ROS-Dependent ER Stress and Autophagy Mediate the Anti-Tumor Effects of Tributyltin (IV) Ferulate in Colon Cancer Cells

 , ,
, ,  ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. The Cytotoxic Effects of TBT-F Are Dependent on ROS Production
2.2. The Role of the Protective Factor Nrf2 and the Effect of Nrf2 Silencing
2.3. TBT-F Induces ER Stress Markers and NAC Prevents These Effects
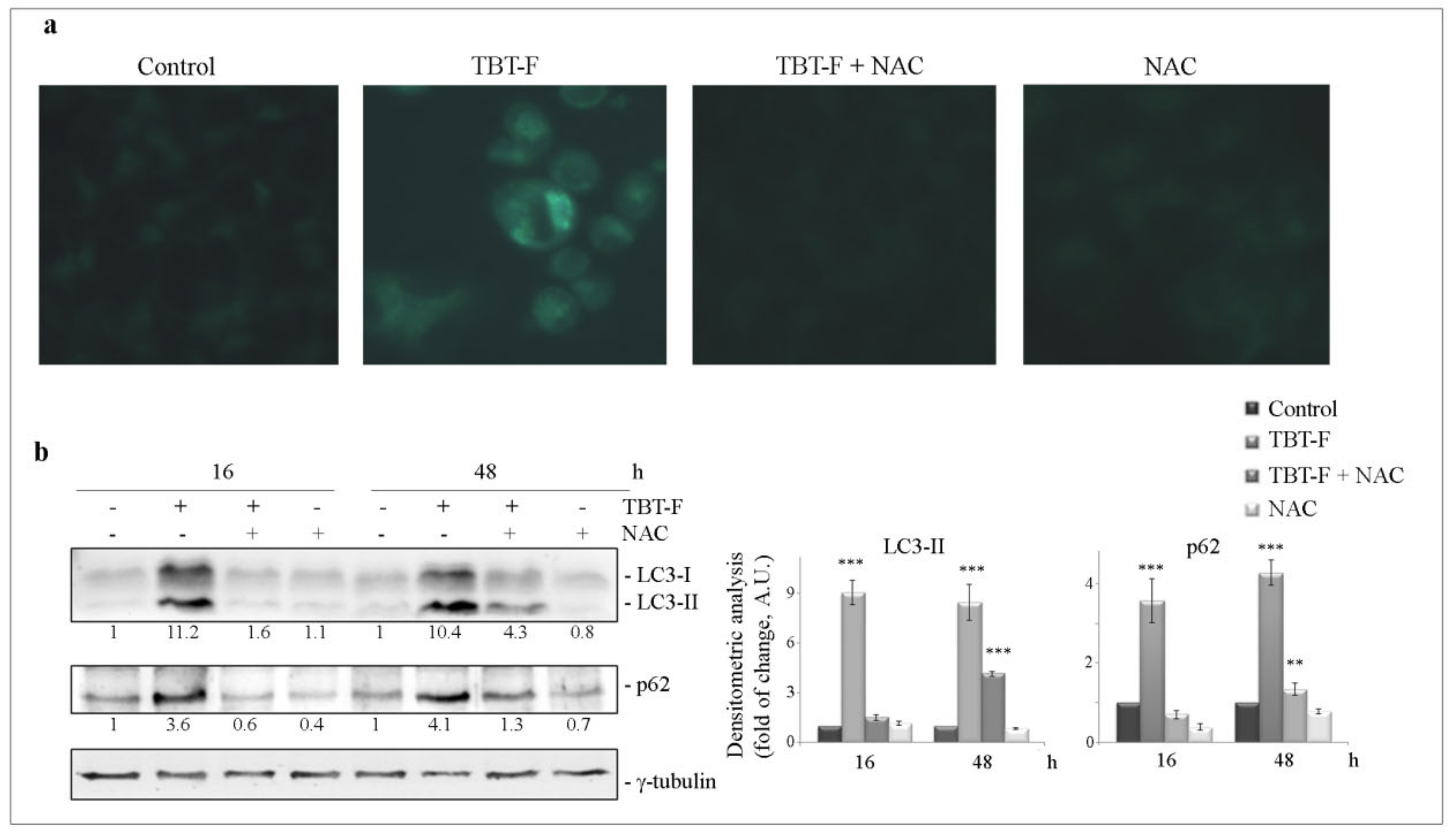
2.4. TBT-F Activates ROS-Dependent Autophagy Correlated with ER Stress
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture Conditions
4.3. Evaluation of Cell Viability
4.4. Lactate Dehydrogenase Release Assay
4.5. Clonogenic Assay
4.6. Intracellular ROS Measurement
4.7. Monodansylcadaverine Labeling
4.8. Western Blot Analysis
4.9. Gene Silencing
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Emanuele, S.; D’Anneo, A.; Calvaruso, G.; Cernigliaro, C.; Giuliano, M.; Lauricella, M. The Double-Edged Sword Profile of Redox Signaling: Oxidative Events as Molecular Switches in the Balance between Cell Physiology and Cancer. Chem. Res. Toxicol. 2018, 31, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Cernigliaro, C.; D’Anneo, A.; Carlisi, D.; Giuliano, M.; Marino Gammazza, A.; Barone, R.; Longhitano, L.; Cappello, F.; Emanuele, S.; Distefano, A.; et al. Ethanol-Mediated Stress Promotes Autophagic Survival and Aggressiveness of Colon Cancer Cells via Activation of Nrf2/HO-1 Pathway. Cancers 2019, 11, 505. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Phys. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. Regulation of the Keap1–Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 2016, 1, 54–61. [Google Scholar] [CrossRef]
- Li, T.; Jiang, D.; Wu, K. p62 promotes bladder cancer cell growth by activating KEAP1/NRF2-dependent antioxidative response. Cancer Sci. 2020, 111, 1156–1164. [Google Scholar] [CrossRef]
- Yasuda, D.; Ohe, T.; Takahashi, K.; Imamura, R.; Kojima, H.; Okabe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Nagano, T.; et al. Inhibitors of the protein–protein interaction between phosphorylated p62 and Keap1 attenuate chemoresistance in a human hepatocellular carcinoma cell line. Free Radic. Res. 2020, 1–13. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.V.; LeDoux, S.P.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA Damage via Augmented Oxidative Stress Regulates Endoplasmic Reticulum Stress and Autophagy: Crosstalk, Links and Signaling. PLoS ONE 2013, 8, e83349. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Higa, A.; Chevet, E. Redox signaling loops in the unfolded protein response. Cell. Signal. 2012, 24, 1548–1555. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef]
- Oh, H.J.; Lee, S.; Park, P.H. ER stress contributes to autophagy induction by adiponectin in macrophages: Implication in cell survival and suppression of inflammatory response. Cytokine 2020, 127, 154959. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef]
- Emanuele, S.; Lauricella, M.; D’Anneo, A.; Carlisi, D.; De Blasio, A.; Di Liberto, D.; Giuliano, M. p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles. Int. J. Mol. Sci. 2020, 21, 5029. [Google Scholar] [CrossRef] [PubMed]
- Pellerito, C.; Emanuele, S.; Ferrante, F.; Celesia, A.; Giuliano, M.; Fiore, T. Tributyltin(IV) ferulate, a novel synthetic ferulic acid derivative, induces autophagic cell death in colon cancer cells: From chemical synthesis to biochemical effects. J. Inorg. Biochem. 2020, 205, 110999. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Bowitz Larsen, K.; Atesoh Awuh, J.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.; Lee, G.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Gao, J.; Yu, H.; Guo, W.; Kong, Y.; Gu, L.; Li, Q.; Yang, S.; Zhang, Y.; Wang, Y. The anticancer effects of ferulic acid is associated with induction of cell cycle arrest and autophagy in cervical cancer cells. Cancer Cell Int. 2018, 18, 102. [Google Scholar] [CrossRef]
- Zduńska, K.; Dana, A.; Kolodziejczak, A.; Rotsztejn, H. Antioxidant Properties of Ferulic Acid and Its Possible Application. Skin Pharmacol. Physiol. 2018, 31, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Kawami, T.; Ishida, A.; Yamazaki, T. Tributyltin induces oxidative stress and neuronal injury by inhibiting glutathione S-transferase in rat organotypic hippocampal slice cultures. Neurochem. Int. 2012, 60, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Gennari, A.; Viviani, B.; Galli, C.L.; Marinovich, M.; Pieters, R.; Corsini, E. Organotins Induce Apoptosis by Disturbance of [Ca2+]i and Mitochondrial Activity, Causing Oxidative Stress and Activation of Caspases in Rat Thymocytes. Toxicol. Appl. Pharmacol. 2000, 169, 185–190. [Google Scholar] [CrossRef]
- Coutinho, J.V.S.; Freitas-Lima, L.C.; Freitas, F.F.C.T.; Freitas, F.P.S.; Podratz, P.L.; Magnago, R.P.L.; Porto, M.L.; Meyrelles, S.S.; Vasquez, E.C.; Brandão, P.A.A.; et al. Tributyltin chloride induces renal dysfunction by inflammation and oxidative stress in female rats. Toxicol. Lett. 2016, 260, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Kanimozhi, V.; Palanivel, K.; Akbarsha, M.A.; Kadalmani, B. Tributyltin-mediated hepatic, renal and testicular tissue damage in male Syrian hamster (Mesocricetus auratus): A study on impact of oxidative stress. SpringerPlus 2016, 5, 1523. [Google Scholar] [CrossRef]
- Hou, L.; Sun, F.; Huang, R.; Sun, W.; Zhang, D.; Wang, Q. Inhibition of NADPH oxidase by apocynin prevents learning and memory deficits in a mouse Parkinson’s disease model. Redox Biol. 2019, 22, 101134. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Zhou, Z.; Xu, J.; Bao, X.; Shi, J.; Liu, B.; Chen, Y.; Li, J. Nuclear Nrf2 Activity in Laryngeal Carcinoma is Regulated by SENP3 After Cisplatin-Induced Reactive Oxygen Species Stress. J. Cancer 2019, 10, 3427–3434. [Google Scholar] [CrossRef]
- Pellerito, L. Organotin(IV)n+ complexes formed with biologically active ligands: Equilibrium and structural studies, and some biological aspects. Coord. Chem. Rev. 2002, 224, 111–150. [Google Scholar] [CrossRef]
- Nakanishi, T.; Nishikawa, J.; Tanaka, K. Molecular targets of organotin compounds in endocrine disruption: Do organotin compounds function as aromatase inhibitors in mammals? Environ. Sci. 2006, 13, 89–100. [Google Scholar]
- Grun, F. Covalent Interactions of Organotins with Nuclear Receptors. In Biochemical and Biological Effects of Organotins; Pagliarani, A., Trombetti, F., Ventrella, V., Eds.; Bentham Science Publishers: Sharjah, UAE, 2012; pp. 53–69. ISBN 978-1-60805-265-3. [Google Scholar]
- Frias, D.P.; Gomes, R.L.N.; Yoshizaki, K.; Carvalho-Oliveira, R.; Matsuda, M.; de Junqueira, M.S.; Teodoro, W.R.; de Vasconcellos, P.C.; de Pereira, D.C.A.; da Conceição, P.R.; et al. Nrf2 positively regulates autophagy antioxidant response in human bronchial epithelial cells exposed to diesel exhaust particles. Sci. Rep. 2020, 10, 3704. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Li, Q.; Zhao, X.; Wang, H.; Li, N.; Fang, Y.; Wang, K.; Jia, Y.; Zhu, P.; Gu, J.; et al. Shiga toxins induce autophagic cell death in intestinal epithelial cells via the endoplasmic reticulum stress pathway. Autophagy 2015, 11, 344–354. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Zhang, T.T.; Song, D.D.; Zhou, J.H.; Han, R.; Qin, Z.H.; Sheng, R. Endoplasmic reticulum chaperone GRP78 is involved in autophagy activation induced by ischemic preconditioning in neural cells. Mol. Brain 2015, 8, 20. [Google Scholar] [CrossRef]
- Lu, H.Y.; Chen, X.Q.; Tang, W.; Wang, Q.X.; Zhang, J. GRP78 silencing enhances hyperoxia-induced alveolar epithelial cell apoptosis via CHOP pathway. Mol. Med. Rep. 2017, 16, 1493–1501. [Google Scholar] [CrossRef][Green Version]
- Yun, S.; Han, Y.S.; Lee, J.H.; Kim, S.; Lee, S.H. Enhanced Susceptibility to 5-Fluorouracil in Human Colon Cancer Cells by Silencing of GRP78. Anticancer Res. 2017, 37, 2975–2984. [Google Scholar] [CrossRef]
- B’chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- B’chir, W.; Chaveroux, C.; Carraro, V.; Averous, J.; Maurin, A.C.; Jousse, C.; Muranishi, Y.; Parry, L.; Fafournoux, P.; Bruhat, A. Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell. Signal. 2014, 26, 1385–1391. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef]
- Lauricella, M.; Lo Galbo, V.; Cernigliaro, C.; Maggio, A.; Palumbo Piccionello, A.; Calvaruso, G.; Carlisi, D.; Emanuele, S.; Giuliano, M.; D’Anneo, A. The Anti-Cancer Effect of Mangifera indica L. Peel Extract is Associated to γH2AX-mediated Apoptosis in Colon Cancer Cells. Antioxidants 2019, 8, 422. [Google Scholar] [CrossRef]
- Emanuele, S.; Notaro, A.; Palumbo Piccionello, A.; Maggio, A.; Lauricella, M.; D’Anneo, A.; Cernigliaro, C.; Calvaruso, G.; Giuliano, M. Sicilian Litchi Fruit Extracts Induce Autophagy versus Apoptosis Switch in Human Colon Cancer Cells. Nutrients 2018, 10, 1490. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, D.; D’Anneo, A.; Martinez, R.; Emanuele, S.; Buttitta, G.; Di Fiore, R.; Vento, R.; Tesoriere, G.; Lauricella, M. The oxygen radicals involved in the toxicity induced by parthenolide in MDA-MB-231 cells. Oncol. Rep. 2014, 32, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Notaro, A.; Emanuele, S.; Geraci, F.; D’Anneo, A.; Lauricella, M.; Calvaruso, G.; Giuliano, M. WIN55,212-2-Induced Expression of Mir-29b1 Favours the Suppression of Osteosarcoma Cell Migration in a SPARC-Independent Manner. Int. J. Mol. Sci. 2019, 20, 5235. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celesia, A.; Morana, O.; Fiore, T.; Pellerito, C.; D’Anneo, A.; Lauricella, M.; Carlisi, D.; De Blasio, A.; Calvaruso, G.; Giuliano, M.; et al. ROS-Dependent ER Stress and Autophagy Mediate the Anti-Tumor Effects of Tributyltin (IV) Ferulate in Colon Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8135. https://doi.org/10.3390/ijms21218135
Celesia A, Morana O, Fiore T, Pellerito C, D’Anneo A, Lauricella M, Carlisi D, De Blasio A, Calvaruso G, Giuliano M, et al. ROS-Dependent ER Stress and Autophagy Mediate the Anti-Tumor Effects of Tributyltin (IV) Ferulate in Colon Cancer Cells. International Journal of Molecular Sciences. 2020; 21(21):8135. https://doi.org/10.3390/ijms21218135
Chicago/Turabian StyleCelesia, Adriana, Ornella Morana, Tiziana Fiore, Claudia Pellerito, Antonella D’Anneo, Marianna Lauricella, Daniela Carlisi, Anna De Blasio, Giuseppe Calvaruso, Michela Giuliano, and et al. 2020. "ROS-Dependent ER Stress and Autophagy Mediate the Anti-Tumor Effects of Tributyltin (IV) Ferulate in Colon Cancer Cells" International Journal of Molecular Sciences 21, no. 21: 8135. https://doi.org/10.3390/ijms21218135
APA StyleCelesia, A., Morana, O., Fiore, T., Pellerito, C., D’Anneo, A., Lauricella, M., Carlisi, D., De Blasio, A., Calvaruso, G., Giuliano, M., & Emanuele, S. (2020). ROS-Dependent ER Stress and Autophagy Mediate the Anti-Tumor Effects of Tributyltin (IV) Ferulate in Colon Cancer Cells. International Journal of Molecular Sciences, 21(21), 8135. https://doi.org/10.3390/ijms21218135