Ischemic Heart Disease Pathophysiology Paradigms Overview: From Plaque Activation to Microvascular Dysfunction

 , ,
, , 
 ,
, 
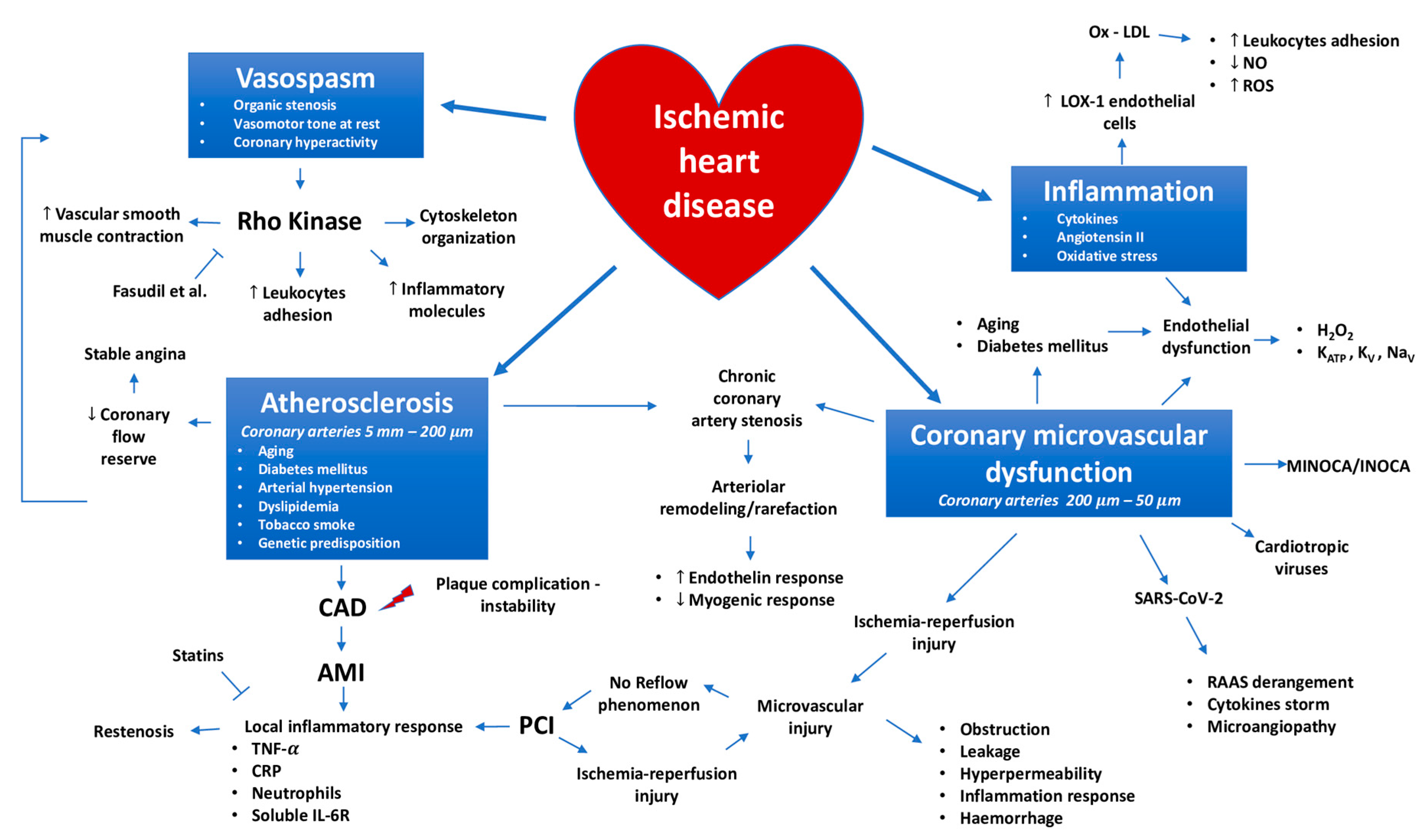{kind=link}
{kind=link}
Abstract
1. Introduction
2. Coronary Macrocirculation Involvement in the Ischemic Heart Disease Pathophysiology
2.1. Ischemic Heart Disease and the Hydraulic Paradigm: Role of Coronary Artery Disease and Vasospasm
2.2. The Biological Paradigm: The Bidirectional Link between Inflammation and Myocardial Ischemia
2.2.1. Inflammation as Cause of Ischemic Heart Disease
2.2.2. Inflammation as Consequence of Ischemic Heart Disease
2.3. Coronary Circulation Involvement during HIV Infection and Antiretroviral Therapy
3. Role of Coronary Microcirculation in the Pathophysiology of Ischemic Heart Disease
3.1. The Mechanistic Point of View of Coronary Microvascular Dysfunction Pathophysiology
3.2. The Functional Point of View of Coronary Microvascular Dysfunction Pathophysiology
3.3. Microvascular–Myocardial Interaction in the Regulation of Coronary Blood Flow
4. Conclusions
Funding
Conflicts of Interest
References
- Moran, A.E.; Forouzanfar, M.H.; Roth, G.A.; Mensah, G.A.; Ezzati, M.; Murray, C.J.; Naghavi, M. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: The Global Burden of Disease 2010 study. Circulation 2014, 129, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbacher, C.P.; Chilian, W.M. Heterogeneity of coronary vasomotion. Basic Res. Cardiol. 1998, 93, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Wolin, M.S.; Ahmad, M.; Gupte, S.A. Oxidant and redox signaling in vascular oxygen sensing mechanisms: Basic concepts, current controversies, and potential importance of cytosolic NADPH. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 289, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Tune, J.D. Withdrawal of vasoconstrictor influences in local metabolic coronary vasodilation. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, 2044–2046. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dzeja, P.P.; Bast, P.; Pucar, D.; Wieringa, B.; Terzic, A. Defective metabolic signaling in adenylate kinase AK1 gene knock-out hearts compromises post-ischemic coronary reflow. J. Biol. Chem. 2007, 282, 31366–31372. [Google Scholar] [CrossRef] [PubMed]
- Ottaviani, G.; Lavezzi, A.M.; Rossi, L.; Matturri, L. Proliferating cell nuclear antigen (PCNA) and apoptosis in hyperacute and acute myocardial infarction. Eur. J. Histochem. 1999, 43, 7–14. [Google Scholar]
- Wong, B.; Kruse, G.; Kutikova, L.; Ray, K.K.; Mata, P.; Bruckert, E. Cardiovascular Disease Risk Associated with Familial Hypercholesterolemia: A Systematic Review of the Literature. Clin. Ther. 2016, 38, 1696–1709. [Google Scholar] [CrossRef]
- Feldman, R.D. Heart Disease in Women: Unappreciated Challenges, GPER as a New Target. Int. J. Mol. Sci. 2016, 17, 760. [Google Scholar] [CrossRef]
- Ostergaard, L.; Kristiansen, S.B.; Angleys, H.; Frøkiær, J.; Michael Hasenkam, J.; Jespersen, S.N.; Bøtker, H.E. The role of capillary transit time heterogeneity in myocardial oxygenation and ischemic heart disease. Basic Res. Cardiol. 2014, 109, 409. [Google Scholar] [CrossRef]
- Crea, F.; Battipaglia, I.; Andreotti, F. Sex differences in mechanisms, presentation and management of ischaemic heart disease. Atherosclerosis 2015, 241, 157–168. [Google Scholar] [CrossRef]
- Eckhouse, S.R.; Jones, J.A.; Spinale, F.G. Gene targeting in ischemic heart disease and failure: Translational and clinical studies. Biochem. Pharmacol. 2013, 85, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Severino, P.; Bruno, N.; Stio, R.; Caira, C.; D’Ambrosi, A.; Brasolin, B.; Ohanyan, V.; Mancone, M. Role of ion channels in coronary microcirculation: A review of the literature. Future Cardiol. 2013, 9, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.C.; Allam, A.H.; Lombardi, G.P.; Wann, L.S.; Sutherland, M.L.; Sutherland, J.D.; Soliman, M.A.; Frohlich, B.; Mininberg, D.T.; Monge, J.M.; et al. Atherosclerosis across 4000 years of human history: The Horus study of four ancient populations. Lancet 2013, 381, 1211–1222. [Google Scholar] [CrossRef]
- Duncker, D.J.; Bache, R.J. Regulation of coronary blood flow during exercise. Physiol. Rev. 2008, 88, 1009–1086. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, F.; Carpenito, M.; Verolino, G.; Chello, C.; Nusca, A.; Lusini, M.; Spadaccio, C.; Nappi, F.; Di Sciascio, G.; Nenna, A. Changes of the coronary arteries and cardiac microvasculature with aging: Implications for translational research and clinical practice. Mech. Ageing Dev. 2019, 184, 111161. [Google Scholar] [CrossRef]
- Nanayakkara, S.; Marwick, T.H.; Kaye, D.M. The ageing heart: The systemic and coronary circulation. Heart 2018, 104, 370–376. [Google Scholar] [CrossRef]
- Prinzmetal, M.; Kennarner, R.; Merliss, B.; Wads, T.; Bor, N. Angina pectoris. I. A variant form of angina pectoris; preliminary report. Am. J. Med. 1959, 27, 375–388. [Google Scholar] [CrossRef]
- James, T.M. Angina without coronary disease (sic). Circulation 1970, 42, 189–191. [Google Scholar] [CrossRef]
- Maseri, A. Mechanisms of myocardial ischemia. Cardiovasc. Drugs Ther. 1990, 4 (Suppl. 4), 827–831. [Google Scholar] [CrossRef]
- Marzilli, M.; Huqi, A. Coronary vasospasm and coronary atherosclerosis: Do we have to choose? J. Am. Coll. Cardiol. 2012, 59, 663–664. [Google Scholar] [CrossRef][Green Version]
- Yamagishi, M.; Miyatake, K.; Tamai, J.; Nakatani, S.; Koyama, J.; Nissen, S.E. Intravascular ultrasound detection of atherosclerosis at the site of focal vasospasm in angiographically normal or minimally narrowed coronary segments. J. Am. Coll. Cardiol. 1994, 23, 352–357. [Google Scholar] [CrossRef]
- Ludmer, P.L.; Selwyn, A.P.; Shook, T.L.; Wayne, R.R.; Mudge, G.H.; Alexander, R.W.; Ganz, P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N. Engl. J. Med. 1986, 315, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H.; Satoh, K. 2015 ATVB Plenary Lecture: Translational research on rho-kinase in cardiovascular medicine. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1756–1769. [Google Scholar] [CrossRef]
- Noma, K.; Oyama, N.; Liao, J.K. Physiological role of ROCKs in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2006, 290, C661–C668. [Google Scholar] [CrossRef]
- Pearson, J.T.; Jenkins, M.J.; Edgley, A.J.; Sonobe, T.; Joshi, M.; Waddingham, M.T.; Fujii, Y.; Schwenke, D.O.; Tsuchimochi, H.; Yoshimoto, M.; et al. Acute Rho-kinase inhibition improves coronary dysfunction in vivo, in the early diabetic microcirculation. Cardiovasc. Diabetol. 2013, 12, 111. [Google Scholar] [CrossRef]
- Wu, N.; Li, W.; Shu, W.; Lv, Y.; Jia, D. Inhibition of Rho-kinase by fasudil restores the cardioprotection of ischemic postconditioninng in hypercholesterolemic rat heart. Mol. Med. Rep. 2014, 10, 2517–2524. [Google Scholar] [CrossRef]
- Yap, J.; Cabrera-Fuentes, H.A.; Irei, J.; Hausenloy, D.J.; Boisvert, W.A. Role of Macrophages in Cardioprotection. Int. J. Mol. Sci. 2019, 20, 2474. [Google Scholar] [CrossRef]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis: An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Simon, D.I. Inflammation and thrombosis: The clot thickens. Circulation 2001, 103, 1718–1720. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and Atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Maseri, A.; Liuzzo, G.; Biasucci, L.M. Pathogenic mechanisms in unstable angina. Heart 1999, 82, 12–14. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Ammirati, E.; Moroni, F.; Magnoni, M.; Camici, P.G. The role of T and B cells in human atherosclerosis and atherothrombosis. Clin. Exp. Immunol. 2015, 179, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Paragh, G.; Seres, I.; Harangi, M.; Fulop, P. Dynamic interplay between metabolic syndrome and immunity. Adv. Exp. Med. Biol. 2014, 824, 171–190. [Google Scholar] [CrossRef] [PubMed]
- Frostegard, J. Immune mechanisms in atherosclerosis, especially in diabetes type 2. Front. Endocrinol. (Lausanne) 2013, 4, 162. [Google Scholar] [CrossRef]
- IL6R Genetics Consortium Emerging Risk Factors Collaboration; Sarwar, N.; Butterworth, A.S.; Freitag, D.F.; Gregson, J.; Willeit, P.; Gorman, D.N.; Gao, P.; Saleheen, D.; Rendon, A.; et al. Interleukin-6 receptor pathways in coronary heart disease: A collaborative meta-analysis of 82 studies. Lancet 2012, 379, 1205–1213. [Google Scholar] [CrossRef]
- Andreotti, F.; Burzotta, F.; Mazza, A.; Manzoli, A.; Robinson, K.; Maseri, A. Homocysteine and arterial occlusive disease: A concise review. Cardiologia 1999, 44, 341–345. [Google Scholar]
- Biasucci, L.M.; Liuzzo, G.; Buffon, A.; Maseri, A. The variable role of inflammation in acute coronary syndromes and in restenosis. Semin. Interv. Cardiol. 1999, 4, 105–110. [Google Scholar] [CrossRef]
- Gupta, S.; Leatham, E.W.; Carrington, D.; Mendall, M.A.; Kaski, J.C.; Camm, A.J. Elevated Chlamydia pneumoniae antibodies, cardiovascular events, and azithromycin in male survivors of myocardial infarction. Circulation 1997, 96, 404–407. [Google Scholar] [CrossRef]
- Gurfinkel, E.; Bozovich, G.; Daroca, A.; Beck, E.; Mautner, B. Randomised trial of roxithromycin in non-Q-wave coronary syndromes: ROXIS pilot study. ROXIS Study Group. Lancet 1997, 350, 404–407. [Google Scholar] [CrossRef]
- Steinberg, D. Atherogenesis in perspective: Hypercholesterolemia and inflammation as partners in crime. Nat. Med. 2002, 8, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Kohno, H.; Hasegawa, A.; Toshima, S.; Amaki, T.; Kurabayashi, M.; Nagai, R.; Suzuki, T.; Amaki, T.; Nagai, R.; et al. Diagnostic implications of circulating oxidized low density lipoprotein levels as a biochemical risk marker of coronary artery disease. Clin. Biochem. 2002, 35, 347–353. [Google Scholar] [CrossRef]
- Mehta, J.L.; Li, D. Identification, regulation and function of a novel lectin-like oxidized low-density lipoprotein receptor. J. Am. Coll. Cardiol. 2002, 39, 1429–1435. [Google Scholar] [CrossRef]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Ehara, S.; Ueda, M.; Naruko, T.; Haze, K.; Itoh, A.; Otsuka, M.; Komatsu, R.; Matsuo, T.; Itabe, H.; Takano, T.; et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation 2001, 103, 1955–1960. [Google Scholar] [CrossRef]
- Mollace, V.; Gliozzi, M.; Musolino, V.; Carresi, C.; Muscoli, S.; Mollace, R.; Tavernese, A.; Gratteri, S.; Palma, E.; Morabito, C.; et al. Oxidized LDL attenuates protective autophagy and induces apoptotic cell death of endothelial cells: Role of oxidative stress and LOX-1 receptor expression. Int. J. Cardiol. 2015, 184, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Shahanawaz, J.; Shmookler Reis, R.J.; Varughese, K.I.; Sawamura, T.; Mehta, J.L. Cross-talk between LOX-1 and PCSK9 in vascular tissues. Cardiovasc. Res. 2015, 107, 556–567. [Google Scholar] [CrossRef]
- Wang, X.; Ding, Z.; Lin, J.; Guo, Z.; Mehta, J.L. LOX-1 in macrophage migration in response to ox-LDL and the involvement of calpains. Biochem. Biophys. Res. Commun. 2015, 467, 135–139. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. CLEAR Harmony Trial. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Sabatine, M.S. PCSK9 inhibitors: Clinical evidence and implementation. Nat. Rev. Cardiol. 2019, 16, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, K.; Kolodgie, F.D.; Lutter, C.; Mori, H.; Romero, M.E.; Finn, A.V.; Virmani, R. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 191–204. [Google Scholar] [CrossRef]
- Zakynthinos, E.; Pappa, N. Inflammatory biomarkers in coronary artery disease. J. Cardiol. 2009, 53, 317–333. [Google Scholar] [CrossRef]
- Blake, G.J.; Ridker, P.M. Inflammatory bio-markers and cardiovascular risk prediction. J. Intern. Med. 2002, 252, 283–294. [Google Scholar] [CrossRef]
- Shishehbor, M.H.; Bhatt, D.L. Inflammation and atherosclerosis. Curr. Atheroscler. Rep. 2004, 6, 131–139. [Google Scholar] [CrossRef]
- Kovanen, P.T.; Kaartinen, M.; Paavonen, T. Infiltrates of activated mast cells at the site of coronary atheromatous erosion or rupture in myocardial infarction. Circulation 1995, 92, 1084–1088. [Google Scholar] [CrossRef]
- Van der Wal, A.C.; Becker, A.E.; van der Loos, C.M.; Das, P.K. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994, 89, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.R.; Falk, E.; Palacios, I.F.; Newell, J.B.; Fuster, V.; Fallon, J.T. Macrophage infiltration in acute coronary syndromes: Implications for plaque rupture. Circulation 1994, 90, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Theroux, P. Pathophysiology of Coronary Artery Disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef]
- Arras, M.; Hoche, A.; Bohle, R.; Eckert, P.; Riedel, W.; Schaper, J. Tumor necrosis factor-alpha in macrophages of heart, liver, kidney, and in the pituitary gland. Cell Tissue Res. 1996, 285, 39–49. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Perrard, J.L.; Mendoza, L.H.; Burns, A.R.; Lindsey, M.L.; Ballantyne, C.M.; Michael, L.H.; Smith, C.W.; Entman, M.L. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation 1998, 98, 687–698. [Google Scholar] [CrossRef]
- Aker, S.; Belosjorow, S.; Konietzka, I.; Duschin, A.; Martin, C.; Heusch, G.; Schulz, R. Serum but not myocardial TNF-alpha concentration is increased in pacing-induced heart failure in rabbits. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R463–R469. [Google Scholar] [CrossRef] [PubMed]
- Barron, H.V.; Cannon, C.P.; Murphy, S.A.; Braunwald, E.; Gibson, C.M. Association between white blood cell count, epicardial blood flow, myocardial perfusion, and clinical outcomes in the setting of acute myocardial infarction: A thrombolysis in myocardial infarction 10 substudy. Circulation 2000, 102, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Bursi, F.; Weston, S.A.; Killian, J.M.; Gabriel, S.E.; Jacobsen, S.J.; Roger, V.L. C-reactive protein and heart failure after myocardial infarction in the community. Am. J. Med. 2007, 120, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.; Golmohammadi, A. Correlation between neutrophilia and congestive heart failure after acute myocardial infarction. Med. J. Ardabil Uni. Med. Sci. 2006, 5, 352–357. [Google Scholar]
- Kirtane, A.J.; Bui, A.; Murphy, S.A.; Barron, H.V.; Gibson, C.M. Association of peripheral neutrophilia with adverse angiographic outcomes in ST-elevation myocardial infarction. Am. J. Cardiol. 2004, 93, 532–536. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.; Morrow, D.A.; Cannon, C.P.; Guo, W.; Murphy, S.A.; Gibson, C.M.; Sabatine, M.S. Association between baseline neutrophil count, clopidogrel therapy, and clinical and angiographic outcomes in patients with ST-elevation myocardial infarction receiving fibrinolytic therapy. Eur. Heart J. 2008, 29, 984–991. [Google Scholar] [CrossRef][Green Version]
- Ritschel, V.N.; Seljeflot, I.; Arnesen, H.; Halvorsen, S.; Eritsland, J.; Fagerland, M.W.; Andersen, G.Ø. Circulating Levels of IL-6 Receptor and gp130 and Long-Term Clinical Outcomes in ST-Elevation Myocardial Infarction. J. Am. Heart Assoc. 2016, 13, 5. [Google Scholar] [CrossRef]
- Paul, M.R.; Brendan, M.E.; Tom, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Stefan, D.A.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Pan, W.Q.; He, Y.H.; Su, Q.; Yang, J.; Fang, Y.H.; Ding, F.H.; Yan, X.X.; Liu, Z.H.; Wang, X.Q.; Yang, K.; et al. Association of decreased serum vasostatin-2 level with ischemic chronic heart failure and with MACE in 3-year follow-up: Vasostatin-2 prevents heart failure in myocardial infarction rats. Int. J. Cardiol. 2016, 221, 1–11. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Erbel, C.; Linden, F.; Domschke, G.; Akhavanpoor, M.; Doesch, A.O.; Buss, S.J.; Giannitsis, E.; Katus, H.A.; Korosoglou, G. Galectin-3 binding protein plasma levels are associated with long-term mortality in coronary artery disease independent of plaque morphology. Atherosclerosis 2016, 251, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Pourafkari, L.; Visnjevac, O.; Ghaffari, S.; Nader, N.D. Statin drugs mitigate cellular inflammatory response after ST elevation myocardial infarction, but do not affect in-hospital mortality. J. Cardiovasc. Thorac. Res. 2016, 8, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Basoli, A.; Cametti, C.; Satriani, F.G.; Mariani, P.; Severino, P. Hemocompatibility of stent materials: Alterations in electrical parameters of erythrocyte membranes. Vasc. Health Risk Manag. 2012, 8, 197–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Čermák, T.; MuŽáková, V.; Matějka, J.; Skalický, J.; Laštovička, P.; Líbalová, M.; Kanďár, R.; Novotný, V.; Čegan, A. Fatty acid profile in erythrocyte membranes and plasma phospholipids affects significantly the extent of inflammatory response to coronary stent implantation. Physiol. Res. 2016, 65, 941–951. [Google Scholar] [CrossRef]
- Huang, G.Y.; Yang, L.J.; Wang, X.H.; Wang, Y.L.; Xue, Y.Z.; Yang, W.B. Relationship between platelet-leukocyte aggregation and myocardial perfusion in patients with ST-segment elevation myocardial infarction after primary percutaneous coronary intervention. Heart Lung 2016, 45, 429–433. [Google Scholar] [CrossRef]
- Francone, M.; Bucciarelli-Ducci, C.; Carbone, I.; Canali, E.; Scardala, R.; Calabrese, F.A.; Sardella, G.; Mancone, M.; Catalano, C.; Fedele, F.; et al. Impact of primary coronary angioplasty delay on myocardial salvage, infarct size, and microvascular damage in patients with ST-segment elevation myocardial infarction: Insight from cardiovascular magnetic resonance. J. Am. Coll. Cardiol. 2009, 54, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Lavalle, C.; Mariani, M.V.; Piro, A.; Straito, M.; Severino, P.; Della Rocca, D.G.; Forleo, G.B.; Romero, J.; Di Biase, L.; Fedele, F. Electrocardiographic features, mapping and ablation of idiopathic outflow tract ventricular arrhythmias. J. Interv. Card. Electrophysiol. 2020, 57, 207–218. [Google Scholar] [CrossRef]
- Oliver, M.; Opie, L. Effects of glucose and fatty acids on myocardial ischemia and arrhythmias. Lancet 1994, 343, 155–158. [Google Scholar] [CrossRef]
- Carnevale, R.; Sciarretta, S.; Valenti, V.; Di Nonno, F.; Calvieri, C.; Nocella, C.; Frati, G.; Forte, M.; d’Amati, G.; Pignataro, M.G.; et al. Low-grade endotoxemia enhances artery thrombus growth via Toll-like receptor 4: Implication for myocardial infarction. Eur. Heart J. 2020, 41, 3156–3165. [Google Scholar] [CrossRef]
- Freiberg, M.S.; Chang, C.C.; Kuller, L.H.; Skanderson, M.; Lowy, E.; Kraemer, K.L.; Butt, A.A.; Bidwell Goetz, M.; Leaf, D.; Oursler, K.A.; et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern. Med. 2013, 173, 614–622. [Google Scholar] [CrossRef]
- Marcus, J.L.; Leyden, W.A.; Chao, C.R.; Chow, F.C.; Horberg, M.A.; Hurley, L.B.; Klein, D.B.; Quesenberry, C.P., Jr.; Towner, W.J.; Silverberg, M.J. HIV infection and incidence of ischemic stroke. AIDS 2014, 28, 1911–1919. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.A.; Chang, C.C.; Kuller, L.; Goetz, M.B.; Leaf, D.; Rimland, D.; Gibert, C.L.; Oursler, K.K.; Rodriguez-Barradas, M.C.; Lim, J.; et al. Risk of heart failure with human immunodeficiency virus in the absence of prior diagnosis of coronary heart disease. Arch. Intern. Med. 2011, 171, 737–743. [Google Scholar] [CrossRef] [PubMed]
- D’Ascenzo, F.; Cerrato, E.; Appleton, D.; Moretti, C.; Calcagno, A.; Abouzaki, N.; Vetrovec, G.; Lhermusier, T.; Carrie, D.; Das Neves, B.; et al. Percutaneous coronary intervention and surgical revascularization in HIV Database (PHD) Study Investigators. Prognostic indicators for recurrent thrombotic events in HIV-infected patients with acute coronary syndromes: Use of registry data from 12 sites in Europe, South Africa and the United States. Thromb. Res. 2014, 134, 558–564. [Google Scholar] [CrossRef]
- Peyracchia, M.; De Lio, G.; Montrucchio, C.; Omedè, P.; d’Ettore, G.; Calcagno, A.; Vullo, V.; Cerrato, E.; Pennacchi, M.; Sardella, G.; et al. Evaluation of coronary features of HIV patients presenting with ACS: The CUORE, a multicenter study. Atherosclerosis 2012, 274, 218–226. [Google Scholar] [CrossRef]
- Fedele, F.; Bruno, N.; Mancone, M. Cardiovascular risk factors and HIV disease. AIDS Rev. 2011, 13, 119–129. [Google Scholar] [PubMed]
- Freiberg, M.S.; So-Armah, K. HIV and Cardiovascular Disease: We Need a Mechanism, and We Need a Plan. J. Am. Heart Assoc. 2016, 24, 4. [Google Scholar] [CrossRef]
- Severino, P.; Calcagno, S.; Sardella, G.; d’Ettorre, G.; Vullo, V.; Cinque, A.; Salvi, N.; Bruno, P.; Mancone, M.; Fedele, F. Pathophysiology of acute coronary syndrome in HIV positive patients: Insight from virtual histology analysis. J. Am. Coll. Cardiol. 2015, 65, A43. [Google Scholar] [CrossRef][Green Version]
- Mattingly, A.S.; Unsal, A.B.; Purdy, J.B.; Gharib, A.M.; Rupert, A.; Kovacs, J.A.; McAreavey, D.; Hazra, R.; Abd-Elmoniem, K.Z.; Hadigan, C. T-Cell Activation and E-Selectin Are Associated with Coronary Plaque in HIV-Infected Young Adults. Pediatr. Infect. Dis. J. 2017, 36, 63–65. [Google Scholar] [CrossRef]
- Hunegnaw, R.; Vassylyeva, M.; Dubrovsky, L.; Pushkarsky, T.; Sviridov, D.; Anashkina, A.A.; Üren, A.; Brichacek, B.; Vassylyev, D.G.; Adzhubei, A.A.; et al. Interaction Between HIV-1 Nef and Calnexin: From Modeling to Small Molecule Inhibitors Reversing HIV-Induced Lipid Accumulation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Feigl, E.O. Coronary physiology. Physiol. Rev. 1983, 63, 1–205. [Google Scholar] [CrossRef]
- Matsumotoa, T.; Kajiyab, F. Coronary microcirculation: Physiology and mechanics. Fluid Dyn. Res. 2005, 37, 60–81. [Google Scholar] [CrossRef]
- Bergantz, L.; Subra, F.; Deprez, E.; Delelis, O.; Richetta, C. Interplay between Intrinsic and Innate Immunity during HIV Infection. Cells 2019, 8, 922. [Google Scholar] [CrossRef]
- Friis-Moller, N.; Sabin, C.A.; Weber, R.; d’Arminio Monforte, A.; El-Sadr, W.M.; Reiss, P.; Thiébaut, R.; Morfeldt, L.; De Wit, S.; Pradier, C.; et al. Data Collection on Adverse Events of Anti-HIV Drugs (DAD) Study Group. Combination antiretroviral therapy and the risk of myocardial infarction. N. Engl. J. Med. 2003, 349, 1993–2003. [Google Scholar] [CrossRef] [PubMed]
- Nou, E.; Lo, J.; Hadigan, C.; Grinspoon, S.K. Pathophysiology and management of cardiovascular disease in patients with HIV. Lancet Diabetes Endocrinol. 2016, 4, 598–610. [Google Scholar] [CrossRef]
- Hsue, P.Y.; Waters, D.D. HIV infection and coronary heart disease: Mechanisms and management. Nat. Rev. Cardiol. 2019, 16, 745–759. [Google Scholar] [CrossRef]
- Wang, T.; Yi, R.; Green Linden, A.; Chelvanambi, S.; Seimetz, M.; Clauss, M. Increased cardiovascular disease risk in the HIV-positive population on ART: Potential role of HIV-Nef and Tat. Cardiovasc. Pathol. 2015, 24, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Rieckmann, P.; Sinowatz, F.; Weih, K.A.; Arthur, L.O.; Goebel, F.D.; Burd, P.R.; Coligan, J.E.; Clouse, K.A. Potent stimulation of monocytic endothelin-1 production by HIV-1 glycoprotein 120. J. Immunol. 1993, 150, 4601–4609. [Google Scholar]
- Ho, J.E.; Scherzer, R.; Hecht, F.M.; Maka, K.; Selby, V.; Martin, J.N.; Ganz, P.; Deeks, S.G.; Hsue, P.Y. The association of CD4+ T-cell counts and cardiovascular risk in treated HIV disease. AIDS 2012, 26, 1115–1120. [Google Scholar] [CrossRef]
- Hann, D.B.; Ramaswamy, C.; Kaplan, R.C.; Kizer Jorge, R.; Anastos, K.; Daskalakis, D.; Zimmerman, R.; Braunstein, S.L. Trends in cardiovascular disease mortality among persons with HIV in New York City, 2001–2012. Clin. Infect. Dis. 2016, 63, 1122–1129. [Google Scholar] [CrossRef]
- Subramanian, S.; Tawakol, A.; Burdo, T.H.; Abbara, S.; Wei, J.; Vijayakumar, J.; Corsini, E.; Abdelbaky, A.; Zanni, M.V.; Hoffmann, U.; et al. Arterial inflammation in patients with HIV. JAMA 2012, 308, 379–386. [Google Scholar] [CrossRef]
- Scherzer, R.; Shah, S.J.; Secemsky, E.; Butler, J.; Grunfeld, C.; Shlipak, M.G.; Hsue, P.Y. Association of Biomarker Clusters with Cardiac Phenotypes and Mortality in Patients with HIV Infection. Circ. Heart Fail. 2018, 11, e004312. [Google Scholar] [CrossRef] [PubMed]
- Leucker, T.M.; Weiss, R.G.; Schär, M.; Bonanno, G.; Mathews, L.; Jones, S.R.; Brown, T.T.; Moore, R.; Afework, Y.; Gerstenblith, G.; et al. Coronary Endothelial Dysfunction Is Associated With Elevated Serum PCSK9 Levels in People With HIV Independent of Low-Density Lipoprotein Cholesterol. J. Am. Heart Assoc. 2018, 7, e009996. [Google Scholar] [CrossRef] [PubMed]
- Masiá, M.; Robledano, C.; Ortiz de la Tabla, V.; Antequera, P.; López, N.; Gutiérrez, F. Increased carotid intima-media thickness associated with antibody responses to varicella-zoster virus and cytomegalovirus in HIV-infected patients. PLoS ONE 2013, 8, e64327. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A.; White, H.D. Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018). J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Camici, P.G.; Bairey Merz, C.N. Coronary microvascular dysfunction: An update. Eur. Heart J. 2014, 35, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Holmes, D.R.; Herrmann, J.; Gersh, B.J. Microcirculatory dysfunction in ST-elevation myocardial infarction: Cause, consequence, or both? Eur. Heart J. 2007, 28, 788–797. [Google Scholar] [CrossRef]
- Marzilli, M. Coronary microcirculation: The new frontier in coronary artery disease. Heart Metab. 2008, 38, 23–25. [Google Scholar]
- Caceres, V.H.U.; Lin, J.; Zhu, X.Y.; Favreau, F.D.; Gibson, M.E.; Crane, J.A.; Lerman, A.; Lerman, L.O. Early experimental hypertension preserves the myocardial microvasculature but aggravates cardiac injury distal to chronic coronary artery obstruction. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H693–H701. [Google Scholar] [CrossRef]
- Sorop, O.; Merkus, D.; de Beer, V.J.; Houweling, B.; Pistea, A.; McFalls, E.O.; Boomsma, F.; van Beusekom, H.M.; van der Giessen, W.J.; VanBavel, E.; et al. Functional and structural adaptations of coronary microvessels distal to a chronic coronary artery stenosis. Circ. Res. 2008, 102, 795–803. [Google Scholar] [CrossRef]
- Duncker, D.J.; Koller, A.; Merkus, D.; Canty, J.M., Jr. Regulation of coronary blood flow in health and ischemic heart disease. Prog. Cardiovasc. Dis. 2015, 57, 409–422. [Google Scholar] [CrossRef]
- Chen, Q.; Rosenson, R.S. Systematic Review of Methods Used for the Microvascular Assessment of Peripheral Arterial Disease. Cardiovasc. Drugs Ther. 2018, 32, 301–310. [Google Scholar] [CrossRef]
- Foà, A.; Agostini, V.; Rapezzi, C.; Olivotto, I.; Corti, B.; Potena, L.; Biagini, E.; Martin Suarez, S.; Rotellini, M.; Cecchi, F.; et al. Histopathological comparison of intramural coronary artery remodeling and myocardial fibrosis in obstructive versus end-stage hypertrophic cardiomyopathy. Int. J. Cardiol. 2019, 291, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Cicalini, S.; Petrosillo, N.; Russo, M.A.; Fedele, F.; Frustaci, A. High prevalence of intramural coronary infection in patients with drug-resistant cardiac syndrome X: Comparison with chronic stable angina and normal controls. Heart 2010, 96, 1926–1931. [Google Scholar] [CrossRef]
- Ong, P.; Safdar, B.; Seitz, A.; Hubert, A.; Beltrame, J.F.; Prescott, E. Diagnosis of coronary microvascular dysfunction in the clinic. Cardiovasc. Res. 2020, 116, 841–855. [Google Scholar] [CrossRef] [PubMed]
- Niccoli, G.; Scalone, G.; Lerman, A.; Crea, F. Coronary microvascular obstruction in acute myocardial infarction. Eur. Heart J. 2016, 37, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Tse, H.F. Microvascular obstruction after percutaneous coronary intervention. Catheter. Cardiovasc. Interv. 2010, 75, 369–377. [Google Scholar] [CrossRef]
- Mariani, M.V.; Magnocavallo, M.; Straito, M.; Piro, A.; Severino, P.; Iannucci, G.; Chimenti, C.; Mancone, M.; Rocca, D.G.D.; Forleo, G.B.; et al. Direct oral anticoagulants versus vitamin K antagonists in patients with atrial fibrillation and cancer a meta-analysis. J. Thromb. Thrombolysis. 2020. [Google Scholar] [CrossRef]
- Crump, R.; Shandling, A.H.; Van Natta, B.; Ellestad, M. Prevalence of patent foramen ovale in patients with acute myocardial infarction and angiographically normal coronary arteries. Am. J. Cardiol. 2000, 85, 1368–1370. [Google Scholar] [CrossRef]
- Bahrmann, P.; Werner, G.S.; Heusch, G.; Ferrari, M.; Poerner, T.C.; Voss, A.; Figulla, H.R. Detection of coronary microembolization by Doppler ultrasound in patients with stable angina pectoris undergoing elective percutaneous coronary interventions. Circulation 2007, 115, 600–608. [Google Scholar] [CrossRef][Green Version]
- Bahrmann, P.; Figulla, H.R.; Wagner, M.; Ferrari, M.; Voss, A.; Werner, G.S. Detection of coronary microembolisation by Doppler ultrasound during percutaneous coronary interventions. Heart 2005, 91, 1186–1192. [Google Scholar] [CrossRef][Green Version]
- Padro, T.; Manfrini, O.; Bugiardini, R.; Canty, J.; Cenko, E.; De Luca, G.; Duncker, D.J.; Eringa, E.C.; Koller, A.; Tousoulis, D.; et al. ESC Working Group on Coronary Pathophysiology and Microcirculation position paper on ‘coronary microvascular dysfunction in cardiovascular disease’. Cardiovasc. Res. 2020, 116, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhou, X.; Ji, W.J.; Lu, R.Y.; Zhang, Y.; Zhang, Y.D.; Ma, Y.Q.; Zhao, J.H.; Li, Y.M. Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: Therapeutic potential of DNase-based reperfusion strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500–H509. [Google Scholar] [CrossRef]
- Calcagno, S.; Infusino, F.; Salvi, N.; Taccheri, T.; Colantonio, R.; Bruno, E.; Birtolo, L.I.; Severino, P.; Lavalle, C.; Pucci, M.; et al. The Role of Ranolazine for the Treatment of Residual Angina beyond the Percutaneous Coronary Revascularization. J. Clin. Med. 2020, 9, 2110. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Jacoby, D.; Moon, J.C.; McKenna, W.J. Update on hypertrophic cardiomyopathy and a guide to the guidelines. Nat. Rev. Cardiol. 2016, 13, 651–675. [Google Scholar] [CrossRef]
- Spoladore, R.; Fisicaro, A.; Faccini, A.; Camici, P.G. Coronary microvascular dysfunction in primary cardiomyopathies. Heart 2014, 100, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Camici, P.G.; Olivotto, I.; Rimoldi, O.E. The coronary circulation and blood flow in left ventricular hypertrophy. J. Mol. Cell Cardiol. 2012, 52, 857–864. [Google Scholar] [CrossRef]
- Infusino, F.; Calcagno, S.; Cimino, S.; Pucci, M.; Salvi, N.; Maestrini, V.; Severino, P.; De Carlo, C.; Colantonio, R.; Sardella, G.; et al. Left ventricular wall stress is associated with myocardial functional recovery in patients with severe aortic stenosis and systolic dysfunction undergoing transcatheter aortic valve replacement. J. Cardiovasc. Med. (Hagerstown) 2020. [Google Scholar] [CrossRef]
- Camici, P.G.; d’Amati, G.; Rimoldi, O. Coronary microvascular dysfunction: Mechanisms and functional assessment. Nat. Rev. Cardiol. 2015, 12, 48–62. [Google Scholar] [CrossRef]
- Camici, P.G.; Crea, F. Coronary microvascular dysfunction. N. Engl. J. Med. 2007, 356, 830–840. [Google Scholar] [CrossRef]
- Cecchi, F.; Olivotto, I.; Gistri, R.; Lorenzoni, R.; Chiriatti, G.; Camici, P.G. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003, 349, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.E.; Jerosch-Herold, M.; Hudsmith, L.E.; Robson, M.D.; Francis, J.M.; Doll, H.A.; Selvanayagam, J.B.; Neubauer, S.; Watkins, H. Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: New insights from multiparametric magnetic resonance imaging. Circulation 2007, 115, 2418–2425. [Google Scholar] [CrossRef] [PubMed]
- Szekeres, M.; Nádasy, G.L.; Dörnyei, G.; Szénási, A.; Koller, A. Remodeling of Wall Mechanics and the Myogenic Mechanism of Rat Intramural Coronary Arterioles in Response to a Short-Term Daily Exercise Program: Role of Endothelial Factors. J. Vasc. Res. 2018, 55, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Severino, P.; D’Amato, A.; Pucci, M.; Infusino, F.; Birtolo, L.I.; Mariani, M.V.; Lavalle, C.; Maestrini, V.; Mancone, M.; Fedele, F. Ischemic Heart Disease and Heart Failure: Role of Coronary Ion Channels. Int. J. Mol. Sci. 2020, 21, 3167. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Skyschally, A.; Kleinbongard, P. Coronary microembolization and microvascular dysfunction. Int. J. Cardiol. 2018, 258, 17–23. [Google Scholar] [CrossRef]
- Kloner, R.A.; King, K.S.; Harrington, M.G. No-reflow phenomenon in the heart and brain. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H550–H562. [Google Scholar] [CrossRef]
- Heusch, G. Coronary microvascular obstruction: The new frontier in cardioprotection. Basic Res. Cardiol. 2019, 114, 45. [Google Scholar] [CrossRef]
- Hollander, M.R.; de Waard, G.A.; Konijnenberg, L.S.; Meijer-van Putten, R.M.; van den Brom, C.E.; Paauw, N.; de Vries, H.E.; van de Ven, P.M.; Aman, J.; Van Nieuw-Amerongen, G.P.; et al. Dissecting the Effects of Ischemia and Reperfusion on the Coronary Microcirculation in a Rat Model of Acute Myocardial Infarction. PLoS ONE 2016, 11, e0157233. [Google Scholar] [CrossRef]
- Ganame, J.; Messalli, G.; Dymarkowski, S.; Rademakers, F.E.; Desmet, W.; Van de Werf, F.; Bogaert, J. Impact of myocardial haemorrhage on left ventricular function and remodelling in patients with reperfused acute myocardial infarction. Eur. Heart J. 2009, 30, 1440–1449. [Google Scholar] [CrossRef]
- Gao, X.-M.; Wu, Q.-Z.; Kiriazis, H.; Su, Y.; Han, L.-P.; Pearson, J.T.; Taylor, A.J.; Du, X.J. Microvascular leakage in acutemyocardial infarction: Characterization by histology, biochemistry, and magnetic resonanceimaging. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1068–H1075. [Google Scholar] [CrossRef]
- Abela, G.S.; Kalavakunta, J.K.; Janoudi, A.; Leffler, D.; Dhar, G.; Salehi, N.; Cohn, J.; Shah, I.; Karve, M.; Kotaru, V.P.; et al. Frequency ofcholesterol crystals in culprit coronary artery aspirate during acutemyocardial infarction and their relation to inflammation and myocardialinjury. Am. J. Cardiol. 2017, 120, 1699–1707. [Google Scholar] [CrossRef]
- Yu, H.; Kalogeris, T.; Korthuis, R.J. Reactive species-induced microvascular dysfunction in ischemia/reperfusion. Free Radic. Biol. Med. 2019, 135, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Calvieri, C.; Masselli, G.; Monti, R.; Spreca, M.; Gualdi, G.F.; Fedele, F. Intramyocardial hemorrhage: An enigma for cardiac MRI? Biomed. Res. Int. 2015, 2015, 859073. [Google Scholar] [CrossRef]
- Kelm, N.Q.; Beare, J.E.; LeBlanc, A.J. Evaluation of Coronary Flow Reserve After Myocardial Ischemia Reperfusion in Rats. J. Vis. Exp. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Dai, L.; Wang, Y.; Zhang, X.; Li, C.; Jiang, S.; Li, Y.; Ding, Z.; Liu, L. HSPA12B Attenuated Acute Myocardial Ischemia/reperfusion Injury via Maintaining Endothelial Integrity in a PI3K/Akt/mTOR-dependent Mechanism. Sci. Rep. 2016, 6, 1. [Google Scholar] [CrossRef]
- Caricati-Neto, A.; Errante, P.R.; Menezes-Rodrigues, F.S. Recent Advances in Pharmacological and Non-Pharmacological Strategies of Cardioprotection. Int. J. Mol. Sci. 2019, 20, 4002. [Google Scholar] [CrossRef] [PubMed]
- Bulluck, H.; Chan, M.H.H.; Bryant, J.A.; Chai, P.; Chawla, A.; Chua, T.S.; Chung, Y.C.; Fei, G.; Ho, H.H.; Ho, A.F.W.; et al. Platelet inhibition to target reperfusion injury trial: Rationale and study design. Clin. Cardiol. 2019, 42, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Ardioprotection Cost Action (CA16225). Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, B.; Fuster, V.; Macaya, C.; Jiménez-Borreguero, J.; Iñiguez, A.; Fernández-Ortiz, A.; Sanz, G.; Sánchez-Brunete, V. Modulación del sistema betaadrenérgico durante el infarto agudo de miocardio: Justificación para un nuevo ensayo clínico [Modulation of the beta-adrenergic system during acute myocardial infarction: Rationale for a new clinical trial]. Rev. Esp. Cardiol. 2011, 64 (Suppl. 2), 28–33. [Google Scholar] [CrossRef]
- Pasupathy, S.; Tavella, R.; Grover, S.; Raman, B.; Procter, N.E.K.; Du, Y.T.; Mahadavan, G.; Stafford, I.; Heresztyn, T.; Holmes, A.; et al. Early use of N-acetylcysteine (NAC) with nitrate therapy in patients undergoing primary percutaneous coronary intervention for ST-segment-elevation myocardial infarction reduces myocardial infarct size (the NACIAM trial). Circulation 2017, 136, 894–903. [Google Scholar] [CrossRef]
- Stone, G.W.; Martin, J.L.; de Boer, M.J.; Margheri, M.; Bramucci, E.; Blankenship, J.C.; Metzger, D.C.; Gibbons, R.J.; Lindsay, B.S.; Weiner, B.H.; et al. MIHOT-II Trial Investigators. Effect of supersaturated oxygen delivery on infarct size after percutaneous coronary intervention in acute myocardial infarction. Circ. Cardiovasc. Interv. 2009, 2, 366–375. [Google Scholar] [CrossRef]
- Gao, X.M.; Su, Y.; Moore, S.; Han, L.P.; Kiriazis, H.; Lu, Q.; Zhao, W.B.; Ruze, A.; Fang, B.B.; Duan, M.J.; et al. Relaxin mitigates microvascular damage and inflammation following cardiac ischemia-reperfusion. Basic Res. Cardiol. 2019, 114, 30. [Google Scholar] [CrossRef] [PubMed]
- Marzilli, M.; Merz, C.N.; Boden, W.E.; Bonow, R.O.; Capozza, P.G.; Chilian, W.M.; DeMaria, A.N.; Guarini, G.; Huqi, A.; Morrone, D.; et al. Obstructive coronary atherosclerosis and ischemic heart disease: An elusive link! J. Am. Coll. Cardiol. 2012, 60, 951–956. [Google Scholar] [CrossRef]
- Marzilli, M.; Crea, F.; Morrone, D.; Bonow, R.O.; Brown, D.L.; Camici, P.G.; Chilian, W.M.; DeMaria, A.; Guarini, G.; Huqi, A.; et al. Myocardial ischemia: From disease to syndrome. Int. J. Cardiol. 2020, 314, 32–35. [Google Scholar] [CrossRef]
- Naya, M.; Murthy, V.L.; Blankstein, R.; Sitek, A.; Hainer, J.; Foster, C.; Gaber, M.; Fantony, J.M.; Dorbala, S.; Di Carli, M.F. Quantitative relationship between the extent and morphology of coronary atherosclerotic plaque and downstream myocardial perfusion. J. Am. Coll. Cardiol. 2011, 58, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Smilowitz, N.R.; Sampson, B.A.; Abrecht, C.R.; Siegfried, J.S.; Hochman, J.S.; Reynolds, H.R. Women have less severe and extensive coronary atherosclerosis in fatal cases of ischemic heart disease: An autopsy study. Am. Heart J. 2011, 161, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Planer, D.; Mehran, R.; Ohman, E.M.; White, H.D.; Newman, J.D.; Xu, K.; Stone, G.W. Prognosis of patients with non-ST-segment-elevation myocardial infarction and nonobstructive coronary artery disease: Propensity-matched analysis from the Acute Catheterization and Urgent Intervention Triage Strategy trial. Circ. Cardiovasc. Interv. 2014, 7, 285–293. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial Function and Dysfunction. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Knapp, M.; Tu, X.; Wu, R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol. Sin. 2019, 40, 1–8. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; Infusino, F.; Maestrini, V.; Mancone, M.; Fedele, F. Myocardial Ischemia and Diabetes Mellitus: Role of Oxidative Stress in the Connection between Cardiac Metabolism and Coronary Blood Flow. J. Diabetes Res. 2019, 2019, 1–19. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; De Marchis, M.; Palmirotta, R.; Volterrani, M.; Mancone, M.; Fedele, F. Diabetes Mellitus and Ischemic Heart Disease: The Role of Ion Channels. Int. J. Mol. Sci. 2018, 19, 802. [Google Scholar] [CrossRef] [PubMed]
- Kibel, A.; Selthofer-Relatic, K.; Drenjancevic, I.; Bacun, T.; Bosnjak, I.; Kibel, D.; Gros, M. Coronary microvascular dysfunction in diabetes mellitus. J. Int. Med. Res. 2017, 45, 1901–1929. [Google Scholar] [CrossRef] [PubMed]
- Athithan, L.; Gulsin, G.S.; McCann, G.P.; Levelt, E. Diabetic cardiomyopathy: Pathophysiology, theories and evidence to date. World J. Diabetes 2019, 10, 490–510. [Google Scholar] [CrossRef] [PubMed]
- Kunadian, V.; Chieffo, A.; Camici, P.G.; Berry, C.; Escaned, J.; Maas, A.; Prescott, E.; Karam, N.; Appelman, Y.; Fraccaro, C.; et al. An EAPCI Expert Consensus Document on Ischaemia with Non-Obstructive Coronary Arteries in Collaboration with European Society of Cardiology Working Group on Coronary Pathophysiology and Microcirculation Endorsed by Coronary Vasomotor Disorders International Study Group. EuroIntervention 2020. [Google Scholar] [CrossRef]
- McEntegart, M.; Eteiba, H.; Watkin, S.; Shaukat, A.; Lindsay, M.; Robertson, K.; Hood, S.; Yii, E.; Sidik, N.; Harvey, A.; et al. Systemic microvascular dysfunction in microvascular and vasospastic angina. Eur. Heart J. 2018, 39, 4086–4097. [Google Scholar] [CrossRef]
- Pacheco, C.C.; Quesada, O.; Pepine, C.J.; Bairey Merz, C. Why names matter for women: MINOCA/INOCA (myocardial infarction/ischemia and no obstructive coronary artery disease). Clin. Cardiol. 2018, 41, 185–193. [Google Scholar] [CrossRef]
- Rozanski, A.; Qureshi, E.; Bauman, M.; Reed, G.; Pillar, G.; Diamond, G.A. Peripheral arterial responses to treadmill exercise among healthy subjects and atherosclerotic patients. Circulation 2001, 103, 2084–2089. [Google Scholar] [CrossRef]
- Yun, J.S.; Park, Y.M.; Cha, S.A.; Ahn, Y.B.; Ko, S.H. Progression of cardiovascular autonomic neuropathy and cardiovascular disease in type 2 diabetes. Cardiovasc. Diabetol. 2018, 17, 109. [Google Scholar] [CrossRef]
- Evans, P.C.; Ed Rainger, G.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Saglietto, A.; D’Ascenzo, F.; Marini, C.; Schiavone, M.; Ghionzoli, N.; Pirrotta, F.; Troiano, F.; Cannillo, M.; et al. Reduction in heart failure hospitalization rate during coronavirus disease 19 pandemic outbreak. ESC Heart Fail. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hendren, N.S.; Drazner, M.H.; Bozkurt, B.; Cooper, L.T. Description and Proposed Management of the Acute COVID-19 Cardiovascular Syndrome. Circulation 2020, 141, 1903–1914. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Colmenero, I.; Santonja, C.; Alonso-Riaño, M.; Noguera-Morel, L.; Hernández-Martín, A.; Andina, D.; Wiesner, T.; Rodríguez-Peralto, J.L.; Requena, L.; Torrelo, A. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: Histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br. J. Dermatol. 2020, 183, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yang, B.; et al. Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely correlated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Piro, A.; Magnocavallo, M.; Della Rocca, D.G.; Neccia, M.; Manzi, G.; Mariani, M.V.; Straito, M.; Bernardini, A.; Severino, P.; Iannucci, G.; et al. Management of cardiac implantable electronic device follow-up in COVID-19 pandemic: Lessons learned during Italian lockdown. J. Cardiovasc. Electrophysiol. 2020. [Google Scholar] [CrossRef]
- Vabret, N.; Samstein, R.; Fernandez, N.; Merad, M. Sinai Immunology Review Project; Trainees; Faculty. Advancing scientific knowledge in times of pandemics. Nat. Rev. Immunol. 2020, 20, 338. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Montone, R.A.; Iannaccone, G.; Meucci, M.C.; Gurgoglione, F.; Niccoli, G. Myocardial and Microvascular Injury Due to Coronavirus Disease 2019. Eur. Cardiol. 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Coats, A.J.S.; Zheng, Z.; Adamo, M.; Ambrosio, G.; Anker, S.D.; Butler, J.; Xu, D.; Mao, J.; Khan, M.S.; et al. Management of heart failure patients with COVID -19: A joint position paper of the Chinese Heart Failure Association and National Heart Failure Committee and the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 941–956. [Google Scholar] [CrossRef]
- Guo, J.; Huang, Z.; Lin, L.; Lv, J. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease: A Viewpoint on the Potential Influence of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers on Onset and Severity of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Am. Heart Assoc. 2020, 9, e016219. [Google Scholar] [CrossRef] [PubMed]
- Fedele, F.; Infusino, F.; Severino, P.; Mancone, M. The role of cardiologists in the coronavirus disease 2019 pandemic. Kardiol. Pol. 2020, 78, 808–809. [Google Scholar] [CrossRef] [PubMed]
- Birtolo, L.I.; Maestrini, V.; Severino, P.; Chimenti, C.; Agnes, G.; Tocci, M.; Colaiacomo, M.C.; Francone, M.; Mancone, M.; Fedele, F. Coronavirus disease 2019 in Rome: Was it circulating before December? J. Cardiovasc. Med. (Hagerstown) 2020, 21, 835–836. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1–7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, D.; Italia, L.; Adamo, M.; Inciardi, R.M.; Lombardi, C.M.; Solomon, S.D.; Metra, M. COVID-19 and heart failure: From infection to inflammation and angiotensin II stimulation. Searching for evidence from a new disease. Eur. J. Heart Fail. 2020, 22, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Infusino, F.; Marazzato, M.; Mancone, M.; Fedele, F.; Mastroianni, C.M.; Severino, P.; Ceccarelli, G.; Santinelli, L.; Cavarretta, E.; Marullo, A.G.M.; et al. Diet Supplementation, Probiotics, and Nutraceuticals in SARS-CoV-2 Infection: A Scoping Review. Nutrients 2020, 12, 1718. [Google Scholar] [CrossRef]
- Staedtke, V.; Bai, R.Y.; Kim, K.; Darvas, M.; Davila, M.L.; Riggins, G.J.; Rothman, P.B.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Disruption of aself-amplifying catecholamine loop reduces cytokine release syndrome. Nature 2018, 564, 273–277. [Google Scholar] [CrossRef]
- Castiglione, V.; Chiriacò, M.; Emdin, M.; Taddei, S.; Vergaro, G. Statin therapy in COVID-19 infection. Eur. Heart J. Cardiovasc. Pharmacother. 2020, 6, 258–259. [Google Scholar] [CrossRef]
- Bifulco, M.; Gazzerro, P. Statin therapy in COVID-19 infection: Much more than a single pathway. Eur. Heart J. Cardiovasc. Pharmacother. 2020. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef]
- Sanchis-Gomar, F.; Lavie, C.J.; Perez-Quilis, C.; Henry, B.M.; Lippi, G. Angiotensin-Converting Enzyme 2 and Antihypertensives (Angiotensin Receptor Blockers and Angiotensin-Converting Enzyme Inhibitors) in Coronavirus Disease 2019. Mayo Clin. Proc. 2020, 95, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Recovery Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Pelliccia, F.; Kaski, J.C.; Crea, F.; Camici, P.G. Pathophysiology of Takotsubo Syndrome. Circulation 2017, 135, 2426–2441. [Google Scholar] [CrossRef]
- Y-Hassan, S. Post-ischemic myocardial stunning was the starting point of Takotsubo syndrome: Restitution is justified after falling down. Int. J. Cardiol. 2015, 198, 174–175. [Google Scholar] [CrossRef]
- Galiuto, L.; De Caterina, A.R.; Porfidia, A.; Paraggio, L.; Barchetta, S.; Locorotondo, G.; Rebuzzi, A.G.; Crea, F. Reversible coronary microvascular dysfunction: A common pathogenetic mechanism in apical ballooning or Tako-Tsubo syndrome. Eur. Heart J. 2010, 31, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.K. Acute stress-induced (takotsubo) cardiomyopathy. Heart 2018, 104, 96–102. [Google Scholar] [CrossRef]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2020, ehaa575. [Google Scholar] [CrossRef]
- Tamis-Holland, J.E.; Jneid, H.; Reynolds, H.R.; Agewall, S.; Brilakis, E.S.; Brown, T.M.; Lerman, A.; Cushman, M.; Kumbhani, D.J.; Arslanian-Engoren, C.; et al. Contemporary diagnosis and management of patients with myocardial infarction in the absence of obstructive coronary artery disease: A scientific statement from the American Heart Association. Circulation 2019, 139, e891–e908. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Baumgart, D.; Camici, P.; Chilian, W.; Gregorini, L.; Hess, O.; Indolfi, C.; Rimoldi, O. Alpha-adrenergic coronary vasoconstriction and myocardial ischemia in humans. Circulation 2000, 101, 689–694. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Scally, C.; Abbas, H.; Ahearn, T.; Srinivasan, J.; Mezincescu, A.; Rudd, A.; Spath, N.; Yucel-Finn, A.; Yuecel, R.; Oldroyd, K.; et al. Myocardial and Systemic Inflammation in Acute Stress-Induced (Takotsubo) Cardiomyopathy. Circulation 2019, 139, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Neishi, Y.; Mochizuki, S.; Miyasaka, T.; Kawamoto, T.; Kume, T.; Sukmawan, R.; Tsukiji, M.; Ogasawara, Y.; Kajiya, F.; Akasaka, T.; et al. Evaluation of bioavailability of nitric oxide in coronary circulation by direct measurement of plasma nitric oxide concentration. Proc. Natl. Acad. Sci. USA 2005, 102, 11456–11461. [Google Scholar] [CrossRef] [PubMed]
- Komaru, T.; Kanatsuka, H.; Shirato, K. Coronary microcirculation: Physiology and pharmacology. Pharmacol. Ther. 2000, 86, 217–261. [Google Scholar] [CrossRef]
- Chilian, W.M. Coronary microcirculation in health and disease. Summary of an NHLBI workshop. Circulation 1997, 95, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.E.; Mittleman, M.A.; Maclure, M.; Sherwood, J.B.; Tofler, G.H. Triggering myocardial infarction by sexual activity. Low absolute risk and prevention by regular physical exertion. Determinants of Myocardial Infarction Onset Study Investigators. JAMA 1996, 275, 1405–1409. [Google Scholar] [CrossRef]
- Hoffman, J.I.; Spaan, J.A. Pressure-flow relations in coronary circulation. Physiol. Rev. 1990, 70, 331–390. [Google Scholar] [CrossRef]
- Gewirtz, H. Coronary circulation: Pressure/flow parameters for assessment of ischemic heart disease. J. Nucl. Cardiol. 2019, 26, 459–470. [Google Scholar] [CrossRef]
- Pries, A.R.; Secomb, T.W. Origins of heterogeneity in tissue perfusion and metabolism. Cardiovasc. Res. 2009, 81, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Pries, A.R.; Cornelissen, A.J.; Sloot, A.A.; Sloot, A.; Hinkeldey, M.; Dreher, M.R.; Höpfner, M.; Dewhirst, M.W.; Secomb, T.W. Structural adaptation and heterogeneity of normal and tumor microvascular networks. PLoS Comput. Biol. 2009, 5, e1000394. [Google Scholar] [CrossRef] [PubMed]
- Pries, A.R.; Badimon, L.; Bugiardini, R.; Camici, P.G.; Dorobantu, M.; Duncker, D.J.; Escaned, J.; Koller, A.; Piek, J.J.; de Wit, C. Coronary vascular regulation, remodelling, and collateralization: Mechanisms and clinical implications on behalf of the working group on coronary pathophysiology and microcirculation. Eur. Heart J. 2015, 36, 3134–3146. [Google Scholar] [CrossRef]
- Jones, E.; Eteiba, W.; Merz, N.B. Cardiac syndrome X and microvascular coronary dysfunction. Trends Cardiovasc. Med. 2012, 22, 161–168. [Google Scholar] [CrossRef]
- Gulati, M.; Cooper-DeHoff, R.M.; McClure, C.; Johnson, B.D.; Shaw, L.J.; Handberg, E.M.; Zineh, I.; Kelsey, S.F.; Arnsdorf, M.F.; Black, H.R.; et al. Adverse cardiovascular outcomes in women with nonobstructive coronary artery disease: A report from the Women’s Ischemia Syndrome Evaluation Study and the St James Women Take Heart Project. Arch. Intern. Med. 2009, 169, 843–850. [Google Scholar] [CrossRef]
- Pepine, C.J.; Anderson, R.D.; Sharaf, B.L.; Reis, S.E.; Smith, K.M.; Handberg, E.M.; Johnson, B.D.; Sopko, G.; Bairey Merz, C.N. Coronary microvascular reactivity to adenosine predicts adverse outcome in women evaluated for suspected ischemia results from the National Heart, Lung and Blood Institute WISE (Women’s Ischemia Syndrome Evaluation) study. J. Am. Coll. Cardiol. 2010, 55, 2825–2832. [Google Scholar] [CrossRef]
- Reis, S.E.; Holubkov, R.; Smith, A.J.C.; Kelsey, S.F.; Sharaf, B.L.; Reichek, N.; Rogers, W.J.; Merz, C.N.; Sopko, G.; Pepine, C.J.; et al. Coronary microvascular dysfunction is highly prevalent in women with chest pain in the absence of coronary artery disease: Results from the NHLBI WISE study. Am. Heart J. 2001, 141, 735–741. [Google Scholar] [CrossRef]
- Von Mering, G.O.; Arant, C.B.; Wessel, T.R.; McGorray, S.P.; Bairey Merz, C.N.; Sharaf, B.L.; Smith, K.M.; Olson, M.B.; Johnson, B.D.; Sopko, G.; et al. Abnormal coronary vasomotion as a prognostic indicator of cardiovascular events in women: Results from the National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation 2004, 109, 722–725. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Weir, E.K. The metabolic basis of vascular oxygen sensing: Diversity, compartmentalization, and lessons from cancer. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H928–H930. [Google Scholar] [CrossRef][Green Version]
- Weir, E.K.; Lopez-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen sensing mechanisms. N. Engl. J. Med. 2005, 353, 2042–2055. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Thebaud, B.; Weir, E.K.; Archer, S.L. Hypoxic pulmonary vasoconstriction: Redox regulation of O2-sensitive K+ channels by a mitochondrial O2-sensor in resistance artery smooth muscle cells. J. Mol. Cell Cardiol. 2004, 37, 1119–1136. [Google Scholar] [CrossRef]
- Saitoh, S.I.; Zhang, C.; Tune, J.D.; Potter, B.; Kiyooka, T.; Rogers, P.A.; Knudson, J.D.; Dick, G.M.; Swafford, A.; Chilian, W.M. Hydrogen peroxide: A feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2614–2621. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.A.; Chilian, W.M.; Bratz, I.N.; Bryan, R.M.; Dick, G.M. H2O2 activates redox- and 4-aminopyridine-sensitive KV channels in coronary vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1404–H1411. [Google Scholar] [CrossRef]
- Pung, Y.F.; Chilian, W.M. Corruption of coronary collateral growth in metabolic syndrome: Role of oxidative stress. World J. Cardiol. 2010, 2, 421–427. [Google Scholar] [CrossRef]
- Ohanyan, V.; Yin, L.; Bardakjian, R.; Kolz, C.; Enrick, M.; Hakobyan, T.; Kmetz, J.; Bratz, I.; Luli, J.; Nagane, M.; et al. Requisite Role of Kv1.5 Channels in Coronary Metabolic Dilation. Circ. Res. 2015, 117, 612–621. [Google Scholar] [CrossRef]
- Fedele, F.; Mancone, M.; Chilian, W.M.; Severino, P.; Canali, E.; Logan, S.; De Marchis, M.L.; Volterrani, M.; Palmirotta, R.; Guadagni, F. Role of genetic polymorphisms of ion channels in the pathophysiology of coronary microvascular dysfunction and ischemic heart disease. Basic Res. Cardiol. 2013, 108, 387. [Google Scholar] [CrossRef]
- Severino, P.; D’Amato, A.; Netti, L.; Pucci, M.; Mariani, M.V.; Cimino, S.; Birtolo, L.I.; Infusino, F.; De Orchi, P.; Palmirotta, R.; et al. Susceptibility to ischaemic heart disease: Focusing on genetic variants for ATP-sensitive potassium channel beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef]
- Nieminen, M.S.; Buerke, M.; Parissis, J.; Ben-Gal, T.; Pollesello, P.; Kivikko, M.; Karavidas, A.; Severino, P.; Comín-Colet, J.; Wikström, G.; et al. Pharmaco-economics of levosimendan in cardiology: A European perspective. Int. J. Cardiol. 2015, 199, 337–341. [Google Scholar] [CrossRef]
- Severino, P.; Netti, L.; Mariani, M.V.; Maraone, A.; D’Amato, A.; Scarpati, R.; Infusino, F.; Pucci, M.; Lavalle, C.; Maestrini, V.; et al. Prevention of Cardiovascular Disease: Screening for Magnesium Deficiency. Cardiol. Res. Pract. 2019, 2019, 4874921. [Google Scholar] [CrossRef]
- Severino, P.; Mariani, M.V.; Fedele, F. Futility in cardiology: The need for a change in perspectives. Eur. J. Heart Fail. 2019, 21, 1483–1484. [Google Scholar] [CrossRef]
- Severino, P.; Maestrini, V.; Mariani, M.V.; Birtolo, L.I.; Scarpati, R.; Mancone, M.; Fedele, F. Structural and myocardial dysfunction in heart failure beyond ejection fraction. Heart Fail. Rev. 2020, 25, 9–17. [Google Scholar] [CrossRef]
- Severino, P.; Mariani, M.V.; Maraone, A.; Piro, A.; Ceccacci, A.; Tarsitani, L.; Maestrini, V.; Mancone, M.; Lavalle, C.; Pasquini, M.; et al. Triggers for Atrial Fibrillation: The Role of Anxiety. Cardiol. Res. Pract. 2019, 18, 1208505. [Google Scholar] [CrossRef] [PubMed]
- Severino, P.; Mather, P.J.; Pucci, M.; D’Amato, A.; Mariani, M.V.; Infusino, F.; Birtolo, L.I.; Maestrini, V.; Mancone, M.; Fedele, F. Advanced Heart Failure and End-Stage Heart Failure: Does a Difference Exist. Diagnostics 2019, 9, 170. [Google Scholar] [CrossRef]
- Fedele, F.; Mancone, M.; Adamo, F.; Severino, P. Heart Failure with Preserved, Mid-Range, and Reduced Ejection Fraction: The Misleading Definition of the New Guidelines. Cardiol. Rev. 2017, 25, 4–5. [Google Scholar] [CrossRef]
- Fedele, F.; Severino, P.; Calcagno, S.; Mancone, M. Heart failure: TNM-like classification. J. Am. Coll. Cardiol. 2014, 63, 1959–1960. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Severino, P.; D'Amato, A.; Pucci, M.; Infusino, F.; Adamo, F.; Birtolo, L.I.; Netti, L.; Montefusco, G.; Chimenti, C.; Lavalle, C.; et al. Ischemic Heart Disease Pathophysiology Paradigms Overview: From Plaque Activation to Microvascular Dysfunction. Int. J. Mol. Sci. 2020, 21, 8118. https://doi.org/10.3390/ijms21218118
Severino P, D'Amato A, Pucci M, Infusino F, Adamo F, Birtolo LI, Netti L, Montefusco G, Chimenti C, Lavalle C, et al. Ischemic Heart Disease Pathophysiology Paradigms Overview: From Plaque Activation to Microvascular Dysfunction. International Journal of Molecular Sciences. 2020; 21(21):8118. https://doi.org/10.3390/ijms21218118
Chicago/Turabian StyleSeverino, Paolo, Andrea D'Amato, Mariateresa Pucci, Fabio Infusino, Francesco Adamo, Lucia Ilaria Birtolo, Lucrezia Netti, Giulio Montefusco, Cristina Chimenti, Carlo Lavalle, and et al. 2020. "Ischemic Heart Disease Pathophysiology Paradigms Overview: From Plaque Activation to Microvascular Dysfunction" International Journal of Molecular Sciences 21, no. 21: 8118. https://doi.org/10.3390/ijms21218118
APA StyleSeverino, P., D'Amato, A., Pucci, M., Infusino, F., Adamo, F., Birtolo, L. I., Netti, L., Montefusco, G., Chimenti, C., Lavalle, C., Maestrini, V., Mancone, M., Chilian, W. M., & Fedele, F. (2020). Ischemic Heart Disease Pathophysiology Paradigms Overview: From Plaque Activation to Microvascular Dysfunction. International Journal of Molecular Sciences, 21(21), 8118. https://doi.org/10.3390/ijms21218118