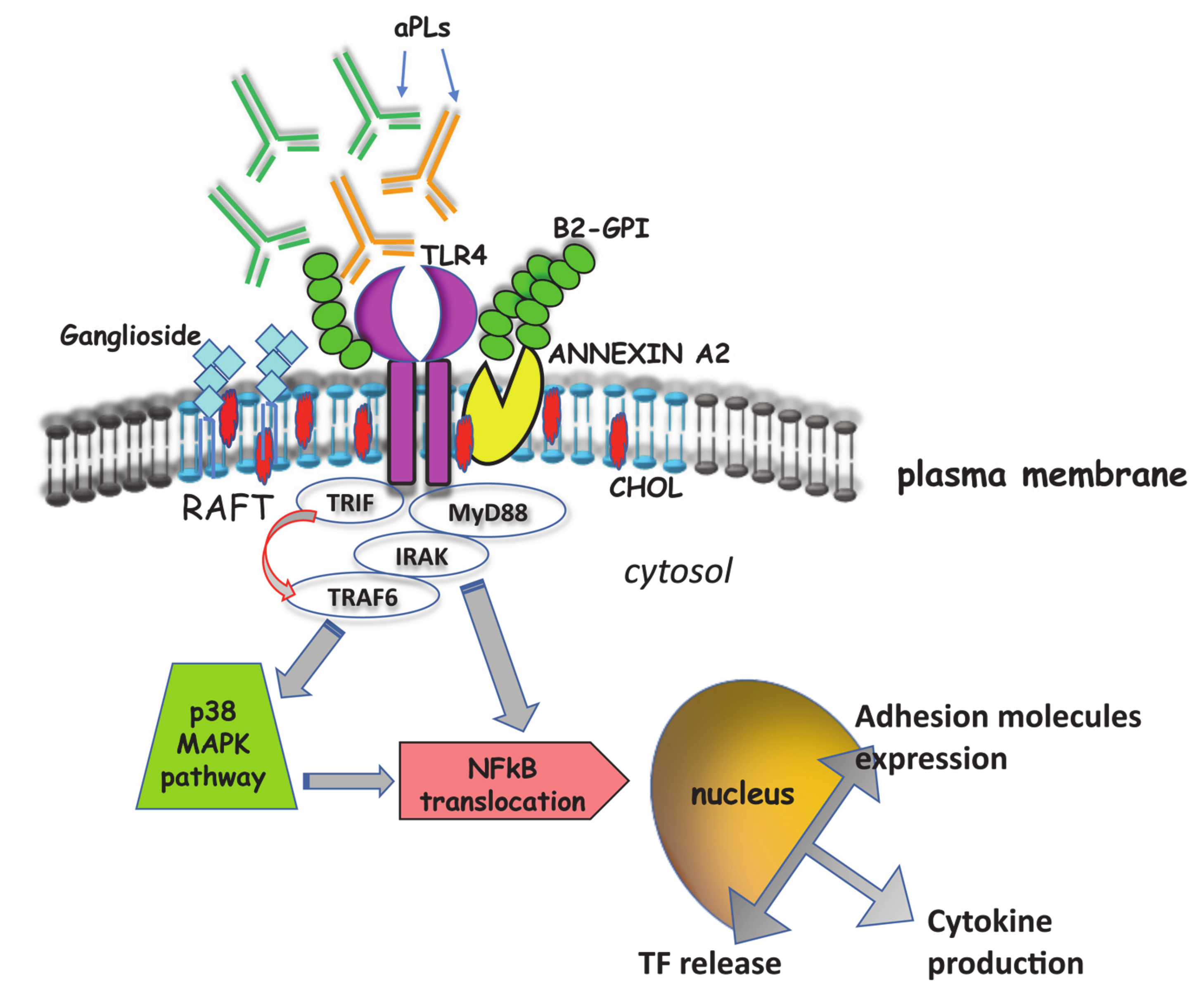
4. Signal Transduction Pathways Triggered by aPL
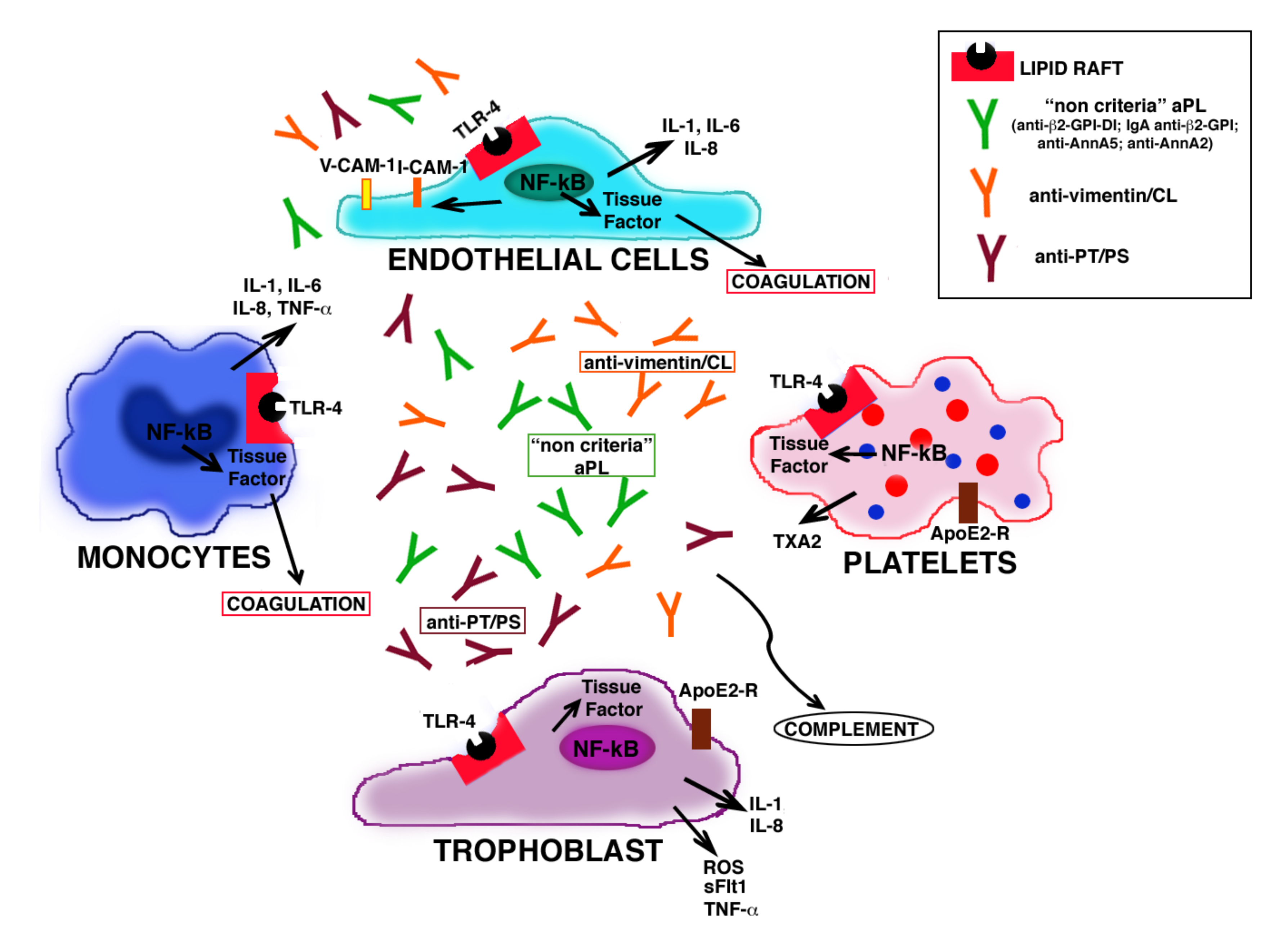
5. Signaling and Pregnancy Complications
6. Role of Antiphospholipid Antibodies in Seronegative APS
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APS | antiphospholipid syndrome |
| OAPS | obstetric antiphospholipid antibodies syndrome |
| IUGR | intrauterine growth restriction |
| CAPS | catastrophic antiphospholipid antibodies syndrome |
| SN-APS | seronegative antiphospholipid antibodies syndrome |
| aPL | antiphospholipid antibodies |
| β2-GPI | beta2-glycoprotein I |
| anti-β2GPI DI | anti-beta2-glycoprotein I domain I |
| CL | Cardiolipin |
| PT/PS | prothrombin/phosphatidylserine |
| LBPA | lysobisphosphatidic acid |
| PE | phosphatidylethanolamine |
| TLC | thin layer chromatographic plates |
| PTMs | post-translational modifications |
| ROS | reactive oxygen species |
| ECs | endothelial cells |
| TLR-4 | Toll-like receptor 4 |
| ApoER2 | apolipoprotein E receptor 2 |
| MyD88 | myeloid differentiation factor 88 |
| IRAK | interleukin-1 receptor-associated kinase 1 |
| MAPK | mitogen-activated protein kinase |
| NF-κB | nuclear factor kappa B |
| TF | Tissue Factor |
| V-CAM | vascular cell adhesion molecule-1 |
| I-CAM | intercellular cell adhesion molecule-1 |
| mTOR | mechanistic target of rapamycin |
| NETs | neutrophil extracellular traps |
References
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; Groot, P.G.D.E.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Piette, J.C.; Font, J.; Khamashta, M.A.; Shoenfeld, Y.; Camps, M.T.; Jacobsen, S.; Lakos, G.; Tincani, A.; Kontopoulou-Griva, I.; et al. Antiphospholipid syndrome: Clinical and immunologic manifestations and patterns of disease expression in a cohort of 1000 patients. Arthritis Rheum. 2002, 46, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Irastorza, G.; Crowther, M.; Branch, W.; Khamashta, M.A. Antiphospholipid syndrome. Lancet 2010, 376, 1498–1509. [Google Scholar] [CrossRef]
- Negrini, S.; Pappalardo, F.; Murdaca, G.; Indiveri, F.; Puppo, F. The antiphospholipid syndrome: From pathophysiology to treatment. Clin. Exp. Med. 2017, 17, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Yelnik, C.M.; Urbanski, G.; Drumez, E.; Sobanski, V.; Maillard, H.; Lanteri, A.; Morell-Dubois, S.; Caron, C.; Dubucquoi, S.; Launay, D.; et al. Persistent triple antiphospho lipid antibody positivity as a strong risk factor of frst thrombosis, in a long-term follow-up study of patients without history of thrombosis or obstetrical morbidity. Lupus 2017, 26, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Otomo, K.; Atsumi, T.; Amengual, O.; Fujieda, Y.; Kato, M.; Oku, K.; Horita, T.; Yasuda, S.; Koike, T. Efficacy of the antiphospholipid score for the diagnosis of antiphospholipid syndrome and its predictive value for thrombotic events. Arthritis Rheum. 2012, 64, 504–512. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Wong, R.C.W. Antiphospholipid antibody testing for the antiphospholipid syndrome: A comprehensive practical review including a synopsis of challenges and recent guidelines. Pathology 2014, 46, 481–495. [Google Scholar] [CrossRef]
- Misasi, R.; Capozzi, A.; Longo, A.; Recalchi, S.; Lococo, E.; Alessandri, C.; Conti, F.; Valesini, G.; Sorice, M. “New” Antigenic Targets and Methodological Approaches for Refining Laboratory Diagnosis of Antiphospholipid Syndrome. J. Immunol. Res. 2015, 2015, 858542. [Google Scholar] [CrossRef]
- De Laat, H.B.; Derksen, R.H.W.M.; De Groot, P.G. beta2-glycoprotein I, the playmaker of the antiphospholipid syndrome. Clin. Immunol. 2004, 112, 161–168. [Google Scholar] [CrossRef]
- Kaburaki, J.; Kuwana, M.; Yamamoto, M.; Kawai, S.; Ikeda, Y. Clinical significance of anti-annexin V antibodies in patients with systemic lupus erythematosus. Am. J. Hematol. 1997, 54, 209–213. [Google Scholar] [CrossRef]
- Salle, V.; Mazie`re, J.C.; Smail, A.; Cévallos, R.; Mazière, C.; Fuentes, V.; Tramier, B.; Makdassi, R.; Choukroun, G.; Vittecoq, O.; et al. Anti-annexin II antibodies in systemic autoimmune diseases and antiphospholipid syndrome. J. Clin. Immunol. 2008, 28, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Arvieux, J.; Darnige, L.; Caron, C.; Reber, G.; Bensa, J.C.; Colomb, M.G. Development of an ELISA for autoantibodies to prothrombin showing their prevalence in patients with lupus anticoagulants. Thromb. Haemost. 1995, 74, 1120–1125. [Google Scholar] [CrossRef]
- Oosting, J.D.; Derksen, R.H.W.M.; Bobbink, I.W.G.; Hackeng, T.M.; Bouma, B.N.; De Groot, P.G. Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C, or protein S: An explanation for their pathogenic mechanism? Blood 1993, 81, 2618–2625. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Arcieri, P.; Griggi, T.; Circella, A.; Misasi, R.; Lenti, L.; Di Nucci, G.D.; Mariani, G. Inhibition of protein S by autoantibodies in patients with acquired protein S deficiency. Thromb. Haemost. 1996, 75, 555–559. [Google Scholar] [CrossRef]
- Sciascia, S.; Sanna, G.; Murru, V.; Roccatello, D.; Khamashta, M.A.; Bertolaccini, M.L. Anti-prothrombin (aPT) and anti- phosphatidylserine/prothrombin (aPS/PT) antibodies and the risk of thrombosis in the antiphospholipid syndrome. A sys- tematic review. Thromb. Haemost. 2013, 111, 354–364. [Google Scholar] [CrossRef]
- Ortona, E.; Capozzi, A.; Colasanti, T.; Conti, F.; Alessandri, C.; Longo, A.; Garofalo, T.; Margutti, P.; Misasi, R.; Khamashta, M.A.; et al. Vimentin/cardiolipin complex as a new antigenic target of the antiphospholipid syndrome. Blood 2010, 116, 2960–2967. [Google Scholar] [CrossRef]
- Sanmarco, M.; Gayet, S.; Alessi, M.C.; Audrain, M.; de Maistre, E.; Gris, J.C.; de Groot, P.G.; Hachulla, E.; Harlé, J.R.; Sié, P.; et al. Antiphosphatidylethanolamine antibodies are associated with an increased odds ratio for thrombosis—A multicenter study with the partici- pation of the European Forum on antiphospholipid antibodies. Thromb. Haemost. 2007, 97, 949–954. [Google Scholar] [PubMed]
- Alessandri, C.; Bombardieri, M.; Di Prospero, L.; Conigliaro, P.; Conti, F.; Labbadia, G.; Misasi, R.; Sorice, M.; Valesini, G. Anti-lysobisphosphatidic acid antibodies in patients with antiphospholipid syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 2005, 140, 173–180. [Google Scholar] [CrossRef]
- Hughes, G.R.V.; Khamashta, M.A. Seronegative antiphospholipid syndrome. Ann. Rheum. Dis. 2003, 62, 1127. [Google Scholar] [CrossRef]
- Sciascia, S.; Amigo, M.; Roccatello, D.; Khamashta, M.A. Diagnosing antiphospholipid syndrome:’extra-criteria’manifestations and technical advances. Nat. Rev. Rheumatol. 2017, 13, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Barbui, T.; Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Antiphospholipid antibodies: Involvement of protein cofactors. Haematologica 1993, 78, 1–4. [Google Scholar]
- Pierangeli, S.S.; Chen, P.P.; Raschi, E.; Scurati, S.; Grossi, C.; Borghi, M.O.; Palomo, I.; Harris, E.N.; Meroni, P.L. Antiphospholipid antibodies and the antiphospholipid syndrome: Pathogenic mechanisms. Semin. Thromb. Hemost. 2008, 34, 236–250. [Google Scholar] [CrossRef]
- Jimenez, S.; Tassies, D.; Espinosa, G.; García-Criado, A.; Plaza, J.; Monteagudo, J.; Cervera, R.; Reverter, J.C. Double heterozygosity polymorphisms for platelet glycoproteins Ia/IIa and IIb/IIIa increases arterial thrombosis and arteriosclerosis in patients with the antiphospholipid syndrome or with systemic lupus erythematosus. Ann. Rheum. Dis. 2008, 67, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Arachchillage, D.R.J.; Laffan, M. Pathogenesis and management of antiphospholipid syndrome. Br. J. Haematol. 2017, 178, 181–195. [Google Scholar] [CrossRef]
- Galli, M.; Comfurius, P.; Maassen, C.; Hemker, H.C.; De Baets, M.H.; Van Breda-Vriesman, P.J.; Barbui, T.; Zwaal, R.F.; Bevers, E.M. Anticardiolipin anti- bodies (ACA) directed not to cardiolipin but to a plasma protein cofactor. Lancet 1990, 335, 1544–1547. [Google Scholar] [CrossRef]
- Robbins, D.L.; Leung, S.; Miller-Blair, D.J.; Ziboh, V. Effect of anticardiolipin/beta2-glycoprotein I complexes on production of thromboxane A2 by platelets from patients with the antiphospholipid syndrome. J. Rheumatol. 1998, 25, 51–56. [Google Scholar]
- Sikara, M.P.; Routsias, J.G.; Samiotaki, M.; Panayotou, G.; Moutsopoulos, H.M.; Vlachoyiannopoulos, P.G. {beta}2 Glycoprotein I ({beta}2GPI) binds platelet factor 4 (PF4): Implications for the pathogenesis of antiphospholipid syndrome. Blood 2010, 115, 713–723. [Google Scholar] [CrossRef]
- Zucker, M.B.; Katz, I.R. Platelet factor 4: Production, structure, and physiologic and immunologic action. Proc. Soc. Exp. Biol. Med. 1991, 198, 693–702. [Google Scholar] [CrossRef]
- Amengual, O.; Atsumi, T.; Khamashta, M.A.; Hughes, G.R. The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb. Haemost. 1998, 79, 276–281. [Google Scholar] [CrossRef]
- Rand, J.H.; Wu, X.X.; Andree, H.A.; Lockwood, C.J.; Guller, S.; Scher, J.; Harpel, P.C. Pregnancy loss in the antiphospholipid-antibody syndrome—a possible thrombogenic mechanism. N. Engl. J. Med. 1997, 337, 154–160. [Google Scholar] [CrossRef]
- Rand, J.H.; Wu, X.X.; Quinn, A.S.; Ashton, A.W.; Chen, P.P.; Hathcock, J.J.; Andree, H.A.M.; Taatjes, D.J. Hydroxychloroquine protects the annexin A5 anticoagulant shield from disruption by antiphospholipid antibodies: Evidence for a novel effect for an old antimalarial drug. Blood 2010, 115, 2292–2299. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.; Borghi, M.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339. [Google Scholar] [CrossRef]
- Linnemann, B. Antiphospholipid Syndrome-an update. Vasa 2018, 47, 451–464. [Google Scholar] [CrossRef]
- Meroni, P.L.; Raschi, E.; Camera, M.; Testoni, C.; Nicoletti, F.; Tincani, A.; Khamashta, M.A.; Balestrieri, G.; Tremoli, E.; Hess, D.C. Endothelial activation by aPL: A potential pathogenetic mechanism for the clinical manifestations of the syndrome. J. Autoimmun. 2000, 15, 237–240. [Google Scholar] [CrossRef]
- Raschi, E.; Testoni, C.; Bosisio, D.; Borghi, M.O.; Koike, T.; Mantovani, A.; Meroni, P.L. Role of the MyD88 transduction signaling pathway in endothelial activation by antiphospholipid antibodies. Blood 2003, 101, 3495–3500. [Google Scholar] [CrossRef] [PubMed]
- Kitchens, C.S. Thrombotic storm: When thrombosis begets thrombosis. Am. J. Med. 1998, 104, 381–385. [Google Scholar] [CrossRef]
- Canaud, G.; Bienaim, E.F.; Tabarin, F.; Bataillon, G.; Seilhean, D.; Noël, L.H.; Dragon-Durey, M.A.; Snanoudj, R.; Friedlander, G.; Halbwachs-Mecarelli, G.L.; et al. Inhibition of the mTORC pathway in the antiphospholipid syndrome. N. Engl. J. Med. 2014, 371, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Nimpf, J.; Bevers, E.M.; Bomans, P.H.; Till, U.; Wurm, H.; Kostner, G.M.; Zwaal, R.F. Prothrombinase activity of human platelets is inhibited by beta 2-glycoprotein-I. Biochim. Biophys. Acta 1986, 884, 142–149. [Google Scholar] [CrossRef]
- Nimpf, J.; Wurm, H.; Kostner, G.M. Interaction of b2-glycoprotein-I with human blood platelets: Influence upon the ADP-induced aggregation. Thromb. Haemost. 1985, 54, 397–401. [Google Scholar]
- Shi, T.; Giannakopoulos, B.; Iverson, G.M.; Cockerill, K.A.; Linnik, M.D.; Krilis, S.A. Domain V of beta2-glycoprotein I binds factor XI/XIa and is cleaved at Lys317-Thr318. J. Biol. Chem. 2005, 280, 907–912. [Google Scholar] [CrossRef]
- Agar, C.; de Groot, P.G.; Mo¨rgelin, M.; Monk, S.D.D.C.; van Os, G.M.A.; Levels, J.H.M.; de Laat, B.; Urbanus, R.T.; Herwald, H.; van der Poll, T.; et al. β2-glycoprotein I: A novel component of innate immunity. Blood 2011, 117, 6939–6947. [Google Scholar] [CrossRef]
- Balasubramanian, K.; Schroit, A.J. Characterization of phosphatidylserine-dependent beta2-glycoprotein I macrophage interactions. Implications for apoptotic cell clearance by phagocytes. J. Biol. Chem. 1998, 273, 29272–29277. [Google Scholar] [CrossRef] [PubMed]
- Schultze, H.E.; Heide, K.; Haupt, H. Uber ein bischer unb ekanntesniedermolekularis b2-globulins des human serums. Naturwissens-chaften 1961, 48, 719. [Google Scholar] [CrossRef]
- De Groot, P.G.; Meijers, J. β(2) -Glycoprotein I: Evolution, structure and function. J. Thromb. Haemost. 2011, 9, 1275–1284. [Google Scholar] [CrossRef]
- McNeil, H.P.; Simpson, R.J.; Chesterman, C.N.; Krilis, S.A. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: Beta 2-glycoprotein I (apolipoprotein H). Proc. Natl. Acad. Sci. USA 1990, 87, 4120–4124. [Google Scholar] [CrossRef]
- Matsuura, E.; Igarashi, Y.; Fujimoto, M.; Ichikawa, K.; Koike, T. Anticardiolipin cofactor(s) and differential diagnosis of autoimmune disease. Lancet 1990, 336, 177–178. [Google Scholar] [CrossRef]
- Ioannou, Y.; Zhang, J.Y.; Qi, M.; Gao, L.; Qi, J.C.; Yu, D.M.; Lau, H.; Sturgess, A.D.; Vlachoyiannopoulos, P.G.; Moutsopoulos, H.M.; et al. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen β2-glycoprotein I. Arthritis Rheum. 2011, 63, 2774–2782. [Google Scholar] [CrossRef]
- Giannakopoulos, B.; Krilis, S.A. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 2013, 368, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Iverson, G.M.; Victoria, E.J.; Marquis, D.M. Anti-beta2 glycoprotein I(beta2GPI) autoantibodies recognize an epitope on the first domain of beta2GPI. Proc. Natl. Acad. Sci. USA 1998, 95, 15542–15546. [Google Scholar] [CrossRef]
- Iverson, G.M.; Reddel, S.; Victoria, E.J.; Cockerill, K.A.; Wang, Y.X.; Marti-Renom, M.A.; Sali, A.; Marquis, D.M.; Krilis, S.A.; Linnik, M.D. Use of single point mutations in domain I of beta 2-glycoprotein I to determine fine antigenic specificity of antiphospholipid autoantibodies. J. Immunol. 2002, 169, 7097–7103. [Google Scholar] [CrossRef]
- De Laat, B.; Derksen, R.; Urbanus, R.; De Groot, P. IgG antibodies that recognize epitope Gly40-Arg43 in domain I of beta 2-glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood 2005, 105, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Pericleous, C.; Ruiz-Limon, P.; Romay-Penabad, Z.; Marín, A.C.; Garza-Garcia, A.; Murfitt, L.; Driscoll, P.C.; Latchman, D.S.; Isenberg, D.A.; Giles, I.; et al. Proof-of-concept study demonstrating the pathogenicity of affinity-purified IgG antibodies directed to domain I of β2-glycoprotein I in a mouse model of anti-phospholipid antibody-induced thrombosis. Rheumatology (Oxford, England) 2015, 54, 722–727. [Google Scholar] [CrossRef]
- Ioannou, Y.; Romay-Penabad, Z.; Pericleous, C.; Giles, I.; Papalardo, E.; Vargas, G.; Shilagard, T.; Latchman, D.S.; Isenberg, D.A.; Rahman, A. In vivo inhibition of antiphospholipid antibody induced pathogenicity utilizing the antigenic target peptide domain I of beta2-glycoprotein I: Proof of concept. J. Thromb. Haemost. 2009, 7, 833–842. [Google Scholar] [CrossRef]
- De Laat, B.; Wu, X.X.; Van Lummel, M.; Derksen, R.H.W.M.; De Groot, P.G.; Rand, J.H. Correlation between antiphospholipid antibodies that recognize domain I of β2-glycoprotein I and a reduction in the anticoagulant activity of annexin A5. Blood 2007, 109, 1490–1494. [Google Scholar] [CrossRef]
- Gardiner, C.; Cohen, H.; Jenkins, A.; Machin, S.J.; Mackie, I.J. Detection of acquired resistance to activated protein C associated with antiphospholipid antibodies using a novel clotting assay. Blood Coagul. Fibrinolysis 2006, 17, 477–478. [Google Scholar] [CrossRef]
- Hulstein, J.J.; Lenting, P.J.; De Laat, B.; Derksen, R.H.; Fijnheer, R.; De Groot, P.G. beta2-Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood 2007, 110, 1483–1491. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Liu, X.; Espinola, R.; Zhu, M.; Harris, N.E.; Chen, P.P. Functional analyses of patient-derived IgG monoclonal anticardiolipin antibodies using in vivo thrombosis and in vivo microcirculation models. Thromb. Haemost. 2000, 84, 388–395. [Google Scholar] [CrossRef]
- Giles, I.; Pericleous, C.; Liu, X.; Ehsanullah, J.; Clarke, L.; Brogan, P.; Newton-West, M.; Swerlick, R.; Lambrianides, A.; Chen, P. Thrombin binding predicts the effects of sequence changes in a human monoclonal antiphospholipid antibody on its in vivo biologic actions. J. Immunol. 2009, 182, 4836–4843. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, C.; Durigutto, P.; Sblattero, D.; Borghi, M.O.; Grossi, C.; Guida, F.; Bulla, R.; Macor, P.; Pregnolato, F.; Meroni, P.L.; et al. A non complement-fixing antibody to beta2 glycoprotein I as a novel therapy to control abortions and thrombosis in antiphospholipid syndrome. Blood 2014, 123, 3478–3487. [Google Scholar] [CrossRef]
- Andreoli, L.; Nalli, C.; Motta, M.; Norman, G.L.; Shums, Z.; Encabo, S.; Binder, W.L.; Nuzzo, M.; Frassi, M.; Lojacono, A. Anti-b2-glycoprotein I IgG antibodies from 1-year-old healthy children born to mothers with systemic autoimmune diseases preferentially target domain 4/5: Might it be the reason for their ‘innocent’ profile? Ann. Rheum. Dis. 2011, 70, 380–383. [Google Scholar] [CrossRef]
- Giles, I.P.; Isenberg, D.A.; Latchman, D.S.; Rahman, A. How do antiphospholipid antibodies bind beta2-glycoprotein I? Arthritis Rheum. 2003, 48, 2111–2121. [Google Scholar] [CrossRef]
- Murthy, V.; Willis, R.; Romay-Penabad, Z.; Ruiz-Limón, P.; Martínez-Martínez, L.A.; Jatwani, S.; Jajoria, P.; Seif, A.; Alarcón, G.S.; Papalardo, E.; et al. Value of isolated IgA anti-β2 -glycoprotein I positivity in the diagnosis of the antiphospholipid syndrome. Arthritis Rheum. 2013, 65, 3186–3193. [Google Scholar] [CrossRef]
- Serrano, M.; Martinez-Flores, J.A.; Norman, G.L.; Naranjo, L.; Morales, J.M.; Serrano, A. The IgA Isotype of Anti-β2 Glycoprotein I Antibodies Recognizes Epitopes in Domains 3, 4, and 5 That Are Located in a Lateral Zone of the Molecule (L-Shaped). Front. Immunol. 2019, 7, 1031. [Google Scholar] [CrossRef]
- George, J.; Blank, M.; Levy, Y.; Meroni, P.; Damianovich, M.; Tincani, A.; Shoenfeld, Y. Differential effects of anti-beta2-glycoprotein I antibodies on endothelial cells and on the manifestations of experimental antiphospholipid syndrome. Circulation 1998, 97, 900–906. [Google Scholar] [CrossRef]
- Sutjita, M.; Hohmann, A.; Comacchio, R.; Boey, M.L.; Bradley, J. A common anti-cardiolipin antibody idiotype in autoimmune disease: Identification using a mouse monoclonal antibody directed against a naturally-occurring anti-phospholipid antibody. Clin. Exp. Immunol. 1989, 75, 211–216. [Google Scholar]
- Blank, M.; Shoenfeld, Y.; Cabilly, S.; Heldman, Y.; Fridkin, M.; Katchalski-Katzir, E. Prevention of experimental antiphospholipid syndrome and endothelial cell activation by synthetic peptides. Proc. Natl. Acad. Sci. USA 1999, 96, 5164–5168. [Google Scholar] [CrossRef]
- Agar, C.; Van Os, G.M.A.; Mörgelin, M.; Sprenger, R.R.; Marquart, J.A.; Urbanus, R.T.; Derksen, R.H.W.M.; Meijers, J.C.M.; De Groot, P.J. Beta2-glycoprotein I can exist in 2 conformations: Implications for our understanding of the antiphospholipid syndrome. Blood 2010, 116, 1336–1343. [Google Scholar] [CrossRef]
- Buchholz, I.; Nestler, P.; Koppen, S.; Delcea, M. Lysine residues control the conformational dynamics of beta 2-glycoprotein I. Phys. Chem. Chem. Phys. 2018, 20, 26819–26829. [Google Scholar] [CrossRef]
- Koike, T.; Ichikawa, K.; Kasahara, H.; Atsumi, T.; Tsutsumi, A.; Matsuura, E. Epitopes on beta2-GPI recognized by anticardiolipin antibodies. Lupus 1998, 7, 14–17. [Google Scholar] [CrossRef]
- Ruben, E.; Planer, W.; Chinnaraj, M.; Chen, Z.; Zuo, X.; Pengo, V.; De Filippis, V.; Alluri, R.K.; McCrae, K.R.; Macor, P.; et al. The J-elongated conformation of β2-glycoprotein I predominates in solution: Implications for our understanding of antiphospholipid syndrome. J. Biol. Chem. 2020, 295, 10794–10806. [Google Scholar] [CrossRef] [PubMed]
- Zavala-Cerna, M.G.; Martínez-García, E.A.; Torres-Bugarín, O.; Rubio-Jurado, B.; Riebeling, C.; Nava, A. The clinical significance of posttranslational modification of autoantigens. Clin. Rev. Allergy Immunol. 2014, 47, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.W.; Lee, K.J. Post-translational modifications and their biological functions: Proteomic analysis and sys- tematic approaches. J. Biochem. Mol. Biol. 2004, 37, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Buttari, B.; Profumo, E.; Capozzi, A.; Saso, L.; Sorice, M.; Riganò, R. Post-translational modifications of proteins in antiphospholipid antibody syndrome. Crit. Rev. Clin. Lab. Sci. 2019, 56, 511–525. [Google Scholar] [CrossRef]
- Ioannou, Y.; Zhang, J.Y.; Passam, F.H.; Rahgozar, S.; Qi, J.C.; Giannakopoulos, B.; Qi, M.; Yu, P.; Yu, D.M.; Hogg, P.J.; et al. Naturally occurring free thiols within beta 2-glycoprotein I in vivo: Nitrosylation, redox modification by endothelial cells, and regulation of oxidative stress-induced cell injury. Blood 2010, 116, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- Buttari, B.; Profumo, E.; Mattei, V.; Siracusano, A.; Ortona, E.; Margutti, P.; Salvati, B.; Sorice, M.; Riganò, R. Oxidized beta2-glycoprotein I induces human dendritic cell maturation and promotes a T helper type 1 response. Blood 2005, 106, 3880–3887. [Google Scholar] [CrossRef]
- Sorice, M.; Buttari, B.; Capozzi, A.; Profumo, E.; Facchiano, F.; Truglia, S.; Recalchi, S.; Alessandri, C.; Conti, F.; Misasi, R.; et al. Antibodies to age-β2 glycoprotein I in patients with anti-phospholipid antibody syndrome. Clin. Exp. Immunol. 2016, 184, 174–182. [Google Scholar] [CrossRef]
- Buttari, B.; Profumo, E.; Capozzi, A.; Facchiano, F.; Saso, L.; Sorice, M.; Riganò, R. Advanced glycation end products of human b2 glycoprotein I modulate the maturation and function of DCs. Blood 2011, 117, 6152–6161. [Google Scholar] [CrossRef]
- Nieminen, M.; Henttinen, T.; Merinen, M.; Marttila-Ichihara, F.; Eriksson, J.E.; Jalkanen, S. Vimentin function in lymphocyte adhesion and transcellular migration. Nat. Cell Biol. 2006, 8, 156–162. [Google Scholar] [CrossRef]
- Scally, S.W.; Petersen, J.; Law, S.C.; Dudek, N.L.; Nel, H.J.; Loh, K.L.; Wijeyewickrema, L.C.; Eckle, S.B.G.; Van Heemst, J.; Pike, R.N.; et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 2013, 210, 2569–2582. [Google Scholar] [CrossRef]
- Alessandri, C.; Agmon-Levin, N.; Conti, F.; Perricone, C.; Ortona, E.; Pendolino, M.; Capozzi, A.; Delunardo, F.; Mancini, R.; Truglia, S.; et al. Anti-mutated citrullinated vimentin antibodies in antiphospholipid syndrome: Diagnostic value and relationship with clinical features. Immunol. Res. 2017, 65, 24–531. [Google Scholar] [CrossRef]
- Scholz, P.; Auler, M.; Brachvogel, B.; Benzing, T.; Mallman, P.; Streichert, T.; Klatt, A.R. Detection of multiple annexin autoantibodies in a patient with recurrent miscarriages, fulminant stroke and seronegative antiphospholipid syndrome. Biochem. Med. (Zagreb) 2016, 26, 272–278. [Google Scholar] [CrossRef]
- Bruschi, M.; Petretto, A.; Vaglio, A.; Santucci, L.; Candiano, G.; Ghiggeri, G.M. Annexin A1 and Autoimmunity: From Basic Science to Clinical Applications. Int. J. Mol. Sci. 2018, 19, 1348. [Google Scholar] [CrossRef]
- Müller-Calleja, N.; Lackner, K.J. Mechanisms of Cellular Activation in the Antiphospholipid Syndrome. Semin. Thromb. Hemost. 2018, 44, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Ronda, N.; Raschi, E.; Borghi, M.O. Humoral autoimmunity against endothelium: Theory or reality? Trends Immunol. 2005, 26, 275–281. [Google Scholar] [CrossRef]
- Sorice, M.; Longo, A.; Capozzi, A.; Garofalo, T.; Misasi, R.; Alessandri, C.; Conti, F.; Buttari, B.; Riganò, R.; Ortona, E.; et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum. 2007, 56, 2687–2697. [Google Scholar] [CrossRef]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef]
- Lutters, B.C.; Derksen, R.H.; Tekelenburg, W.L.; Lenting, P.J.; Arnout, J.; De Groot, P. Dimers of β2-glycoprotein I increase platelet deposition to collagen via interaction with phospholipids and the apolipoprotein E receptor 2. J. Biol. Chem. 2003, 278, 33831–33838. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Bitton, A.; Nahary, L.; Arango, M.T.; Benhar, I.; Blank, M.; Shoenfeld, Y.; Chapman, J. Cross-reactivity between annexin A2 and Beta-2-glycoprotein I in animal models of antiphospholipidsyndrome. Immunol. Res. 2017, 65, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Meroni, P.L.; Raschi, E.; Testoni, C.; Parisio, A.; Borghi, M.O. Innate immunity in the antiphospholipid syndrome: Role of the Toll-like receptors in endothelial cell activation by antiphospholipid antibodies. Autoimm. Rev. 2004, 3, 510–515. [Google Scholar] [CrossRef]
- Cuadrado, M.J.; Lopez-Pedrera, C.; Khamashta, M.A.; Camps, M.T.; Tinahones, F.; Torres, A.; Hughes, G.R.; Velasco, F. Thrombosis in primary antiphospholipid syndrome: A pivotal role for monocyte tissue factor expression. Arthritis Rheum. 1997, 40, 834–841. [Google Scholar] [CrossRef]
- Conti, F.; Sorice, M.; Circella, A.; Alessandri, C.; Pittoni, V.; Caronti, B.; Calderaro, C.; Griggi, T.; Misasi, R.; Valesini, G. Beta-2-glycoprotein I expression on monocytes is increased in anti-phospholipid antibody syndrome and correlates with tissue factor expression. Clin. Exp. Immunol. 2003, 132, 509–516. [Google Scholar] [CrossRef]
- Capozzi, A.; Manganelli, V.; Riitano, G.; Recalchi, S.; Truglia, S.; Alessandri, C.; Longo, A.; Garofalo, T.; Misasi, R.; Valesini, G.; et al. Tissue factor over-expression in platelets of patients with anti-phospholipid syndrome: Induction role of anti-β2-GPI antibodies. Clin. Exp. Immunol. 2019, 196, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Prinz, N.; Clemens, N.; Strand, D.; Pütz, I.; Lorenz, M.; Daiber, A.; Stein, P.; Degreif, A.; Radsak, M.; Schild, H.; et al. Antiphospholipid antibodies induce translocation of TLR7 and TLR8 to the endosome in human monocytes and plasmacytoid dendritic cells. Blood 2011, 118, 2322–2332. [Google Scholar] [CrossRef]
- Saito, M.; Makino, Y.; Inoue, K.; Watanabe, Y.; Hoshi, O.; Kubota, T. Anti-DNA antibodies cross-reactive with β 2-glycoprotein I induce monocyte tissue factor through the TLR9 pathway. Immunol. Med. 2020, 1–12. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Blank, M.; Liu, X.; Espinola, R.; Fridkin, M.; Ostertag, M.V.; Harris, E.N.; Shoenfeld, Y. A peptide that shares similarity with bacterial antigens reverses thrombogenic properties of antiphospholipid antibodies in vivo. J. Autoimmun. 2004, 22, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.; Asherson, R.A.; Cervera, R.; Shoenfeld, Y. Antiphospholipid syndrome infectious origin. J. Clin. Immunol. 2004, 24, 12–23. [Google Scholar] [CrossRef]
- Underhill, D.M.; Ozinsky, A. Toll-like receptors: Key mediators of microbe detection. Curr. Opin. Immunol. 2002, 14, 103–110. [Google Scholar] [CrossRef]
- Colasanti, T.; Alessandri, C.; Capozzi, A.; Sorice, M.; Delunardo, F.; Longo, A.; Pierdominici, M.; Conti, F.; Truglia, S.; Siracusano, A.; et al. Autoantibodies specific to a peptide of β2-glycoprotein I cross-react with TLR4, inducing a proinflammatory phenotype in endothelial cells and monocytes. Blood 2010, 120, 3360–3370. [Google Scholar] [CrossRef]
- Lopez-Pedrera, C.; Buendia, P.; Cuadrado, M.J.; Siendones, E.; Anguirre, M.A.; Barbarroja, N.; Montiel-Duarte, C.; Torres, A.; Khamashta, M.; Velasco, F. Antiphospholipid antibodies from patients with the antiphospholipid syndrome induce monocyte tissue factor expression through the simultaneous activation of NF-kB/Rel proteins via the p38 mitogen-activated protein kinase pathway, and of the MEK-1/ERK pathway. Arthritis Rheum. 2006, 54, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zhou, H.; Wang, T.; Xie, Y.; Wang, T.; Wang, X.; Yan, J. Activation of mTOR is involved in anti-beta2GPI/beta2GPI-induced expression of tissue factor and IL-8 in monocytes. Thromb. Res. 2017, 157, 103–110. [Google Scholar] [CrossRef]
- Tambralli, A.; Gockman, K.; Knight, J.S. NETs in APS: Current Knowledge and Future Perspectives. Curr. Rheumatol. Rep. 2020, 22, 67. [Google Scholar] [CrossRef]
- Nilsson, I.M.; Astedt, B.; Hedner, U.; Berezin, D. Intrauterine death and circulating anticoagulant (“antithromboplastin”). Acta Med. Scand. 1975, 197, 153–159. [Google Scholar] [CrossRef]
- Hughes, G.R. The Prosser-White oration 1983. Connective tissue disease and the skin. Clin. Exp. Dermatol. 1984, 9, 535–544. [Google Scholar] [CrossRef]
- Rai, R.S.; Regan, L.; Clifford, K.; Pickering, W.; Dave, M.; Mackie, I.; McNally, T.; Cohen, H. Antiphospholipid antibodies and beta 2-glycoprotein-I in 500 women with recurrent miscarriage: Results of a comprehensive screening approach. Hum. Reprod. 1995, 10, 2001–2005. [Google Scholar] [CrossRef]
- Schreiber, K.; Hunt, B.J. Pregnancy and antiphospholipid syndrome. Semin. Thromb. Hemost. 2016, 42, 780–788. [Google Scholar] [CrossRef]
- Fischetti, F.; Durigutto, P.; Pellis, V.; Debeus, A.; Macor, P.; Bulla, R.; Bossi, F.; Ziller, F.; Sblattero, D.; Meroni, P. Thrombus formation induced by antibodies to beta2-glycoprotein I is complement dependent and requires a priming factor. Blood 2005, 106, 2340–2346. [Google Scholar] [CrossRef]
- Chamley, L.W.; Allen, J.L.; Johnson, P.M. Synthesis of beta2 glycoprotein 1 by the human placenta. Placenta. 1997, 18, 403–410. [Google Scholar] [CrossRef]
- Schreiber, K.; Sciascia, S.; De Groot, P.G.; Devreese, K.; Jacobsen, S.; Ruiz-Irastorza, G.; Salmon, J.E.; Shoenfeld, Y.; Shovman, O.; Hunt, B.J. Antiphospholipid syndrome. Nat. Rev. Dis. Primers 2018, 4, 17103. [Google Scholar] [CrossRef] [PubMed]
- Derksen, J.H.; De Groot, P.G. The obstetric antiphospholipid syndrome. J. Reprod. Immunol. 2008, 77, 41–50. [Google Scholar] [CrossRef]
- Chamley, L.W.; Duncalf, A.M.; Mitchell, M.D.; Johnson, P.M. Action of anticardiolipin and antibodies to beta2-glycoprotein-I on trophoblast proliferation as a mechanism for fetal death. Lancet 1998, 352, 1037–1038. [Google Scholar] [CrossRef]
- Carroll, T.Y.; Mulla, M.J.; Han, C.S.; Brosens, J.J.; Chamley, L.W.; Giles, I.; Pericleous, C.; Rahman, A.; Sfakianaki, A.K.; Paidas, M.J.; et al. Modulation of trophoblast angiogenic factor secretion by antiphospholipid antibodies is not reversed by heparin. Am. J. Reprod. Immunol. 2011, 66, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, V.; Gelber, S.E.; Vukelic, M.; Sacharidou, A.; Herz, J.; Urbanus, R.T.; De Groot, P.G.; Natale, D.R.; Harihara, A.; Redecha, P.; et al. ApoE Receptor 2 Mediation of Trophoblast Dysfunction and Pregnancy Complications Induced by Antiphospholipid Antibodies in Mice. Arthritis Rheumatol. 2016, 68, 730–739. [Google Scholar] [CrossRef]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef]
- Abrahams, V.M.; Chamley, L.W.; Salmon, J.E. Emerging Treatment Models in Rheumatology: Antiphospholipid Syndrome and Pregnancy: Pathogenesis to Translation. Arthritis Rheumatol. 2017, 69, 1710–1721. [Google Scholar] [CrossRef]
- Mulla, M.J.; Myrtolli, K.; Brosens, J.J.; Chamley, L.W.; Kwak-Kim, J.Y.; Paidas, M.J.; Abrahams, V.M. Antiphospholipid antibodies limit trophoblast migration by reducing IL-6 production and STAT3 activity. Am. J. Reprod. Immunol. 2010, 63, 339–348. [Google Scholar] [CrossRef]
- Marchetti, T.; Ruffatti, A.; Wuillemin, C.; De Moerloose, P.; Cohen, M. Hydroxychloroquine restores trophoblast fusion affected by antiphospholipid antibodies. J. Thromb. Haemost. 2014, 12, 910–920. [Google Scholar] [CrossRef]
- Viall, C.A.; Chamley, L.W. Histopathology in the placentae of women with antiphospholipid antibodies: A systematic review of the literature. Autoimmun. Rev. 2015, 14, 446–471. [Google Scholar] [CrossRef]
- Huppertz, B.; Kingdom, J.; Caniggia, I.; Desoye, G.; Black, S.; Korr, H.; Kaufmann, P. Hypoxia favours necrotic versus apoptotic shedding of placental syncytiotrophoblast into the maternal circulation. Placenta 2003, 24, 181–190. [Google Scholar] [CrossRef]
- Oku, K.; Nakamura, H.; Kono, M.; Ohmura, K.; Kato, M.; Bohgaki, T.; Horita, T.; Yasuda, S.; Amengual, O.; Atsumi, T. Complement and thrombosis in the antiphospholipid syndrome. Autoimmun. Rev. 2016, 15, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Girardi, G.; Berman, J.; Redecha, P.; Spruce, L.; Thurman, J.M.; Kraus, D.; Hollmann, T.J.; Casali, P.; Caroll, M.C.; Wetsel, R.A.; et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J. Clin. Investig. 2003, 112, 1644–1654. [Google Scholar] [CrossRef]
- Girardi, G.; Yarilin, D.; Thurman, J.M.; Holers, V.M.; Salmon, J.E. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J. Exp. Med. 2006, 203, 2165–2175. [Google Scholar] [CrossRef]
- Shamonki, J.M.; Salmon, J.E.; Hyjek, E.; Baergen, R.N. Excessive complement activation is associated with placental injury in patients with antiphospholipid antibodies. Am. J. Obstet. Gynecol. 2007, 196, e1–e5. [Google Scholar] [CrossRef]
- Berman, J.; Girardi, G.; Salmon, J.E. TNF-alpha is a critical effector and a target for therapy in antiphospholipid antibody-induced pregnancy loss. J. Immunol. 2005, 174, 485–490. [Google Scholar] [CrossRef]
- Holers, V.M.; Girardi, G.; Mo, L.; Guthridge, J.M.; Molina, H.; Pierangeli, S.S.; Espinola, R.; Xiaowei, L.E.; Mao, D.; Vialpando, C.G.; et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J. Exp. Med. 2002, 195, 211–220. [Google Scholar] [CrossRef]
- Girardi, G.; Redecha, P.; Salmon, J.E. Heparin prevents antiphospholipid antibody-induced fetal loss by inhibiting complement activation. Nat. Med. 2004, 10, 1222–1226. [Google Scholar] [CrossRef]
- Meroni, P.L.; Macor, P.; Durigutto, P.; De Maso, L.; Gerosa, M.; Ferraresso, M.; Borghi, M.O.; Mollnes, T.E.; Tedesco, F. Complement activation in antiphospholipid syndrome and its inhibition to prevent rethrombosis after arterial surgery. Blood 2016, 127, 365–367. [Google Scholar] [CrossRef]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: A newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef]
- Marder, W.; Knight, J.S.; Kaplan, M.J.; Somers, E.C.; Zhang, X.; O’Dell, A.A.; Padmanabhan, V.; Lieberman, R.W. Placental histology and neutrophil extracellular traps in lupus and pre-eclampsia pregnancies. Lupus Sci. Med. 2015, 3, e000134. [Google Scholar] [CrossRef]
- Truglia, S.; Capozzi, A.; Mancuso, S.; Recalchi, S.; Spinelli, F.R.; Perricone, C.; De Carolis, C.; Manganelli, V.; Riitano, G.; Garofalo, T.; et al. A monocentric cohort of obstetric seronegative antiphospholipid syndrome. Front. Immunol. 2018, 9, 1678. [Google Scholar] [CrossRef]
- Conti, F.; Capozzi, A.; Truglia, S.; Lococo, E.; Longo, A.; Misasi, R.; Alessandri, C.; Valesini, G.; Sorice, M. The Mosaic of “Seronegative” Antiphospholipid Syndrome. J. Immunol. Res. 2014, 2014, 389601. [Google Scholar] [CrossRef]
- Conti, F.; Alessandri, C.; Spinelli, F.R.; Capozzi, A.; Martinelli, F.; Recalchi, S.; Misasi, R.; Valesini, G.; Sorice, M. TLC immunostaining for detection of “antiphospholipid” antibodies. Methods. Mol. Biol. 2014, 1134, 95–101. [Google Scholar] [CrossRef]
- Zohoury, N.; Bertolaccini, M.L.; Rodriguez-Garcia, J.L.; Shums, Z.; Ateka-Barrutia, O.; Sorice, M.; Norman, G.L.; Khamashta, M. Closing the Serological Gap in the Antiphospholipid Syndrome: The Value of “Non-criteria” Antiphospholipid Antibodies. J. Rheumatol. 2017, 44, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Oku, K.; Amengual, O.; Zigon, P.; Horita, T.; Yasuda, S.; Atsumi, T. Essential role of the p38 mitogen-activated protein kinase pathway in tissue factor gene expression mediated by the phosphatidylserine-dependent antiprothrombin antibody. Rheumatology (Oxford, England) 2013, 52, 1775–1784. [Google Scholar] [CrossRef]
- Litvinova, E.; Darnige, L.; Kirilovsky, A.; Burnel, Y.; De Luna, G.; Dragon-Durey, M.A. Prevalence and Significance of Non-conventional Antiphospholipid Antibodies in Patients With Clinical APS Criteria. Front. Immunol. 2018, 9, 2971. [Google Scholar] [CrossRef] [PubMed]
- Bertolaccini, M.L.; Amengual, O.; Atsumietal, T.; Binder, W.L.; De Laat, B.; Forastiero, R.; Kutteh, W.H.; Lambert, M.; Matsubayashi, H.; Murthy, V. ‘Non-criteria’ aPL tests: Report of a task force and preconference workshop at the 13th International Congress on Antiphospholipid Antibodies, Galveston, TX, USA, April 2010. Lupus 2011, 20, 191–205. [Google Scholar] [CrossRef]
- Cockrell, E.; Espinola, R.G.; McCrae, K.R. Annexin A2: biology and relevance to the antiphospholipid syndrome. Lupus 2008, 17, 944–952. [Google Scholar] [CrossRef]
- Salle, V.; Schmidt, J.; Smail, A.; Mazière, C.; Conte, M.A.; Bruléc, A.; Mazière, J.C.; Cadet, E.; Herpee, Y.E.; Duhaut, P. Antibodies directed against annexin A2 and obstetric morbidity. J. Reprod. Immunol. 2016, 118, 50–53. [Google Scholar] [CrossRef]
- Romay-Penabad, Z.; Montiel-Manzano, M.G.; Shilagard, T.; Papalardo, E.; Vargas, G.; Deora, A.B.; Wang, M.; Jacovina, A.T.; Garcia-Latorre, E.; Reyes-Maldonado, E.; et al. Annexin A2 is involved in antiphospholipid antibody mediated pathogenic effects in vitro and in vivo. Blood 2009, 114, 3074–3083. [Google Scholar] [CrossRef]
- Hedhli, N.; Falcone, D.J.; Huang, B.; Cesarman-Maus, G.; Kraemer, R.; Zhai, H.; Tsirka, S.E.; Santambrogio, L.; Hajjar, K.A. The Annexin A2/S100A10 System in Health and Disease: Emerging Paradigms. J. Biomed. Biotechnolog. 2012, 2012. [Google Scholar] [CrossRef]
- Conti, F.; Alessandri, C.; Sorice, M.; Capozzi, A.; Longo, A.; Garofalo, T.; Misasi, R.; Bompane, D.; Hughes, G.R.V.; Khamashta, M.A.; et al. Thin-layer chromatography immunostaining in detecting anti-phospholipid antibodies in seronegative anti-phospholipid syndrome. Clin. Exp. Immunol. 2012, 167, 429–437. [Google Scholar] [CrossRef]
- Conti, F.; Priori, R.; Alessandri, C.; Misasi, R.; Capozzi, A.; Pendolino, M.; Truglia, S.; Frisenda, S.; Sorice, M.; Valesini, G. Diagnosis of catastrophic anti-phospholipid syndrome in a patient tested negative for conventional tests. Clin. Exp. Rheumatol. 2017, 35, 678–680. [Google Scholar]
- Nayfe, R.; Uthman, I.; Aoun, J.; Aldin, E.S.; Merashli, M.; Khamashta, M.A. Seronegative antiphospholipid syndrome. Rheumatology 2013, 52, 1358–1367. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Liu, X.W.; Barker, J.H.; Anderson, G.; Harris, E.N. Induction of thrombosis in a mouse model by IgG, IgM and IgA immunoglobulins from patients with the antiphospholipid syndrome. Thromb. Haemost. 1995, 74, 1361–1367. [Google Scholar] [CrossRef]
- Ruiz-García, R.; Serrano, M.; Martínez-Flores, J.Á.; Mora, S.; Morillas, L.; Martín-Mola, M.Á.; Morales, J.M.; Paz-Artal, E.; Serrano, A. Isolated IgA Anti-𝛽2 Glycoprotein I Antibodies in Patients with Clinical Criteria for Antiphospholipid Syndrome. J. Immunol. Res. 2014, 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Ettore, E.; Menichelli, D.; Pani, A.; Violi, F.; Pastori, D. Seronegative antiphospholipid syndrome: Refining the value of “non-criteria” antibodies for diagnosis and clinical management. Haematologica 2020, 105, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Shoenfeld, Y.; Twig, G.; Katz, U.; Sherer, Y. Autoantibody explosion in antiphospholipid syndrome. J. Autoimmun. 2008, 30, 74–83. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Non-Criteria aPL | Clinical Association | References |
|---|---|---|
| Anti-β2-GPI-DI | association with thrombosis and pregnancy complications (more than antibodies directed to other domains) | [53,54,55,56,57,58,59] |
| IgA anti-β2-GPI | correlation with thrombosis, miscarriages, pulmonary hypertension, seizure, thrombocitopenia and livedo reticularis | [62,143,145] |
| Anti-PT/PS | strongly correlation with thrombosis and obstetric manifestations; in association with other anti-PL contribute to assess the risk of thrombosis | [132,133,134,135] |
| TLC-immunostaining detection of anti-CL | TLC-immunostaining could potentially identify the presence of aPL in SN-APS; this is in association with vascular thrombosis | [130,140,141] |
| Anti-Vimentin/CL | correlation with thrombotic events and pregnancy morbidity | [13] |
| Anti-AnnA5 | clinical correlation with pregnancy-related morbidity is still controversial | [135] |
| Anti-AnnA2 | alter pro-fibrinolytic activity; correlate with thrombotic events | [136,137,138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misasi, R.; Longo, A.; Recalchi, S.; Caissutti, D.; Riitano, G.; Manganelli, V.; Garofalo, T.; Sorice, M.; Capozzi, A. Molecular Mechanisms of “Antiphospholipid Antibodies” and Their Paradoxical Role in the Pathogenesis of “Seronegative APS”. Int. J. Mol. Sci. 2020, 21, 8411. https://doi.org/10.3390/ijms21218411
Misasi R, Longo A, Recalchi S, Caissutti D, Riitano G, Manganelli V, Garofalo T, Sorice M, Capozzi A. Molecular Mechanisms of “Antiphospholipid Antibodies” and Their Paradoxical Role in the Pathogenesis of “Seronegative APS”. International Journal of Molecular Sciences. 2020; 21(21):8411. https://doi.org/10.3390/ijms21218411
Chicago/Turabian StyleMisasi, Roberta, Agostina Longo, Serena Recalchi, Daniela Caissutti, Gloria Riitano, Valeria Manganelli, Tina Garofalo, Maurizio Sorice, and Antonella Capozzi. 2020. "Molecular Mechanisms of “Antiphospholipid Antibodies” and Their Paradoxical Role in the Pathogenesis of “Seronegative APS”" International Journal of Molecular Sciences 21, no. 21: 8411. https://doi.org/10.3390/ijms21218411
APA StyleMisasi, R., Longo, A., Recalchi, S., Caissutti, D., Riitano, G., Manganelli, V., Garofalo, T., Sorice, M., & Capozzi, A. (2020). Molecular Mechanisms of “Antiphospholipid Antibodies” and Their Paradoxical Role in the Pathogenesis of “Seronegative APS”. International Journal of Molecular Sciences, 21(21), 8411. https://doi.org/10.3390/ijms21218411