Needle in a Haystack: The Naïve Repertoire as a Source of T Cell Receptors for Adoptive Therapy with Engineered T Cells



Abstract
:1. Introduction
2. Identification of Functional Tumor-Specific TCRs for Highly Personalized ACT
2.1. Clinical Success of TCR-Based ACT According to the Different Tumor-Antigen Classes
2.2. Targeting of Tumor Neoantigens Drives the Next-Generation ACT toward a Personalized Therapy
2.3. Identification of Functional Neoantigen-Specific TCRs
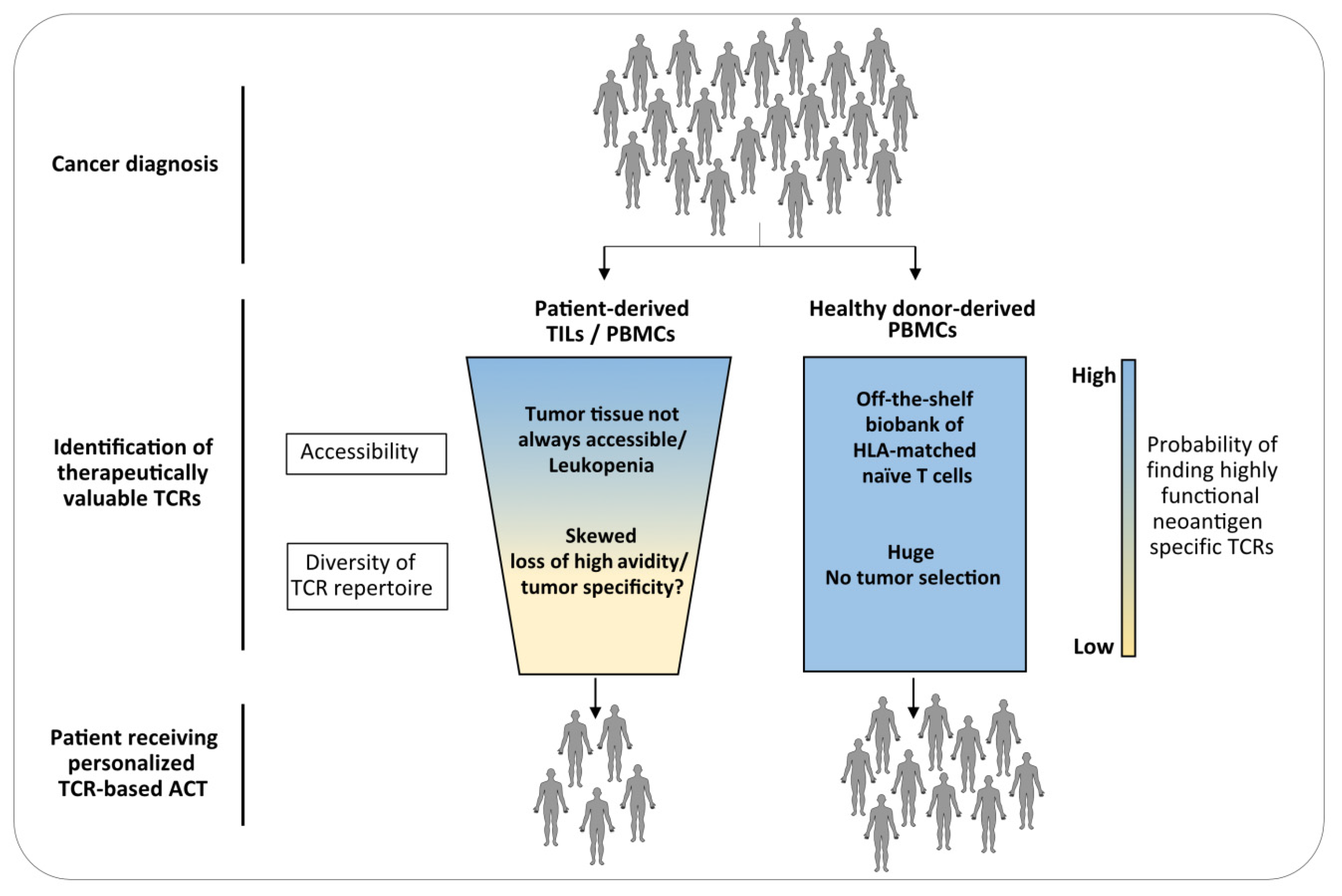
3. The Naïve Repertoire of Healthy Donors as Source for TCRs for ACT
4. Features of the Naïve Repertoire
4.1. Generation of the Naïve TCR Repertoire
4.2. Clone-Size Distribution
4.3. Avidity of the Naïve TCR Repertoire in the Context of Tumor Antigens
5. Identification of TCRs from the Naïve Repertoire: Challenges and Future Perspectives
5.1. Detection of Low-Frequency Antigen-Specific Naïve T Cells
5.2. High Abundance of Low Avidity TCRs
5.3. Preclinical Validation of TCR Functionality
5.4. Prediction of TCR Cross-Reactivity
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACT | Adoptive T cell therapy |
| APC | Antigen presenting cells |
| APL | Altered peptide ligand |
| HLA | Human leucocyte antigen |
| CDR | Complementary-determining region |
| NSPM | Nonsynonymous point mutations |
| MHC | Major histocompatibility complex |
| MS PBMCs | Mass spectrometry peripheral blood mononuclear cells |
| pMHC | Peptide-major histocompatibility complex |
| TAA | Tumor-associated antigen |
| TCR | T cell receptor |
| TILs | Tumor infiltrating lymphocytes |
| WES | Whole exome sequencing |
References
- Li, D.; Li, X.; Zhou, W.L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.D.; et al. Genetically engineered t cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4. [Google Scholar] [CrossRef]
- Neuenhahn, M.; Albrecht, J.; Odendahl, M.; Schlott, F.; Dössinger, G.; Schiemann, M.; Lakshmipathi, S.; Martin, K.; Bunjes, D.; Harsdorf, S.; et al. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia 2017, 31, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Ng, C.Y.C.; Loftin, S.; Smith, C.A.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995. [Google Scholar] [CrossRef]
- Tzannou, I.; Leen, A.M. Preventing stem cell transplantation-associated viral infections using T-cell therapy. Immunotherapy 2015, 7, 793–810. [Google Scholar] [CrossRef] [Green Version]
- Spiess, P.J.; Yang, J.C.; Rosenberg, S.A. In vivo antitumor activity of tumor-infiltrating lymphocytes expanded in recombinant interleukin-2. J. Natl. Cancer Inst. 1987, 79, 1067–1075. [Google Scholar] [PubMed]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Solomon, D.; Avis, F.P.; Chang, A.E.; Freerksen, D.L.; Linehan, W.M.; Lotze, M.T.; Robertson, C.N.; Seipp, C.A.; Simon, P. Immunotherapy of patients with advanced cancer using tumor-infiltrating lymphocytes and recombinant interleukin-2: A pilot study. J. Clin. Oncol. 1988, 6, 839–853. [Google Scholar] [CrossRef]
- Rohaan, M.W.; van den Berg, J.H.; Kvistborg, P.; Haanen, J.B.A.G. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: A viable treatment option. J. Immunother. Cancer 2018, 6, 102. [Google Scholar] [CrossRef]
- Lee, S.; Margolin, K. Tumor-infiltrating lymphocytes in melanoma. Curr. Oncol. Rep. 2012, 14, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.A.; Guillaume, P.; et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8 + T cells in immunotherapy-naive ovarian cancer. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2018, 25, 89–94. [Google Scholar] [CrossRef]
- Kalaora, S.; Wolf, Y.; Feferman, T.; Barnea, E.; Greenstein, E.; Reshef, D.; Tirosh, I.; Reuben, A.; Patkar, S.; Levy, R.; et al. Combined analysis of antigen presentation and T-cell recognition reveals restricted immune responses in melanoma. Cancer Discov. 2018, 8, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of shared and neoantigen-reactive T cells for adoptive cell therapy based on CD137 separation. Front. Immunol. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Inozume, T.; Hanada, K.I.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8++PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ding, Z.-Y. The Ways of Isolating Neoantigen-Specific T Cells. Front. Oncol. 2020, 10, 1347. [Google Scholar] [CrossRef]
- Bianchi, V.; Harari, A.; Coukos, G. Neoantigen-Specific Adoptive Cell Therapies for Cancer: Making T-Cell Products More Personal. Front. Immunol. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic t cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, K.; Müller, T.R.; Gökmen, F.; Grassmann, S.; Effenberger, M.; Poltorak, M.; Stemberger, C.; Schumann, K.; Roth, T.L.; Marson, A.; et al. Orthotopic replacement of T-cell receptor α- and β-chains with preservation of near-physiological T-cell function. Nat. Biomed. Eng. 2019, 3, 974–984. [Google Scholar] [CrossRef]
- Provasi, E.; Genovese, P.; Lombardo, A.; Magnani, Z.; Liu, P.Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Ahmadi, M.; King, J.W.; Xue, S.A.; Voisine, C.; Holler, A.; Wright, G.P.; Waxman, J.; Morris, E.; Stauss, H.J. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 2011, 118, 3528–3537. [Google Scholar] [CrossRef]
- Schober, K.; Müller, T.R.; Busch, D.H. Orthotopic T-Cell Receptor Replacement-An 《Enabler》 for TCR-Based Therapies. Cells 2020, 9, 1367. [Google Scholar] [CrossRef]
- Davis, M.M.; Boniface, J.J.; Reich, Z.; Lyons, D.; Hampl, J.; Arden, B.; Chien, Y. Ligand recognition by alpha beta T cell receptors. Annu. Rev. Immunol. 1998, 16, 523–544. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.-C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Rapoport, A.P.; Stadtmauer, E.A.; Binder-Scholl, G.K.; Goloubeva, O.; Vogl, D.T.; Lacey, S.F.; Badros, A.Z.; Garfall, A.; Weiss, B.; Finklestein, J.; et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 2015, 21, 914–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.T.; Scanlan, M.J.; Sahin, U.; Türeci, O.; Gure, A.O.; Tsang, S.; Williamson, B.; Stockert, E.; Pfreundschuh, M.; Old, L.J. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA 1997, 94, 1914–1918. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalían, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.J.G.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013. [Google Scholar] [CrossRef] [Green Version]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.J.W.; Lu, J.; Spencer, M.; Hopkins, F.; Tran, E.; Rosenberg, S.A.; Long, E.O.; Sun, P.D. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc. Natl. Acad. Sci. USA 2020, 117, 12826–12835. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [Green Version]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.-C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Cafri, G.; Yossef, R.; Pasetto, A.; Deniger, D.C.; Lu, Y.-C.; Parkhurst, M.; Gartner, J.J.; Jia, L.; Ray, S.; Ngo, L.T.; et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat. Commun. 2019, 10, 449. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Yossef, R.; Cafri, G.; Paria, B.C.; Lowery, F.J.; Jafferji, M.; Good, M.L.; Sachs, A.; Copeland, A.R.; Kim, S.P.; et al. Antigen experienced T cells from peripheral blood recognize p53 neoantigens. Clin. Cancer Res. 2020, 26, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- van der Lee, D.I.; Reijmers, R.M.; Honders, M.W.; Hagedoorn, R.S.; de Jong, R.C.M.; Kester, M.G.D.; van der Steen, D.M.; de Ru, A.H.; Kweekel, C.; Bijen, H.M.; et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J. Clin. Investig. 2019, 129, 774–785. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, W.; Zhou, B.; Su, Z.; Gu, X.; Zhou, Z.; Chen, S. TSNAdb: A Database for Tumor-specific Neoantigens from Immunogenomics Data Analysis. Genom. Proteom. Bioinforma. 2018, 16, 276–282. [Google Scholar] [CrossRef]
- Castle, J.C.; Uduman, M.; Pabla, S.; Stein, R.B.; Buell, J.S. Mutation-derived neoantigens for cancer immunotherapy. Front. Immunol. 2019, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Weenink, B.; van Brakel, M.; Wijers, R.; Sillevis Smitt, P.A.E.; French, P.J.; Debets, R. Lack of B and T cell reactivity towards IDH1(R132H) in blood and tumor tissue from LGG patients. J. Neurooncol. 2019, 144, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Zamora, A.E.; Crawford, J.C.; Allen, E.K.; Guo, X.Z.J.; Bakke, J.; Carter, R.A.; Abdelsamed, H.A.; Moustaki, A.; Li, Y.; Chang, T.C.; et al. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8+ T cell responses. Sci. Transl. Med. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.E.; Schotte, R.; Calis, J.J.A.; Behjati, S.; Velds, A.; Hilkmann, H.; El Atmioui, D.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for neoantigen identification. Front. Immunol. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalaora, S.; Barnea, E.; Merhavi-Shoham, E.; Qutob, N.; Teer, J.K.; Shimony, N.; Schachter, J.; Rosenberg, S.A.; Besser, M.J.; Admon, A.; et al. Use of HLA peptidomics and whole exome sequencing to identify human immunogenic neo-antigens. Oncotarget 2016, 7, 5110–5117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strønen, E.; Toebes, M.; Kelderman, S.; Van Buuren, M.M.; Yang, W.; Van Rooij, N.; Donia, M.; Böschen, M.L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Gee, M.H.; Han, A.; Lofgren, S.M.; Beausang, J.F.; Mendoza, J.L.; Birnbaum, M.E.; Bethune, M.T.; Fischer, S.; Yang, X.; Gomez-Eerland, R.; et al. Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell 2018, 172, 549–563.e16. [Google Scholar] [CrossRef] [Green Version]
- Kula, T.; Dezfulian, M.H.; Wang, C.I.; Abdelfattah, N.S.; Hartman, Z.C.; Wucherpfennig, K.W.; Lyerly, H.K.; Elledge, S.J. T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 2019, 178, 1016–1028.e13. [Google Scholar] [CrossRef]
- Joglekar, A.V.; Leonard, M.T.; Jeppson, J.D.; Swift, M.; Li, G.; Wong, S.; Peng, S.; Zaretsky, J.M.; Heath, J.R.; Ribas, A.; et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods 2019, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Bethune, M.T.; Wong, S.; Joglekar, A.V.; Leonard, M.T.; Wang, J.K.; Kim, J.T.; Cheng, D.; Peng, S.; Zaretsky, J.M.; et al. T cell antigen discovery via trogocytosis. Nat. Methods 2019, 16, 183–190. [Google Scholar] [CrossRef]
- Altman, J.D.; Davis, M.M. MHC-peptide tetramers to visualize antigen-specific T cells. Curr. Protoc. Immunol. 2016, 2016, 17.3.1–17.3.44. [Google Scholar] [CrossRef] [PubMed]
- Dössinger, G.; Bunse, M.; Bet, J.; Albrecht, J.; Paszkiewicz, P.J.; Weißbrich, B.; Schiedewitz, I.; Henkel, L.; Schiemann, M.; Neuenhahn, M.; et al. MHC Multimer-Guided and Cell Culture-Independent Isolation of Functional T Cell Receptors from Single Cells Facilitates TCR Identification for Immunotherapy. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.N.; Kishton, R.J.; Restifo, N.P. Developing neoantigen-targeted T cell–based treatments for solid tumors. Nat. Med. 2019, 25, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Rodenko, B.; Toebes, M.; Hadrup, S.R.; van Esch, W.J.E.; Molenaar, A.M.; Schumacher, T.N.M.; Ovaa, H. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 2006, 1, 1120–1132. [Google Scholar] [CrossRef]
- Toebes, M.; Coccoris, M.; Bins, A.; Rodenko, B.; Gomez, R.; Nieuwkoop, N.J.; Van De Kasteele, W.; Rimmelzwaan, G.F.; Haanen, J.B.A.G.; Ovaa, H.; et al. Design and use of conditional MHC class I ligands. Nat. Med. 2006, 12, 246–251. [Google Scholar] [CrossRef]
- Bakker, A.H.; Hoppes, R.; Linnemann, C.; Toebes, M.; Rodenko, B.; Berkers, C.R.; Hadrup, S.R.; Van Esch, W.J.E.; Heemskerk, M.H.M.; Ovaa, H.; et al. Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11, and -B7. Proc. Natl. Acad. Sci. USA 2008, 105, 3825–3830. [Google Scholar] [CrossRef] [Green Version]
- Frøsig, T.M.; Yap, J.; Seremet, T.; Lyngaa, R.; Svane, I.M.; Thor Straten, P.; Heemskerk, M.H.M.; Grotenbreg, G.M.; Hadrup, S.R. Design and validation of conditional ligands for HLA-B*08:01, HLA-B*15:01, HLA-B*35:01, and HLA-B*44:05. Cytom. Part A 2015, 87, 967–975. [Google Scholar] [CrossRef]
- Luimstra, J.J.; Garstka, M.A.; Roex, M.C.J.; Redeker, A.; Janssen, G.M.C.; van Veelen, P.A.; Arens, R.; Frederik Falkenburg, J.H.; Neefjes, J.; Ovaa, H. A flexible MHC class I multimer loading system for large-scale detection of antigen-specific T cells. J. Exp. Med. 2018, 215, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.K.; Ostermeir, K.; Ramnarayan, V.R.; Schuster, H.; Zacharias, M.; Springer, S. Dipeptides promote folding and peptide binding of MHC class I molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 15383–15388. [Google Scholar] [CrossRef] [Green Version]
- Saini, S.K.; Schuster, H.; Ramnarayan, V.R.; Rammensee, H.G.; Stevanović, S.; Springer, S. Dipeptides catalyze rapid peptide exchange on MHC class I molecules. Proc. Natl. Acad. Sci. USA 2015, 112, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Bentzen, A.K.; Marquard, A.M.; Lyngaa, R.; Saini, S.K.; Ramskov, S.; Donia, M.; Such, L.; Furness, A.J.S.; McGranahan, N.; Rosenthal, R.; et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 2016, 34, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Zaretsky, J.M.; Ng, A.H.C.; Chour, W.; Bethune, M.T.; Choi, J.; Hsu, A.; Holman, E.; Ding, X.; Guo, K.; et al. Sensitive Detection and Analysis of Neoantigen-Specific T Cell Populations from Tumors and Blood. Cell Rep. 2019, 28, 2728–2738.e7. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Ma, K.Y.; Schonnesen, A.A.; Zhang, M.; He, C.; Sun, E.; Williams, C.M.; Jia, W.; Jiang, N. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol. 2018, 36, 1156–1159. [Google Scholar] [CrossRef]
- Doubrovina, E.; Oflaz-Sozmen, B.; Prockop, S.E.; Kernan, N.A.; Abramson, S.; Teruya-Feldstein, J.; Hedvat, C.; Chou, J.F.; Heller, G.; Barker, J.N.; et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV + lymphomas after allogeneic hematopoietic cell transplantation. Blood 2012, 119, 2644–2656. [Google Scholar] [CrossRef]
- Hafezi, M.; Bertoletti, A.; Tan, A. Personalized T-cell therapy in liver transplanted patients with hepatitis B virus related hepatocellular carcinoma. Hepatoma Res. 2020, 2020. [Google Scholar] [CrossRef]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immunother. Cancer 2019, 7, 1–17. [Google Scholar] [CrossRef]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Jiang, Y.Z.; Xiong, Y.; Li, Y.J.; Li, X.W.; et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med. J. (Engl.) 2020, 133, 1015–1024. [Google Scholar] [CrossRef]
- Yossef, R.; Robbins, P.F.; Rosenberg, S.A.; Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef] [Green Version]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific in filtrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef]
- Gros, A.; Tran, E.; Parkhurst, M.R.; Ilyas, S.; Pasetto, A.; Groh, E.M.; Robbins, P.F.; Yossef, R.; Garcia-Garijo, A.; Fajardo, C.A.; et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Investig. 2019, 129, 4992–5004. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Petit, P.F.; Van den Eynde, B.J. Apoptosis of tumor-infiltrating T lymphocytes: A new immune checkpoint mechanism. Cancer Immunol. Immunother. 2019, 68, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Saff, R.R.; Spanjaard, E.S.; Hohlbaum, A.M.; Marshak-Rothstein, A. Activation-Induced Cell Death Limits Effector Function of CD4 Tumor-Specific T Cells. J. Immunol. 2004, 172, 6598–6606. [Google Scholar] [CrossRef]
- Ménétrier-Caux, C.; Ray-Coquard, I.; Blay, J.Y.; Caux, C. Lymphopenia in Cancer Patients and its Effects on Response to Immunotherapy: An opportunity for combination with Cytokines? J. Immunother. Cancer 2019, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, M.; Stengl, A.; Schober, K.; Gerget, M.; Kampick, M.; Müller, T.R.; Schumacher, D.; Helma, J.; Leonhardt, H.; Busch, D.H. FLEXamers: A Double Tag for Universal Generation of Versatile Peptide-MHC Multimers. J. Immunol. 2019, 202, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Messaoudi, I.; Guevara Patiño, J.A.; Dyall, R.; LeMaoult, J.; Nikolich-Žugich, J. Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science 2002, 298, 1797–1800. [Google Scholar] [CrossRef] [Green Version]
- Davenport, M.P.; Price, D.A.; McMichael, A.J. The T cell repertoire in infection and vaccination: Implications for control of persistent viruses. Curr. Opin. Immunol. 2007, 19, 294–300. [Google Scholar] [CrossRef]
- Chen, H.; Ndhlovu, Z.M.; Liu, D.; Porter, L.C.; Fang, J.W.; Darko, S.; Brockman, M.A.; Miura, T.; Brumme, Z.L.; Schneidewind, A.; et al. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 2012, 13, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Starenki, D.; Cooper, S.J.; Myers, R.M.; Li, Q. powerTCR: A model-based approach to comparative analysis of the clone size distribution of the T cell receptor repertoire. PLoS Comput. Biol. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Oltz, E.M.; Young, F.; Gorman, J.; Taccioli, G.; Chen, J. VDJ recombination. Immunol. Today 1992, 13, 306–314. [Google Scholar] [CrossRef]
- Jung, D.; Alt, F.W. Unraveling V(D)J Recombination: Insights into Gene Regulation. Cell 2004, 116, 299–311. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.D.; Bunker, J.J.; Erickson, S.A.; Oh-Hora, M.; Bendelac, A. Crossreactive αβ T Cell Receptors Are the Predominant Targets of Thymocyte Negative Selection. Immunity 2015, 43, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Jameson, S.C. Naive T cell homeostasis: From awareness of space to a sense of place. Nat. Rev. Immunol. 2009, 9, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Merkenschlager, M.; Graf, D.; Lovatt, M.; Bommhardt, U.; Zamoyska, R.; Fisher, A.G. How many thymocytes audition for selection? J. Exp. Med. 1997, 186, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Mora, T.; Walczak, A.M. Quantifying lymphocyte receptor diversity. Syst. Immunol. 2018, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Zarnitsyna, V.I.; Evavold, B.D.; Schoettle, L.N.; Blattman, J.N.; Antia, R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front. Immunol. 2013, 4, 485. [Google Scholar] [CrossRef] [Green Version]
- Arstila, T.P.; Casrouge, A.; Baron, V.; Even, J.; Kanellopoulos, J.; Kourilsky, P. A direct estimate of the human alphabeta T cell receptor diversity. Science 1999, 286, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Robins, H.S.; Campregher, P.V.; Srivastava, S.K.; Wacher, A.; Turtle, C.J.; Kahsai, O.; Riddell, S.R.; Warren, E.H.; Carlson, C.S. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009, 114, 4099–4107. [Google Scholar] [CrossRef]
- Qi, Q.; Liu, Y.; Cheng, Y.; Glanville, J.; Zhang, D.; Lee, J.Y.; Olshen, R.A.; Weyand, C.M.; Boyd, S.D.; Goronzy, J.J. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 2014, 111, 13139–13144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laydon, D.J.; Bangham, C.R.M.; Asquith, B. Estimating T-cell repertoire diversity: Limitations of classical estimators and a new approach. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [Green Version]
- Vanhanen, R.; Heikkilä, N.; Aggarwal, K.; Hamm, D.; Tarkkila, H.; Pätilä, T.; Jokiranta, T.S.; Saramäki, J.; Arstila, T.P. T cell receptor diversity in the human thymus. Mol. Immunol. 2016, 76, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.B. The effect of age on thymic function. Front. Immunol. 2013, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- den Braber, I.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mögling, R.; Bregje de Boer, A.; Willems, N.; Schrijver, E.H.R.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of Peripheral Naive T Cells Is Sustained by Thymus Output in Mice but Not Humans. Immunity 2012, 36, 288–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rane, S.; Hogan, T.; Seddon, B.; Yates, A.J. Age is not just a number: Naive T cells increase their ability to persist in the circulation over time. PLoS Biol. 2018, 16, 1–20. [Google Scholar] [CrossRef]
- Quigley, M.F.; Greenaway, H.Y.; Venturi, V.; Lindsay, R.; Quinn, K.M.; Seder, R.A.; Douek, D.C.; Davenport, M.P.; Price, D.A. Convergent recombination shapes the clonotypic landscape of the naive T-cell repertoire. Proc. Natl. Acad. Sci. USA 2010, 107, 19414–19419. [Google Scholar] [CrossRef] [Green Version]
- Pogorelyy, M.V.; Elhanati, Y.; Marcou, Q.; Sycheva, A.L.; Komech, E.A.; Nazarov, V.I.; Britanova, O.V.; Chudakov, D.M.; Mamedov, I.Z.; Lebedev, Y.B.; et al. Persisting fetal clonotypes influence the structure and overlap of adult human T cell receptor repertoires. PLoS Comput. Biol. 2017, 13, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Venturi, V.; Quigley, M.F.; Greenaway, H.Y.; Ng, P.C.; Ende, Z.S.; McIntosh, T.; Asher, T.E.; Almeida, J.R.; Levy, S.; Price, D.A.; et al. A Mechanism for TCR Sharing between T Cell Subsets and Individuals Revealed by Pyrosequencing. J. Immunol. 2011, 186, 4285–4294. [Google Scholar] [CrossRef] [PubMed]
- Alanio, C.; Lemaitre, F.; Law, H.K.W.; Hasan, M.; Albert, M.L. Enumeration of human antigen-specific naive CD8+ T cells reveals conserved precursor frequencies. Blood 2010, 115, 3718–3725. [Google Scholar] [CrossRef] [Green Version]
- de Greef, P.C.; Oakes, T.; Gerritsen, B.; Ismail, M.; Heather, J.M.; Hermsen, R.; Chain, B.; de Boer, R.J. The naive t-cell receptor repertoire has an extremely broad distribution of clone sizes. Elife 2020, 9, 1–24. [Google Scholar] [CrossRef]
- Venturi, V.; Price, D.A.; Douek, D.C.; Davenport, M.P. The molecular basis for public T-cell responses? Nat. Rev. Immunol. 2008, 8, 231–238. [Google Scholar] [CrossRef]
- Murugan, A.; Mora, T.; Walczak, A.M.; Callan, C.G. Statistical inference of the generation probability of T-cell receptors from sequence repertoires. Proc. Natl. Acad. Sci. USA 2012, 109, 16161–16166. [Google Scholar] [CrossRef] [Green Version]
- Venturi, V.; Kedzierska, K.; Price, D.A.; Doherty, P.C.; Douek, D.C.; Turner, S.J.; Davenport, M.P. Sharing of T cell receptors in antigen-specific responses is driven by convergent recombination. Proc. Natl. Acad. Sci. USA 2006, 103, 18691–18696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.Q.; Parker, P.; Ma, K.Y.; He, C.; Shi, Q.; Cui, Z.; Williams, C.M.; Wendel, B.S.; Meriwether, A.I.; Salazar, M.A.; et al. Direct measurement of T cell receptor affinity and sequence from naïve antiviral T cells. Sci. Transl. Med. 2016, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Schmid, D.A.; Irving, M.B.; Posevitz, V.; Hebeisen, M.; Posevitz-Fejfar, A.; Sarria, J.-C.F.; Gomez-Eerland, R.; Thome, M.; Schumacher, T.N.M.; Romero, P.; et al. Evidence for a TCR Affinity Threshold Delimiting Maximal CD8 T Cell Function. J. Immunol. 2010, 184, 4936–4946. [Google Scholar] [CrossRef]
- Zehn, D.; Lee, S.Y.; Bevan, M.J. Complete but curtailed T-cell response to very low-affinity antigen. Nature 2009, 458, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Busch, D.H.; Pamer, E.G. T cell affinity maturation by selective expansion during infection. J. Exp. Med. 1999, 189, 701–710. [Google Scholar] [CrossRef]
- Savage, P.A.; Boniface, J.J.; Davis, M.M. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity 1999, 10, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Reeves, E.; James, E. Antigen processing and immune regulation in the response to tumours. Immunology 2017, 150, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.J.; Andargachew, R.; Martinez, H.A.; Evavold, B.D. Low-affinity CD4+ T cells are major responders in the primary immune response. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hombrink, P.; Raz, Y.; Kester, M.G.D.; De Boer, R.; Weißbrich, B.; Von dem Borne, P.A.; Busch, D.H.; Schumacher, T.N.M.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Mixed functional characteristics correlating with TCR-ligand koff-rate of MHC-tetramer reactive T cells within the naive T-cell repertoire. Eur. J. Immunol. 2013, 43, 3038–3050. [Google Scholar] [CrossRef]
- Allard, M.; Couturaud, B.; Carretero-Iglesia, L.; Duong, M.N.; Schmidt, J.; Monnot, G.C.; Romero, P.; Speiser, D.E.; Hebeisen, M.; Rufer, N. TCR-ligand dissociation rate is a robust and stable biomarker of CD8+ T cell potency. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, D.T.; Alfei, F.; Roelli, P.; Barras, D.; Chennupati, V.; Darbre, S.; Delorenzi, M.; Pinschewer, D.D.; Zehn, D. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J. Exp. Med. 2016, 213, 1819–1834. [Google Scholar] [CrossRef]
- D’Ippolito, E.; Schober, K.; Nauerth, M.; Busch, D.H. T cell engineering for adoptive T cell therapy: Safety and receptor avidity. Cancer Immunol. Immunother. 2019, 68, 1701–1712. [Google Scholar] [CrossRef]
- Fahmy, T.M.; Bieler, J.G.; Edidin, M.; Schneck, J.P. Increased TCR avidity after T cell activation: A mechanism for sensing low-density antigen. Immunity 2001, 14, 135–143. [Google Scholar] [CrossRef]
- Aldridge, S.; Teichmann, S.A. Single cell transcriptomics comes of age. Nat. Commun. 2020, 11, 9–12. [Google Scholar] [CrossRef]
- Zheng, G.X.Y.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Subramaniam, M.; Targ, S.; Nguyen, M.; Maliskova, L.; McCarthy, E.; Wan, E.; Wong, S.; Byrnes, L.; Lanata, C.M.; et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018, 36, 89–94. [Google Scholar] [CrossRef]
- Xu, J.; Falconer, C.; Nguyen, Q.; Crawford, J.; McKinnon, B.D.; Mortlock, S.; Senabouth, A.; Andersen, S.; Chiu, H.S.; Jiang, L.; et al. Genotype-free demultiplexing of pooled single-cell RNA-seq. Genome Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Nauerth, M.; Weissbrich, B.; Knall, R.; Franz, T.; Dossinger, G.; Bet, J.; Paszkiewicz, P.J.; Pfeifer, L.; Bunse, M.; Uckert, W.; et al. TCR-Ligand koff Rate Correlates with the Protective Capacity of Antigen-Specific CD8+ T Cells for Adoptive Transfer. Sci. Transl. Med. 2013, 5, 192ra87. [Google Scholar] [CrossRef] [Green Version]
- Hinrichs, C.S.; Borman, Z.A.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Huang, J.; Klebanoff, C.A.; Johnson, L.A.; Kerkar, S.P.; Yang, S.; et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood 2011, 117, 808–814. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.H.; Kim, T.; Song, S.Y.; Park, S.; Cho, H.H.; Jung, S.-H.; Ahn, J.-S.; Kim, H.-J.; Lee, J.-J.; Kim, H.-O.; et al. Naïve CD8(+) T cell derived tumor-specific cytotoxic effectors as a potential remedy for overcoming TGF-β immunosuppression in the tumor microenvironment. Sci. Rep. 2016, 6, 28208. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Zhiya, Y.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naïve rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, F.K.M.; Ellinger, C.; Kieback, E.; Wilde, S.; Lietz, M.; Schendel, D.J.; Uckert, W. Unbiased Identification of T-Cell Receptors Targeting Immunodominant Peptide-MHC Complexes for T-Cell Receptor Immunotherapy. Hum. Gene Ther. 2017, 28, 1158–1168. [Google Scholar] [CrossRef] [Green Version]
- Rosskopf, S.; Leitner, J.; Paster, W.; Morton, L.T.; Hagedoorn, R.S.; Steinberger, P.; Heemskerk, M.H.M. A Jurkat 76 based triple parameter reporter system to evaluate TCR functions and adoptive T cell strategies. Oncotarget 2018, 9, 17608–17619. [Google Scholar] [CrossRef] [Green Version]
- Corr, M.; Slanetz, A.E.; Boyd, L.F.; Jelonek, M.T.; Khilko, S.; Al-Ramadi, B.K.; Kim, Y.S.; Maher, S.E.; Bothwell, A.L.; Margulies, D.H. T cell receptor-MHC class I peptide interactions: Affinity, kinetics, and specificity. Science 1994, 265, 946–949. [Google Scholar] [CrossRef]
- Hebeisen, M.; Schmidt, J.; Guillaume, P.; Baumgaertner, P.; Speiser, D.E.; Luescher, I.; Rufer, N. Identification of rare high-avidity, tumor-reactive CD8+ T Cells by Monomeric TCR-ligand off-rates measurements on living cells. Cancer Res. 2015, 75, 1983–1991. [Google Scholar] [CrossRef] [Green Version]
- Nauerth, M.; Stemberger, C.; Mohr, F.; Weißbrich, B.; Schiemann, M.; Germeroth, L.; Busch, D.H. Flow cytometry-based TCR-ligand Koff-rate assay for fast avidity screening of even very small antigen-specific T cell populations ex vivo. Cytom. Part A 2016, 89, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Abad, J.D.; Wrzensinski, C.; Overwijk, W.; De Witte, M.A.; Jorritsma, A.; Hsu, C.; Gattinoni, L.; Cohen, C.J.; Paulos, C.M.; Palmer, D.C.; et al. T-cell receptor gene therapy of established tumors in a murine melanoma model. J. Immunother. 2008, 31, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Zhang, W.; Guo, J.; Wei, X.; Phiwpan, K.; Zhang, J.; Zhou, X. Improved Transgenic Mouse Model for Studying HLA Class i Antigen Presentation. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Want, M.Y.; Konstorum, A.; Huang, R.Y.; Jain, V.; Matsueda, S.; Tsuji, T.; Lugade, A.; Odunsi, K.; Koya, R.; Battaglia, S. Neoantigens retention in patient derived xenograft models mediates autologous T cells activation in ovarian cancer. Oncoimmunology 2019, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kim, S.; Hundal, J.; Herndon, J.M.; Li, S.; Petti, A.A.; Soysal, S.D.; Li, L.; McLellan, M.D.; Hoog, J.; et al. Breast cancer neoantigens can induce CD8+ T-cell responses and antitumor immunity. Cancer Immunol. Res. 2017, 5, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, H.; Lindberg, M.F.; Donia, M.; Söderberg, E.M.V.; Andersen, R.; Keller, U.; Ny, L.; Svane, I.M.; Nilsson, L.M.; Nilsson, J.A. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun. 2017, 8, 707. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesisin a dish: Modeling development and disease using organoid technologies. Science 2014, 345. [Google Scholar] [CrossRef]
- Lou, Y.-R.; Leung, A.W. Next generation organoids for biomedical research and applications. Biotechnol. Adv. 2018, 36, 132–149. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [Green Version]
- Schnalzger, T.E.; de Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.T.; Alférez, D.G.; Amant, F.; Annibali, D.; Arribas, J.; Biankin, A.V.; Bruna, A.; Budinská, E.; Caldas, C.; Chang, D.K.; et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat. Rev. Cancer 2017, 17, 254–268. [Google Scholar] [CrossRef]
- Shi, J.; Li, Y.; Jia, R.; Fan, X. The fidelity of cancer cells in PDX models: Characteristics, mechanism and clinical significance. Int. J. Cancer 2020, 146, 2078–2088. [Google Scholar] [CrossRef]
- Biernacki, M.A.; Foster, K.A.; Woodward, K.B.; Coon, M.E.; Cummings, C.; Cunningham, T.M.; Dossa, R.G.; Brault, M.; Stokke, J.; Olsen, T.M.; et al. CBFB-MYH11 fusion neoantigen enables T cell recognition and killing of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 5127–5141. [Google Scholar] [CrossRef] [PubMed]
- Bentzen, A.K.; Hadrup, S.R. T-cell-receptor cross-recognition and strategies to select safe T-cell receptors for clinical translation. Immuno Oncol. Technol. 2019, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sewell, A.K. Why must T cells be cross-reactive? Nat. Rev. Immunol. 2012, 12, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Mason, D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol. Today 1998, 19, 395–404. [Google Scholar] [CrossRef]
- Wooldridge, L.; Ekeruche-Makinde, J.; Van Den Berg, H.A.; Skowera, A.; Miles, J.J.; Tan, M.P.; Dolton, G.; Clement, M.; Llewellyn-Lacey, S.; Price, D.A.; et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 2012, 287, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Rist, M.J.; Hibbert, K.M.; Croft, N.P.; Smith, C.; Neller, M.A.; Burrows, J.M.; Miles, J.J.; Purcell, A.W.; Rossjohn, J.; Gras, S.; et al. T Cell Cross-Reactivity between a Highly Immunogenic EBV Epitope and a Self-Peptide Naturally Presented by HLA-B*18:01+ Cells. J. Immunol. 2015, 194, 4668–4675. [Google Scholar] [CrossRef] [Green Version]
- Zehn, D.; Bevan, M.J. T cells with low avidity for a tissue-restricted antigen routinely evade central and peripheral tolerance and cause autoimmunity. Immunity 2006, 25, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Cole, D.K.; Bulek, A.M.; Dolton, G.; Schauenberg, A.J.; Szomolay, B.; Rittase, W.; Trimby, A.; Jothikumar, P.; Fuller, A.; Skowera, A.; et al. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J. Clin. Investig. 2016, 126, 2191–2204. [Google Scholar] [CrossRef] [Green Version]
- Lang, H.L.E.; Jacobsen, H.; Ikemizu, S.; Andersson, C.; Harlos, K.; Madsen, L.; Hjorth, P.; Sondergaard, L.; Svejgaard, A.; Wucherpfennig, K.; et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 2002, 3, 940–943. [Google Scholar] [CrossRef] [PubMed]
- de Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijen, H.M.; van der Steen, D.M.; Hagedoorn, R.S.; Wouters, A.K.; Wooldridge, L.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Preclinical Strategies to Identify Off-Target Toxicity of High-Affinity TCRs. Mol. Ther. 2018, 26, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, M.E.; Mendoza, J.L.; Sethi, D.K.; Dong, S.; Glanville, J.; Dobbins, J.; Özkan, E.; Davis, M.M.; Wucherpfennig, K.W.; Garcia, K.C. Deconstructing the peptide-MHC specificity of t cell recognition. Cell 2014, 157, 1073–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzen, A.K.; Such, L.; Jensen, K.K.; Marquard, A.M.; Jessen, L.E.; Miller, N.J.; Church, C.D.; Lyngaa, R.; Koelle, D.M.; Becker, J.C.; et al. T cell receptor fingerprinting enables in-depth characterization of the interactions governing recognition of peptide-MHC complexes. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef] [Green Version]
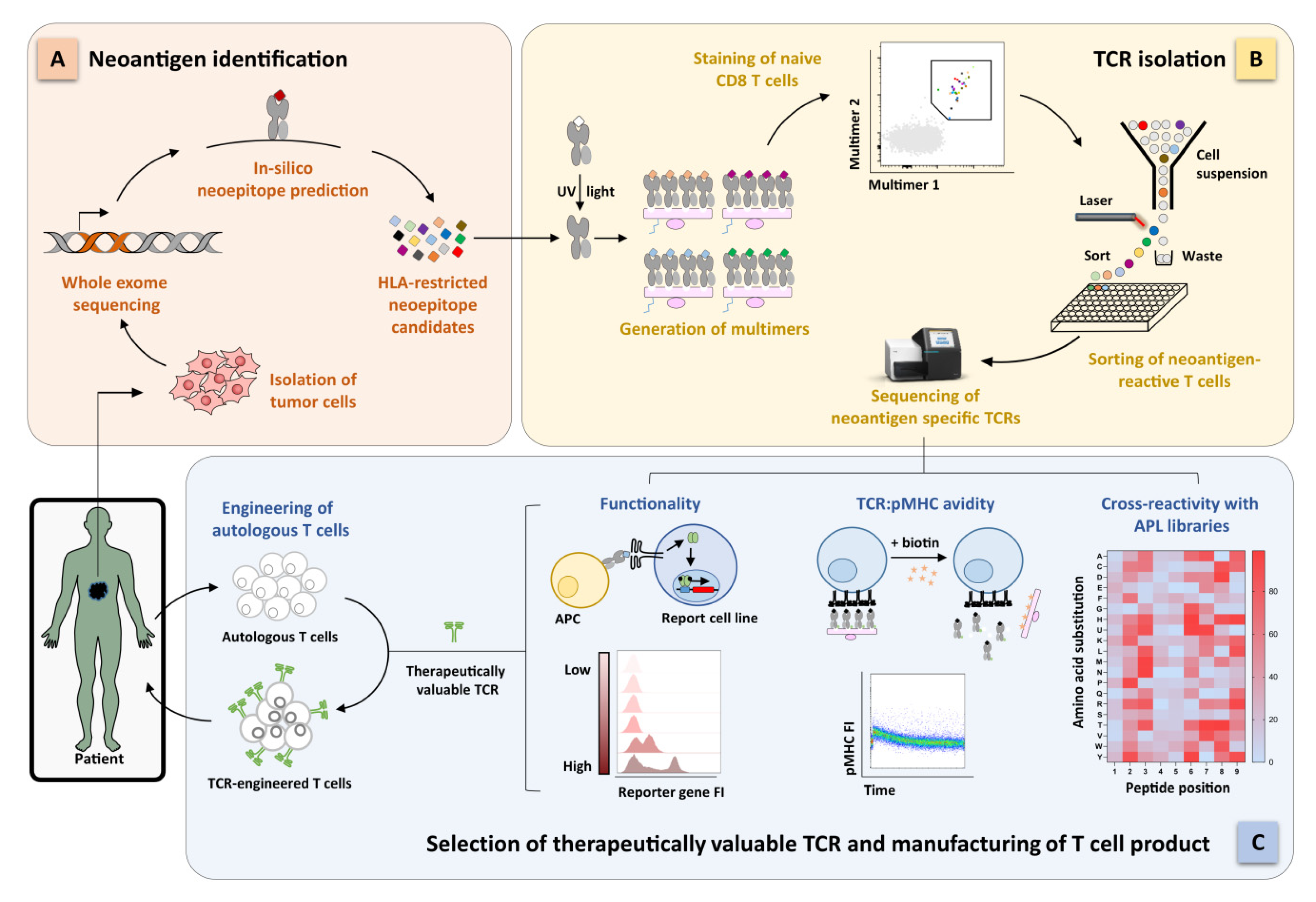

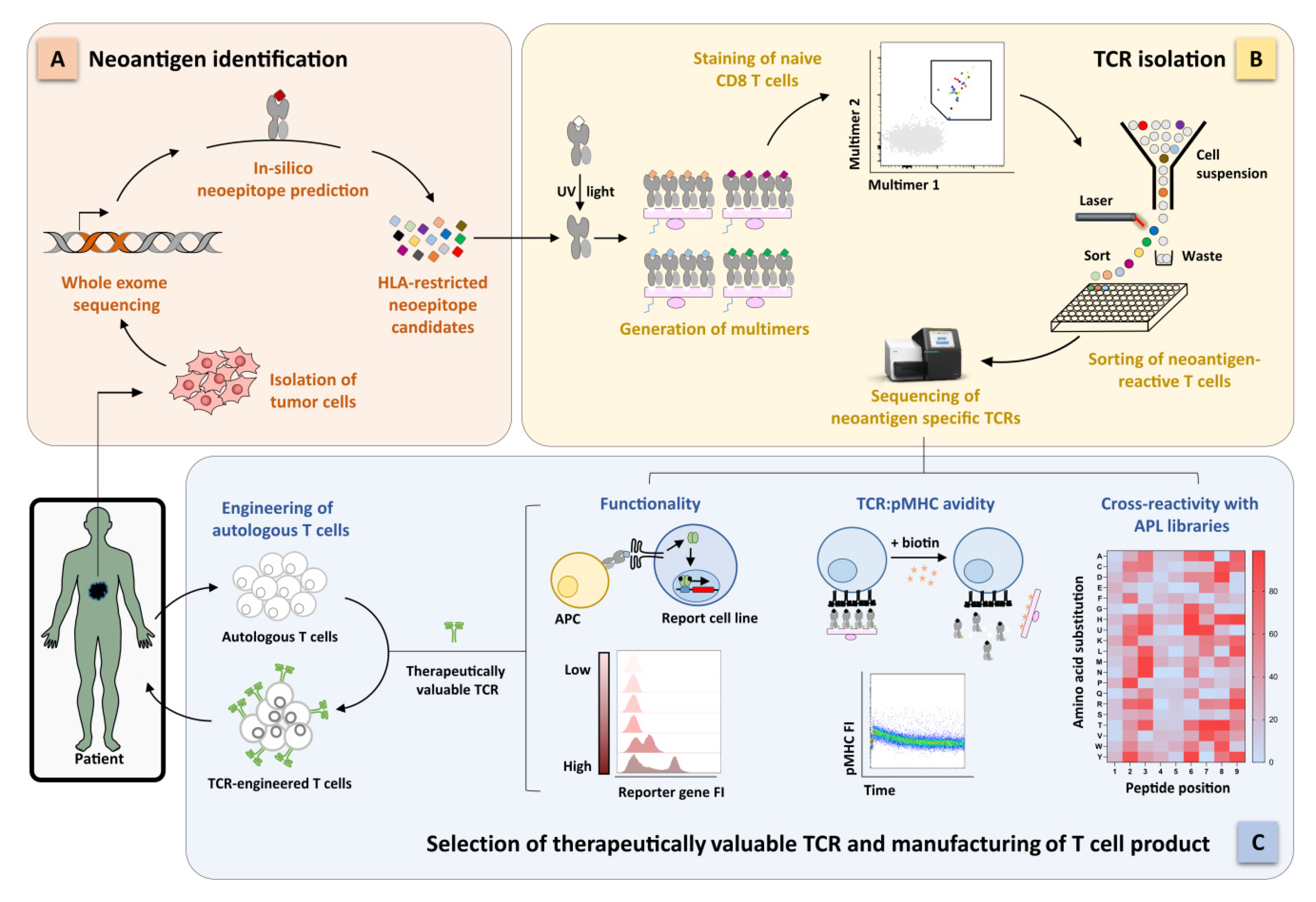{kind=link}
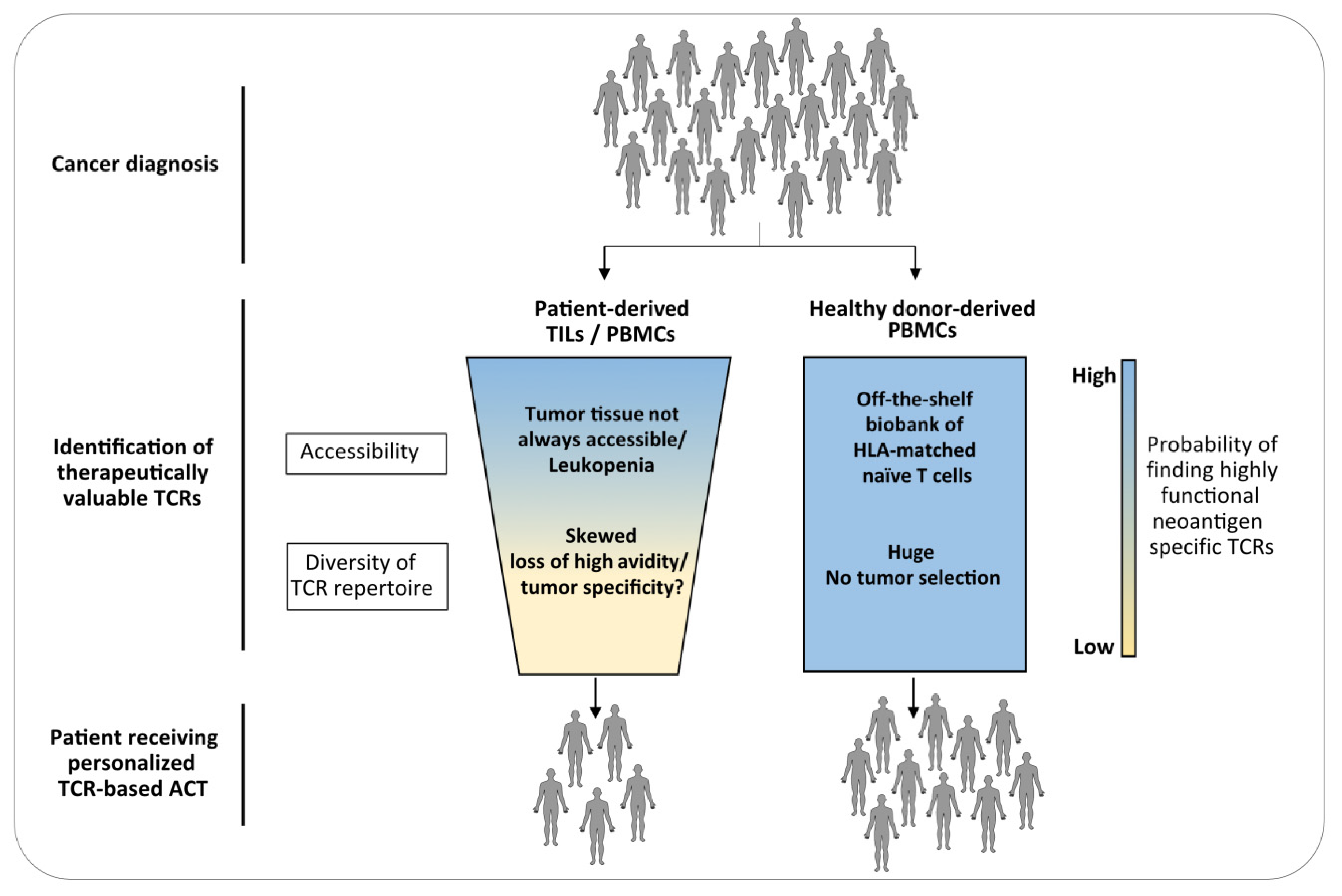{kind=link}
| Strategies | Advantages | Disadvantages | Suitable for Personalized ACT |
|---|---|---|---|
| Identification of Neoantigens | |||
| Whole exome sequencing | Fast and high-throughput | No information on epitope presentation and immunogenicity | Yes |
| Mass cytometry of HLA-ligandome | Identify naturally HLA-presented antigens | Require sophisticated equipment Low sensitivity | No |
| In silico peptide prediction | Easily accessible | Prediction tools are not always accurate, in particular for HLAs with low frequency | Yes |
| pMHC yeast library | Precise neoantigen target and direct TCR identification | Neglects endogenous antigen processing and lacks functional readout | No |
| Engineered APCs | Physiological neoantigen presentation. Functional readout | Dependency on predefined antigen library | No |
| Trogocytosis | Simultaneous identification of TCR and neoantigen | Dependency on predefined antigen library Lack of functional readout | No |
| Identification of Neoantigens-Specific TCRs | |||
| pMHC multimer libraries | High versatility and throughput | Lack of functional readout (pMHC multimer staining does not correlate with T cell functionality) | Yes |
| Autologous T-cell function assays | No HLA restrictions | Highly dependent on phenotypic status at the time of isolation Potential bias by the in vitro culture | No |
| Functional Characterization of TCRs | |||
| Primary T cells | Physiological T cell signaling, identical to infusion product | Variability in cellular phenotype | No |
| Reporter cell lines | High throughput, standardized TCR validation | Less physiological, less sensitive for subtle differences between TCRs | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ippolito, E.; Wagner, K.I.; Busch, D.H. Needle in a Haystack: The Naïve Repertoire as a Source of T Cell Receptors for Adoptive Therapy with Engineered T Cells. Int. J. Mol. Sci. 2020, 21, 8324. https://doi.org/10.3390/ijms21218324
D’Ippolito E, Wagner KI, Busch DH. Needle in a Haystack: The Naïve Repertoire as a Source of T Cell Receptors for Adoptive Therapy with Engineered T Cells. International Journal of Molecular Sciences. 2020; 21(21):8324. https://doi.org/10.3390/ijms21218324
Chicago/Turabian StyleD’Ippolito, Elvira, Karolin I. Wagner, and Dirk H Busch. 2020. "Needle in a Haystack: The Naïve Repertoire as a Source of T Cell Receptors for Adoptive Therapy with Engineered T Cells" International Journal of Molecular Sciences 21, no. 21: 8324. https://doi.org/10.3390/ijms21218324