Targeting the JAK2/STAT3 Pathway—Can We Compare It to the Two Faces of the God Janus?



Abstract
:1. Muscle Cachexia
2. STAT3 Protein
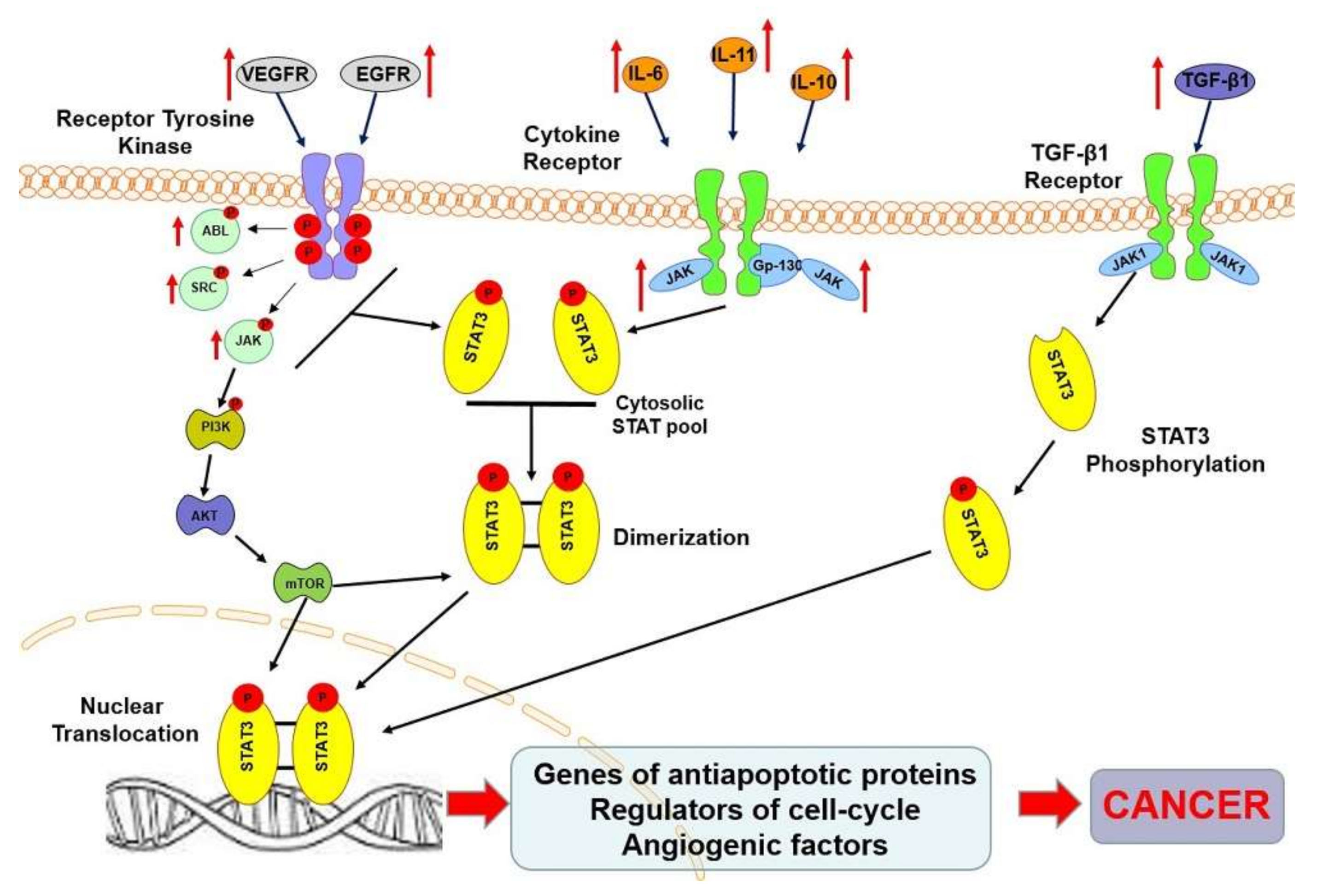
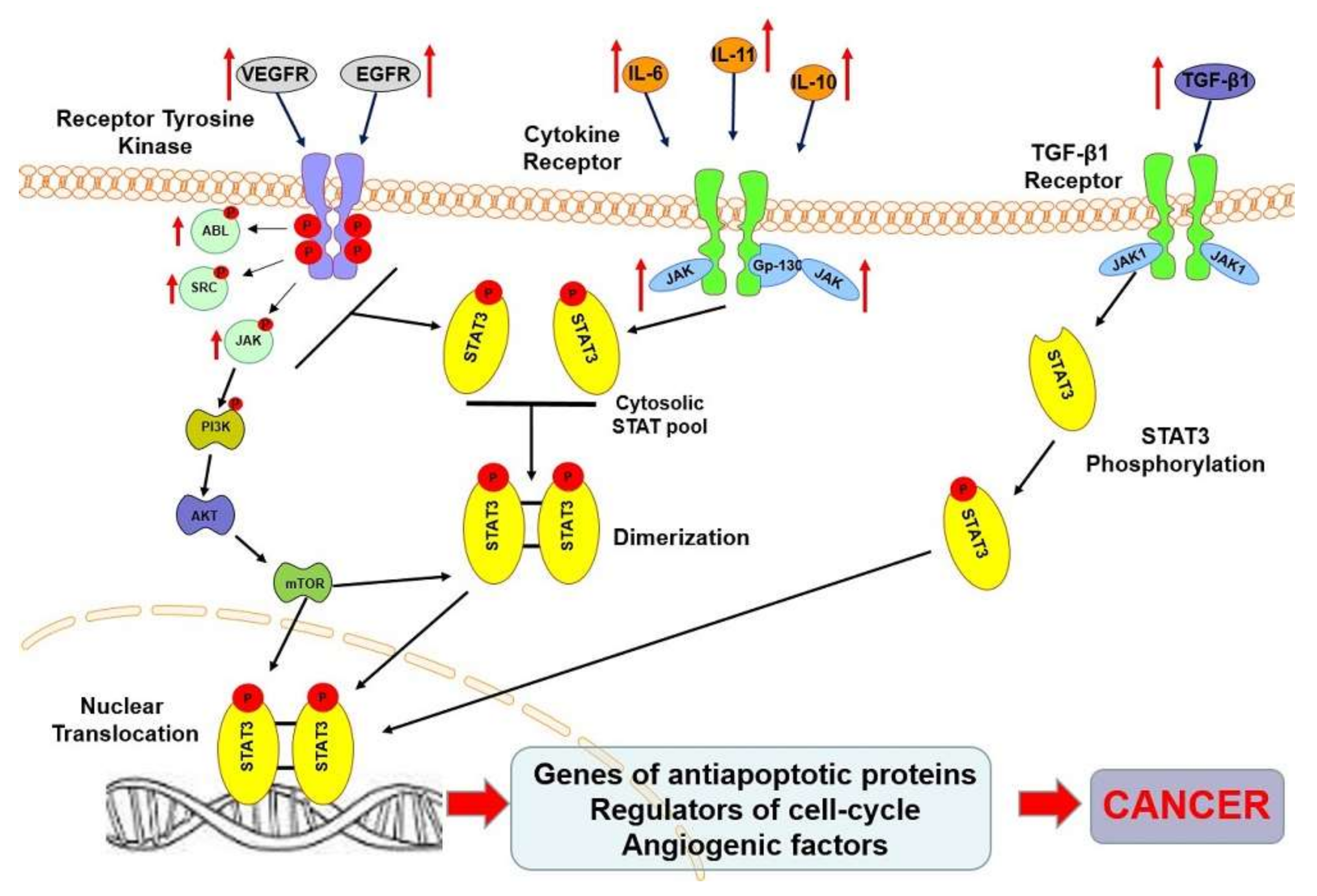
3. STAT3 Protein in Oncogenesis
4. STAT3 Protein in Skeletal Muscle
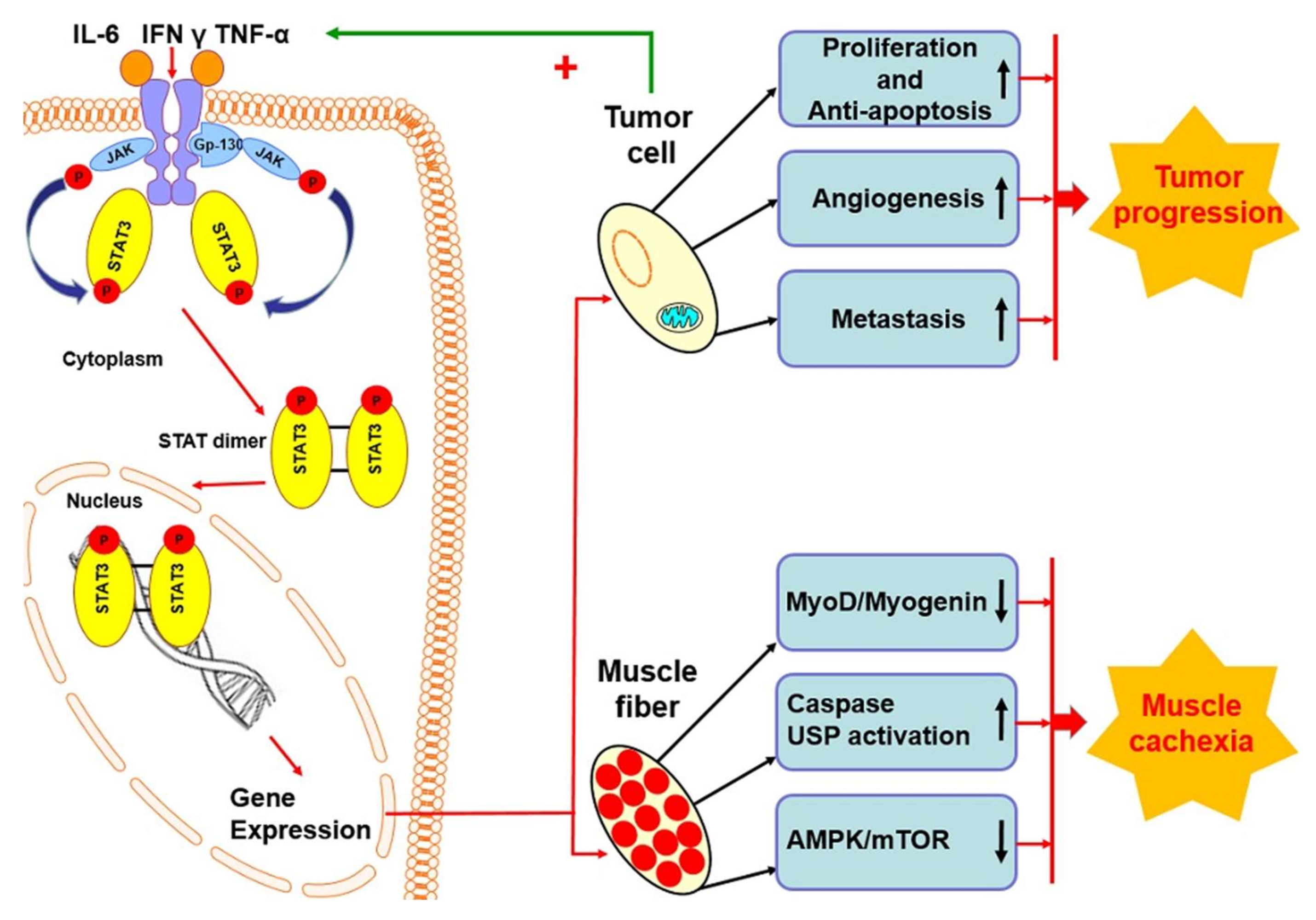
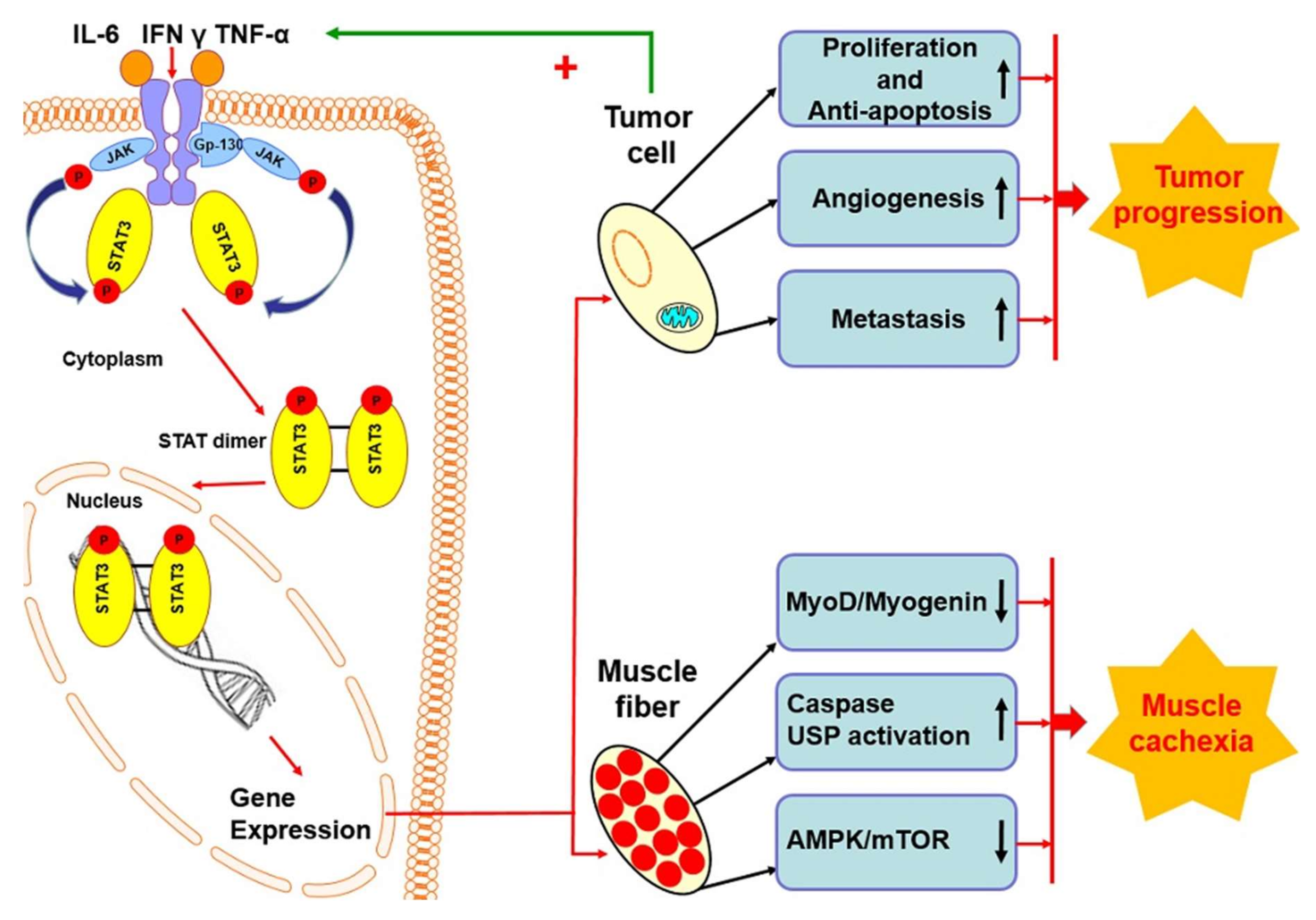
5. STAT3 Protein in Cancer Cachexia
6. Synthetic STAT3 Inhibitors in Anticancer Therapy
7. Natural STAT3 Inhibitors
8. Perspectives
Funding
Conflicts of Interest
References
- Donohoe, C.L.; Ryan, A.M.; Reynolds, J.V. Cancer Cachexia: Mechanisms and clinical implications. Gastroenterol. Res. Pract. 2011, 2011, 601434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siren, P.M.A.; Siren, M.J. Systemic zinc redistribution and dyshomeostasis in cancer cachexia. J. Cachexia Sarcopenia Muscle 2010, 1, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabiec, K.; Burchert, M.; Milewska, M.; Błaszczyk, M.; Grzelkowska-Kowalczyk, K. Systemic and local mechanisms leading to cachexia in cancer. Postępy Hig. Med. Dos. 2013, 67, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Dhanapal, R.; Saraswathi, T.R.; Govind, R.N. Cancer cachexia. J. Oral Maxillofac. Pathol. 2011, 15, 257–260. [Google Scholar] [CrossRef]
- Onesti, J.K.; Guttridge, D.C. Inflammation based regulation of cancer cachexia. Biomed. Res. Int. 2014, 168407. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, B.; Liang, C.; Li, Y.; Song, Y.H. Cytokine signalling in skeletal muscle wasting. Trends Endrocrinol. Metab. 2016, 27, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Khal, J.; Wyke, S.M.; Russell, S.T.; Hine, A.V.; Tisdale, M.J. Expression of the ubiquitin-proteasome pathway and muscle loss in experimental cancer cachexia. Br. J. Cancer 2005, 93, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Scherbakov, N.; Doehner, W. Cachexia as a common characteristic in multiple chronic diseases. J. Cachexia Sarcopenia Muscle 2019, 9, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Tarin, D. Update on clinical and mechanistic aspects of paraneoplastic syndromes. Cancer Metastasis Rev. 2013, 32, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Ezeoke, C.C.; Morley, J. Pathophysiology of anorexia in the cancer cachexia syndrome. J. Cachexia Sarcopenia Muscle 2015, 6, 287–302. [Google Scholar] [CrossRef] [Green Version]
- Akamizu, T.; Kangawa, K. Ghrelin for cachexia. J. Cachexia Sarcopenia Muscle 2010, 1, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, S.; Farsijani, S. Cancer cachexia and diabetes: Similarities in metabolic alterations and possible treatment. Appl. Physiol. Nutr. Metab. 2013, 39. [Google Scholar] [CrossRef]
- Tuca, A.; Jimenez-Fonseca, P.; Gascon, P. Clinical evaluation and optimal management of cancer cachexia. Crit. Rev. Oncol. Hemat. 2013, 88, 625–636. [Google Scholar] [CrossRef]
- Penna, F.; Busquets, S.; Toledo, M.I.; Pin, F.; Massa, D.; Lopez-Soriano, F.J.; Costelli, P.; Argiles, J.M. Erythropoietin administration partially prevent adipose tissue loss in experimental cancer cachexia models. J. Lipid Res. 2013, 54, 3045–3051. [Google Scholar] [CrossRef] [Green Version]
- Gullett, N.P.; Mazurak, V.; Hebbar, G.; Ziegler, T. Nutritional interventions for cancer-induced cachexia. Curr. Probl. Cancer. 2011, 35, 58–90. [Google Scholar] [CrossRef] [Green Version]
- Fagard, R.; Metelev, V.; Ines, S.; Baran-Marszak, F. STAT3 inhibitors for cancer therapy. Have all roads been explored? JAK-STAT 2013, 2, e22882. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Crescenzo, R.; Abate, F.; Lasora, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccartolla, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015, 7, 516–532. [Google Scholar] [CrossRef] [Green Version]
- Brachet-Botineau, M.; Polomski, M.; Neubauer, H.; Juen, L.; Hedou, D.; Viaud-Massurd, M.-C.; Prie, G.; Gouilleux, F. Pharmacological inhibition of oncogenic STAT3 and STAT5 signaling in hematopoietic cancers. Cancers 2020, 12, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siveen, K.S.; Sikka, S.; Surana, R.; Di, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.H.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochem. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009, 16, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhang, Y.; Sun, J. STAT3 activation in infection and infection-associated cancer. Mol. Cell. Endocrinol. 2017, 451, 80–87. [Google Scholar] [CrossRef]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment. JAK-STAT3 Signal. 2013, 2. [Google Scholar] [CrossRef] [Green Version]
- Gaudagin, E.; Mazala, D.; Chen, Y.W. STAT3 in skeletal muscle function and disorders. Int. J. Mol. Sci. 2018, 19, 2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, E.D.; Keseru, G.M.; Gunning, P.T.; Moriggl, R. Targeting STAT3 and STAT5 in cancer. Cancers 2020, 12, 2002. [Google Scholar] [CrossRef]
- Tierney, M.T.; Aydogdu, T.; Sala, D.; Malecova, B.; Gatto, S.; Puri, P.L.; Latella, L.; Sacco, A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 2014, 20, 1182–1186. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Xiao, F.; Wang, G.; Wei, X.; Jiang, L.; Chen, Y.; Zhu, L.; Wang, H.; Diao, Y.; Wang, H.; et al. STAT3 regulates the self-renewal of adult muscle satellite cells during injury-induced muscle regeneration. Cell Rep. 2016, 16, 2102–2115. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.Q.; Xu, H.; Wu, Q.; Liu, X.; Lufei, C.; Xu, X.Q.; Fu, X.Y. STAT3-inducible mouse ESCs: A model to study the role of STAT3 in ESC maintenance and lineage differentiation. Stem Cells Int. 2018, 2018, 8632950. [Google Scholar] [CrossRef]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF- κβ-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [Green Version]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Nunes, A.M.; Wuebbles, R.D.; Sarathy, A.; Fontelonga, T.M.; Deries, M.; Burkin, D.J.; Thorsteinsdottir, S. Impaired fetal muscle development and JAK-STAT activation mark disease onset and progression in a mouse model for merosin-deficient congenital muscular dystrophy. Hum. Mol. Gen. 2017, 26, 2018–2033. [Google Scholar] [CrossRef]
- Wada, E.; Tanihata, J.; Iwamura, A.; Takeda, S.; Hayashi, Y.; Matsuda, R. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet. Muscle 2017, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Li, T.; Azuelos, I.; Giordano, C.; Liang, H.; Hussain, S.; Matecki, S.; Petrof, B. Ventilator-induced diaphragmatic dysfunction in MDX mice. Muscle Nerve 2018, 57, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Chen, J.; Hu, C.; Teng, M.; Jiao, K.; Shen, Z.; Yue, J.; Zhong, L.; Li, Y. The ROS-mediated activation of the IL-6/STAT3 signaling pathway is involved in the 27-hydroxycholesterol-induced cellular senescence in nerve cells. Toxicol. In Vitro 2017, 45, 10–18. [Google Scholar] [CrossRef]
- Benito, C.; Davis, C.M.; Gomez-Sanchez, J.A.; Turmaine, M.; Meijer, D.; Poli, V.; Rhona, M.; Jessen, K.R. STAT3 controls the long-term survival and phenotype of repair Schwann cells during nerve regeneration. J. Neurosci. 2017, 37, 4255–4269. [Google Scholar] [CrossRef]
- Liao, X.H.; Xiang, Y.; Li, H.; Zheng, D.L.; Xu, Y.; Yu, C.X.; Li, J.P.; Zhang, X.Y.; Xing, W.B.; Cao, D.S.; et al. VEGF-A stimulates STAT3 activity via nitrosylation of myocardium to regulate the expression of vascular smooth muscle cell differentiation markers. Sci. Rep. 2017, 7, 2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lin, A.; Jiang, N.; Yan, H.; Ni, Z.; Qian, J.; Fang, W. Interleukin-6 trans-signaling induces vascular endothelial growth factor synthesis partly via Janus kinases-STAT3 pathway in human mesothelial cells. Nephrology 2017, 22, 150–158. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of Il-6 in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metabol. 2012, 303, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Watchorn, T.M.; Waddell, I.; Dowidar, N.; Ross, J.A. Proteolysis-inducing factor regulates hepatic gene expression via the transcription factors NF-(kappa)B and STAT3. FASEB J. 2001, 15, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.A.; Skipworth, R.J.E.; Fearon, K.C. Cachexia, survival, and the acute phase response. Curr. Opin. Supp. Palliat. Care 2008, 2, 267–274. [Google Scholar] [CrossRef]
- White, J.P.; Bayness, J.W.; Welle, S.L.; Kostek, M.C.; Matesic, L.E.; Sato, S.; Carson, J.A. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the ApcMin/+ mouse. PLoS ONE 2011, 6, e24650. [Google Scholar] [CrossRef] [Green Version]
- Mehl, K.A.; Davis, M.; Berger, F.G.; Carson, J.A. Myofiber degeneration/regeneration is induced in the cachectic ApcMin/+ mouse. J. Appl. Physiol. 2005, 99, 2379–2387. [Google Scholar] [CrossRef]
- Dent, J.R.; Byron, H.; Shahriar, T.; Sathe, A.; LaBarge, S.A.; Svensson, K.; McCurdy, C.E.; Schenk, S. Skeletal muscle mitochondrial function and exercise capacity are not impaired in mice with knockout of STAT3. J. Appl. Physiol. 2019, 127, 1117–1127. [Google Scholar] [CrossRef]
- Belizario, J.E.; Fontes-Oliveira, C.C.; Borges, J.P.; Kashiabara, J.A.; Vannier, E. Skeletal muscle wasting and renewal: A pivotal role of myokine Il-6. Springerplus 2016, 5, 619. [Google Scholar] [CrossRef] [Green Version]
- Narsale, A.A.; Carson, J.A. Role of IL-6 in cachexia–therapeutic implications. Curr. Opin. Support. Palliat. Care 2015, 8, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Mendes, M.C.S.; Pimentel, G.D.; Costa, F.O.; Carvalheira, J.B. Molecular and neuroendocrine mechanisms of cancer cachexia. J. Endocrinol. 2015, 226, R29–R43. [Google Scholar] [CrossRef] [Green Version]
- Pijet, B.; Pijet, M.; Litwiniuk, A.; Gajweska, M.; Pająk, B.; Orzechowski, A. TNF-α and IFN-s-Dependent muscle decay is linked to NF-κβ and STAT-1α-stimulated Atrogin1 and MURF1 genes in C2C12 myotubes. Mediat. Inflamm. 2013, 2013, 171437. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.F.; Sanchez, B.J.; Hall, D.T.; Tremblay, A.M.K.; Di Marco, S.; Gallouzi, I.E. STAT3 promotes IFN γ/TNF-α induced muscle wasting in an NF-κβ-dependent and IL-6-independent manner. EMBO Mol. Med. 2017, 9, 622–637. [Google Scholar] [CrossRef]
- Lee, H.; Jeong, A.J.; Ye, S.K. Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep. 2019, 52, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.; Cabell, L.A.; Schaefer, T.S.; McMurray, J.S. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003, 13, 633–636. [Google Scholar] [CrossRef]
- Turkson, J.; Zhang, S.; Palmer, J.; Kay, H.; Stanko, J.; Mora, L.B.; Sebti, S.; Yu, H.; Jove, R. Inhibition of constitutive signal transducer and activator of transcription 3 activation by novel platinum complexes with potent antitumor activity. Mol. Cancer Ther. 2004, 3, 1533–1542. [Google Scholar]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Wang, R.; Wang, S.; Lin, J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4700–4705. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Zhou, F.; Zhang, R.; Claret, F.X. STAT3 inhibitor Stattic exhibits potent antitumor activity and induces chemo- and radio-sensitivity in nasopharyngeal carcinoma. PLoS ONE 2013, 8, e54565. [Google Scholar] [CrossRef] [Green Version]
- Jinn, N.; Zhu, Q.; Yuan, P.; Li, Y.; Mao, L.; Tweardy, D.J. Targeting signal transducer and activator of transcription 3 with G-quartet oligonucleotides: A potential novel therapy for head and neck cancer. Mol. Cancer Ther. 2006, 5, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Handle, F.; Puhr, M.; Schaefer, G.; Lorito, N.; Hoefer, J.; Gruber, M.; Guggenberger, F.; Santer, F.R.; Marques, R.B.; van Weerden, W.M.; et al. The STAT3 inhibitor Galiellalactone reduces IL-6-mediated AR activity in benign and malignant prostate models. Mol. Cancer Ther. 2018, 17, 2722–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkson, J.; Zhang, S.; Mora, L.B.; Burns, A.; Sebti, S.; Jove, R. A novel platinium compound inhibits constitutive Stat3 signaling and induces cell cycle arrest and apoptosis of malignant cells. J. Biol. Chem. 2005, 280, 32979–32988. [Google Scholar] [CrossRef] [Green Version]
- Nagel-Wolfrum, K.; Buerger, C.; Wittig, I.; Butz, K.; Hoppe-Seyler, F.; Groner, B. The interaction of specific peptide aptamers with the DNA binding domain and the dimerization domain of the transcription factor Stat3 inhibits transactivation and induces apoptosis in tumor cells. Mol. Cancer Res. 2004, 2, 170–182. [Google Scholar]
- Trecul, A.; Morceau, F.; Dicato, M.; Diederich, M. Dietary compounds as potent inhibitors of the signal transducers and activators of transcription (STAT)3 regulatory network. Genes Nutr. 2012, 7, 111–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assi, H.H.; Paran, C.; VanderVeen, N.; Savakus, J.; Doherty, R.; Petruzzella, E.; Hoeschele, J.D.; Appelman, H.; Raptis, L.; Mikkelsen, T.; et al. Preclinical characterization of signal molecule inhibitors for primary and metastatic brain cancer therapy. J. Pharmacol. Exp. Ther. 2014, 349, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Sayyah, J.; Sayeski, P.P. Jak2 inhibitors: Rationale and role as therapeutic agents in hematologic malignancies. Curr. Oncol. Rep. 2009, 11, 117–124. [Google Scholar] [CrossRef]
- Horiguchi, A.; Asano, T.; Kuoda, K.; Sato, A.; Asakuma, J.; Ito, K.; Hayakawa, M.; Sumitomo, M.; Asano, T. STAT3 inhibitor WP1066 as a novel therapeutic agent for renal cell carcinoma. Br. J. Cancer 2010, 102, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Tsujita, Y.; Horiguchi, A.; Tasaki, S.; Isono, M.; Asano, T.; Ito, K.; Asano, T.; Mayumi, Y.; Kushibiki, T. STAT3 inhibition by WP1066 suppresses the growth and invasiveness of bladder cancer cells. Oncol. Rep. 2017, 38, 2197–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamaru, A.; Szymanowski, S.; Iwado, E.; Aoki, H.; Yokoyama, T.; Fokt, I.; Hess, K.; Conrad, C.; Madden, T.; Sawaya, R.; et al. A novel inhibitor of the STAT3 pathway induces apoptosis in malignant glioma cells both in vitro and in vivo. Oncogene 2007, 26, 2435–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, S.F.; Kong, L.Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007, 67, 3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.T.; Xue, J.; Chou, P.C.; Zhou, A.; Yang, P.; Conrad, C.A.; Aldape, K.D.; Priebe, W.; Patterson, C.; Sawaya, R.; et al. STAT3 orchestrates the interaction between endothelial and tumor cells and inhibition of STAT3 suppresses brain metastasis of breast cancer cells. Oncotarget 2015, 6, 10016–10029. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 plays a critical role in KRAS- induced pancreatic tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Grant, T.J.; Hua, K.; Singh, A. Molecular pathogenesis of pancreatic cancer. Prog. Mol. Biol. Transl. Sci. 2016, 144, 241–275. [Google Scholar] [CrossRef]
- Guha, S.; Chakraborty, A.; Szymanski, S.; Fokt, I.; Abbruzzese, J.; Kazerooni, R.; Madden, T.; Priebe, W. WP1066, a potent inhibitor of Jak2/STAT3 pathway inhibits pancreatic tumor growth both in vitro and in vivo. Cancer Res. 2007, 67, 2393. [Google Scholar]
- Wang, Y.; Wang, H.; Yao, H.; Li, C.; Fang, J.Y.; Xu, J. Regulation of PD-L1: Emerging routes for targeting tumor immune evasion. Front. Pharmacol. 2018, 9, 536. [Google Scholar] [CrossRef]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013, e166. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, L.; Genini, D.; Laurini, E.; Merulla, J.; Perez, L.; Fermeglia, M.; Carbone, G.M.; Pricl, S.; Catapano, C. Hitting the right spot: Mechanism of action of OPB-31121, a novel and potent inhibitor of Signal Transducer and Activator of Transcription 3 (STAT3). Mol. Oncol. 2015, 9, 1194–1206. [Google Scholar] [CrossRef] [Green Version]
- Ryan, E.T. Antiparasitic Agents. Principles and Practice of Pediatric Infectious Diseases, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Nelson, E.A.; Sharma, S.V.; Settleman, J.; Frank, D.A. A chemical biology approach to developing STAT inhibitors: Molecular strategies for accelerating clinical translation. Oncotarget 2011, 2, 518–524. [Google Scholar] [CrossRef]
- Friedman, J.F.; Kurtis, J.D.; Ramadhan, M.; Opollo, M.; Lanar, D.E.; Duffy, P.E. Malaria is related to decreased nutritional status among male adolescents and adults in the setting of intense perennial transmission. J. Inf. Dis. 2003, 188, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Onwuamaegbu, M.E.; Henein, M.Y.; Stewart Coats, A.J. Cachexia in malaria and heart failure: Therapeutic considerations in clinical practice. Postgrad. Med. J. 2004, 80, 642–649. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.A.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT)3 inhibitor, in patients with refractory solid malignancies. Orig. Artic. Early Drug Dev. 2015, 5, 998–1005. [Google Scholar] [CrossRef]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenii, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, L.; Lahiri, T.; Cammer, M.; Levy, D.E. OPB-51602, a direct inhibitor of STAT3, impairs growth and viability of respiratory Complex I. Cell Papers 2020, 3575125. [Google Scholar] [CrossRef]
- Hirpara, J.L.; Higuchi, K.; Tsunoda, T.; Goh, B.C.; Pervaiz, S. OPB-51602: A novel STAT3 inhibitor that targets mitochondrial respiratory chain and triggers STAT3 dependent ROS production. Exp. Mol. Ther. 2016, 76, 833. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Dong, Y.; Chen, Z.; Eckols, T.K.; Kasembeli, M.M.; Tweardy, D.J.; Mitch, W.E. Pharmacokinetics and pharmacodynamics of TTI-101, a STAT3 inhibitor that blocks muscle proteolysis in rats with chronic kidney disease. Am. J. Physiol. Renal Physiol. 2020, 319, F84–F92. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.M.; Grothey, A. Napabucasin: An update on the first-in-class cancer stemness inhibitor. Drugs 2017, 77, 1091–1103. [Google Scholar] [CrossRef]
- Hitron, M.; Stephenson, J.; Chi, K.N.; Edenfield, W.J.; Leggett, D.; LiWei, L.Y.; Gada, K.; Li, C. A phase 1b study of the cancer stem cell inhibitor BBI608 administered with paclitaxel in patients with advanced malignancies. J. Clin. Oncol. 2014, 32, 2530. [Google Scholar] [CrossRef]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; Chang, W.H.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Shih, S.C.; Chen, Y.J. Caffeic acid phenethyl ester induces apoptosis of human pancreatic cancer cells involving caspase and mitochondrial dysfunction. Pancreatology 2008, 8, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of antitumor effects in preclinical models of human breast cancer. Cancer Lett. 2011, 308, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.J.; Shih, S.C.; Wang, H.Y.; Lin, C.C.; Liu, C.Y.; Wang, T.E.; Chu, C.H.; Chen, Y.J. Caffeic acid phenethyl ester inhibits epithelial-mesenchymal transition of human pancreatic cancer cells. Evid. Based Complement. Altern. Med. 2013, 2013, 270906. [Google Scholar] [CrossRef]
- Lee, E.S.; Uhm, K.O.; Lee, Y.M.; Han, M.S.; Lee, M.S.; Man Park, J.; Suh, P.G.; Park, S.H.; Kim, H.S. CAPE (caffeic acid phenethyl ester) stimulates glucose uptake through AMPK (AMP-activated protein kinase) activation in skeletal muscle cells. Biochem. Biophys. Res. Comm. 2007, 361, 854–858. [Google Scholar] [CrossRef]
- Ozyurt, B.; Iraz, M.; Koca, K.; Ozyurt, H.; Sahin, S. Protective effects of caffeic acid phenethyl ester on skeletal muscle ischemia-reperfusion injury in rats. Mol. Cell Biochem. 2006, 292, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Bello, S.R.; Naliwaiko, K.; Vicentini, M.S.; Rossetti, F.X.; Fernandes, L.C.; Messias-Reason, I.J. Nutrition and cancer capsaicin treatment reduce tumor growth, tumor cell proliferation ex vivo and partially reverses cancer cachexia in Walker 256 tumor-bearing rats. Nutr. Cancer 2019, 71, 111–117. [Google Scholar] [CrossRef]
- Cazenave, S.M.; Olea-Herrero, N.; Vara, D.; Morell, C.; Diaz-Laviada, I. The vanilloid capsaicin induces Il-6 secretion in prostate PC-3 cancer cells. Cytokine 2011, 54, 330–337. [Google Scholar] [CrossRef]
- Janssens, P.L.H.R.; Hursel, R.; Westerterp-Plantenga, M.S. Capsaicin increases the sensation of fullness in energy balance and decreases the desire to eat after dinner in negative energy balance. Appetite 2014, 77, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Yallapu, M.M.; Jaggi, M.; Chauhan, S. Curcumin nanoformulations: A future nanomedicine for cancer. Drug Discov. Today 2012, 17, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Kudo, C.; Yamakoshi, H.; Sato, A.; Ohori, H.; Ishioka, C.; Iwabuchi, Y.; Shibata, H. Novel curcumin analogs, GO-Y030 and GO-Y078, are multi-targeted agents with enhanced abilities for multiple myeloma. Anticancer Res. 2011, 31, 3719–3726. [Google Scholar] [PubMed]
- Chung, S.S.; Vadgama, J.V. Curcumin and epigallocatechin gallate inhibit the cancer stem cell phenotype via down-regulation of STAT3-NFκB signaling. Anticancer Res. 2015, 35, 39–46. [Google Scholar]
- Qiao, Q.; Jiang, Y.; Li, G. Curcumin enhances the response of non-Hodgkin’s lymphoma cells to ionizing radiation through further induction of cell cycle arrest at the G2/M phase and inhibition of mTOR phosphorylation. Oncol. Rep. 2013, 29, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.L.; Liu, Y.Y.; Ma, Y.G.; Vue, Y.X.; Liu, D.G.; Ren, Y.; Xiao-Bai, L.; Yao, L.; Li, Z. Curcumin blocks small cell lung cancer cells migration, invasion, angiogenesis, cell cycle, and neoplasia through the Janus kinase-STAT3 signaling pathway. PLoS ONE 2012, 7, e37960. [Google Scholar] [CrossRef]
- Weissenberger, J.; Priester, M.; Bernreuther, C.; Rakel, S.; Glatzel, M.; Seifert, V.; Kogel, D. Dietary curcumin attenuates glioma growth in a syngeneic mouse model by inhibition of the JAK1/2/STAT3 signaling pathway. Res. Clin. Cancer 2010, 16, 5781–5795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrow, M.G.; Song, L.J.; Altoik, S.; Gray, J.; Haura, E.B.; Kumar, N.B. Curcumin: A novel STAT3 pathway inhibitor for chemoprevention of lung cancer. Eur. J. Cancer Prev. 2012, 21, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhu, Y. Curcumin inhibits human non-small cell lung cancer xenografts by targeting the STAT3 pathway. Am. J. Transl. Res. 2017, 9, 3633–3641. [Google Scholar]
- Busquets, S.; Carbo, N.; Almendro, V.; Quiles, M.T.; Lopez-Soriano, F.; Argiles, J.M. Curcumin, a natural product present in turmeric, decreases tumor growth but does not behave as an anti-cachectic compound in a rat model. Cancer Lett. 2001, 167, 33–38. [Google Scholar] [CrossRef]
- McCarthy Beckett, D.; Pycha, K.; Berg, T. Effects of curcumin on tumor growth and muscle mass in a mouse model of cancer cachexia. Oncol. Nurs. Forum 2008, 35, 455–459. [Google Scholar] [CrossRef]
- Parsons, H.A.; Baracos, V.E.; Hong, D.S.; Abbruzzese, J.; Bruera, E.; Kurzorck, R. The effects of patients with advanced pancreatic cancer. Oncotarget 2016, 7, 20293–20304. [Google Scholar] [CrossRef]
- Siddiqui, R.A.; Hassan, S.; Harvey, K.A.; Rasol, T.; Das, T.; Mukerji, P.; DeMichele, S. Attenuation of proteolysis and muscle wasting by curcumin c3 complex in MAC16 colon tumor-bearing mice. Br. J. Nutr. 2009, 102, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Oelkrug, C.; Lange, C.M.; Wenzel, E.; Fricke, S.; Hartke, M.; Simasi, J.; Schubert, A. Analysis of the tumoricidal and anti-cachectic potential of curcumin. Anticancer Res. 2014, 34, 4781–4788. [Google Scholar]
- Alamdari, N.; O’Neal, P.; Hasselgren, P.O. Curcumin and muscle wasting—A new role for an old drug? Nutrition 2009, 25, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.K.; Sung, B.; Seok, K.; Aggarwal, B.B. Butein suppresses constitutive and inducible signal transducer and activator of transcription (STAT)3 activation and STAT3-regulated gene products through the induction of a protein tyrosine phosphate SHP-1. Mol Pharmacol. 2009, 75, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Ma, H.; Shi, W.; Duan, J.; Wang, Y.; Zhang, C.; Li, C.; Lin, J.; Li, S.; Lv, J.; et al. Inhibition of STAT3 signaling pathway by ursolic acid suppresses the growth of hepatocellular carcinoma. Int. J. Oncol. 2017, 51, 555–562. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Compound | Mechanism of Action | Indication | Clinical Trial ID | Chemical Structure |
|---|---|---|---|---|
| Anamorelin hydrochloride | selective agonist of the ghrelin/growth hormone secretagogue receptor | cancer cachexia, non–small-cell lung cancer (NSCLC) | NCT03743064, NCT03637816, NCT03743051, NCT01387269, NCT01387282, NCT03035409, NCT01395914, NCT00622193 |  PubChem Identifier: CID 9828911, https://pubchem.ncbi.nlm.nih.gov/compound/Anamorelin |
| Relamorelin (RM-131) | selective agonist of the ghrelin/growth hormone secretagogue receptor | anorexia nervosa | NCT01642550 | 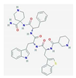 PubChem Identifier: CID 85364156, https://pubchem.ncbi.nlm.nih.gov/compound/Relamorelin |
| NGM120 Monoclonal antibody against GDNP protein alpha-like receptor (GFRAL)–3P10 antibody | GDNF family receptor-α-like (GFRAL)-Ret proto-oncogene (RET) blocker | cancer cachexia | NCT04068896 | |
| Vitamin D | promotion of lipid partitioning and muscle metabolic function | cancer cachexia | NCT03144128 | 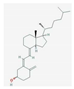 PubChem Identifier: CID 5280795, https://pubchem.ncbi.nlm.nih.gov/compound/Cholecalciferol |
| Branched Chain Amino Acid (BCAA) | regulation of the anabolic pathway of muscle synthesis | sarcopenia in chronic liver disease | NCT04246918 | |
| Omega-3 fatty acids | regulation of cell signaling, cell structure, and fluidity of membranes | cancer cachexia | NCT01596933, NCT00031707 | |
| Beta-hydroxy-beta-methyl butyrate (HMB) | improvement of muscle hypertrophy and strength, aerobic performance, resistance to fatigue, and regenerative capacity | critical illness, cancer cachexia | NCT03464708, NCT03151291 | 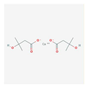 PubChem Identifier: CID 9860341, https://pubchem.ncbi.nlm.nih.gov/compound/Calcium-beta-hydroxy-beta-methylbutyrate |
| Compound | Mechanism of Action | Indication | Clinical trial ID | Chemical Structure |
|---|---|---|---|---|
| WP1066 | cell-permeable JAK2, STAT3, STAT5, and ERK1/2 inhibitor, responsible for the dephosphorylation and nuclear export of constitutively phosphorylated STAT3 | metastatic malignant neoplasms in the brain; metastatic melanoma; recurrent glioblastoma; recurrent brain neoplasm | NCT04334863 NCT01904123 |  PubChem Identifier: CID 11210478, https://pubchem.ncbi.nlm.nih.gov/compound/wp1066 |
| OPB-31121 | potent inhibition of STAT3 and STAT5 phosphorylation without upstream kinase inhibition | advanced cancer, solid tumors, hepatocellular carcinoma | NCT00955812 NCT01406574 | |
| TTI-101 | binaphthol sulfonamide-based inhibitor of STAT3 that specifically targets and binds to the phosphotyrosine peptide-binding site within the Src homology 2 (SH2) domain of STAT3 | breast cancer, head, and neck squamous cell carcinoma, non–small-cell lung cancer, colorectal cancer, gastric adenocarcinoma, melanoma | NCT04068896 | 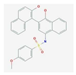 PubChem Identifier: SID 382371065, 432001-19-9, https://pubchem.ncbi.nlm.nih.gov/substance/382371065 |
| Pyrimethamine | synthetic derivative of ethyl-pyrimidine, a competitive inhibitor of dihydrofolate reductase (DHFR)—a key enzyme in the redox cycle for tetrahydrofolate production; a cofactor required for DNA and proteins synthesis | relapsed chronic lymphocytic leukemia, small lymphocytic lymphoma | NCT01066663 | 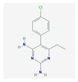 PubChem Identifier: CID 4993, https://pubchem.ncbi.nlm.nih.gov/compound/Pyrimethamine |
| OPB-51602 | inhibition of STAT3 phosphorylation and activation of STAT3 | advanced solid tumors: breast cancer, head, and neck squamous cell carcinoma, non–small-cell lung cancer, hepatocellular cancer, colorectal cancer, gastric adenocarcinoma, melanoma | NCT01423903 NCT01344876 NCT01184807 | |
| Napabucasin (Napa, BBI608) | STAT3 and cancer cell stemness inhibitor | gastrointestinal malignancies, pancreatic cancer, GBM | NCT03721744 NCT02753127 NCT03522649 |  PubChem Identifier: CID 10331844 https://pubchem.ncbi.nlm.nih.gov/compound/Napabucasin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaśkiewicz, A.; Domoradzki, T.; Pająk, B. Targeting the JAK2/STAT3 Pathway—Can We Compare It to the Two Faces of the God Janus? Int. J. Mol. Sci. 2020, 21, 8261. https://doi.org/10.3390/ijms21218261
Jaśkiewicz A, Domoradzki T, Pająk B. Targeting the JAK2/STAT3 Pathway—Can We Compare It to the Two Faces of the God Janus? International Journal of Molecular Sciences. 2020; 21(21):8261. https://doi.org/10.3390/ijms21218261
Chicago/Turabian StyleJaśkiewicz, Anna, Tomasz Domoradzki, and Beata Pająk. 2020. "Targeting the JAK2/STAT3 Pathway—Can We Compare It to the Two Faces of the God Janus?" International Journal of Molecular Sciences 21, no. 21: 8261. https://doi.org/10.3390/ijms21218261
APA StyleJaśkiewicz, A., Domoradzki, T., & Pająk, B. (2020). Targeting the JAK2/STAT3 Pathway—Can We Compare It to the Two Faces of the God Janus? International Journal of Molecular Sciences, 21(21), 8261. https://doi.org/10.3390/ijms21218261