Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions


Abstract
1. Introduction
2. Overview of NMDAR
3. Clinical Findings
3.1. NMDAR Expression in the Central Nervus System of Patients with Depression and Schizophrenia
3.2. Clinical Pharmacological Findings of NMDAR in Depression and Schizophrenia
4. Preclinical Findings
4.1. Behavioural Study
- (1)
- (2)
- (3)
4.2. Signal Transduction Associated with NMDAR
4.3. Neurotransmitter Release Associated with NMDAR
5. Candidate Pathophysiology of Depression and Schizophrenia Associated with NMDAR
5.1. Molecular Mechanism
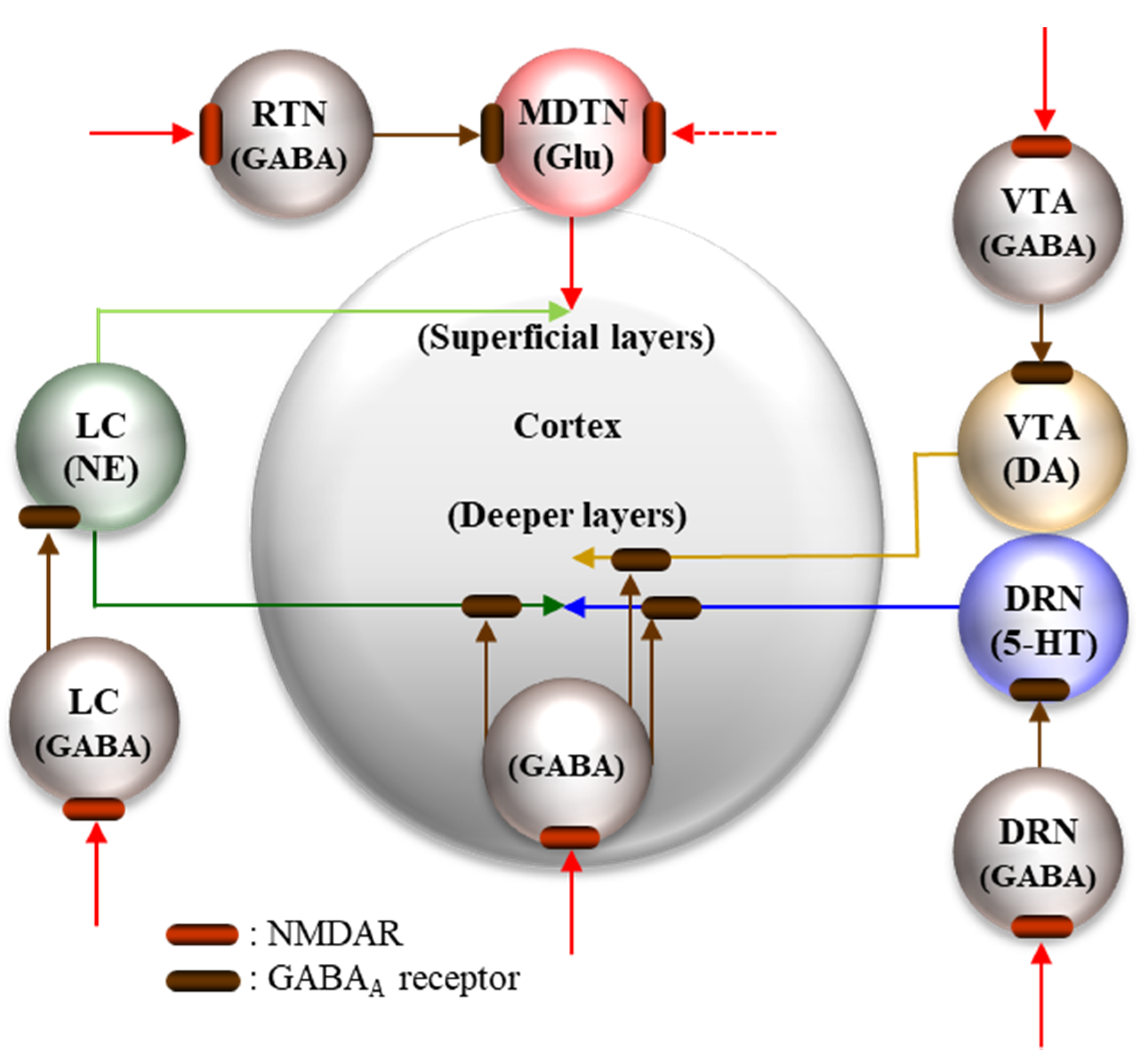
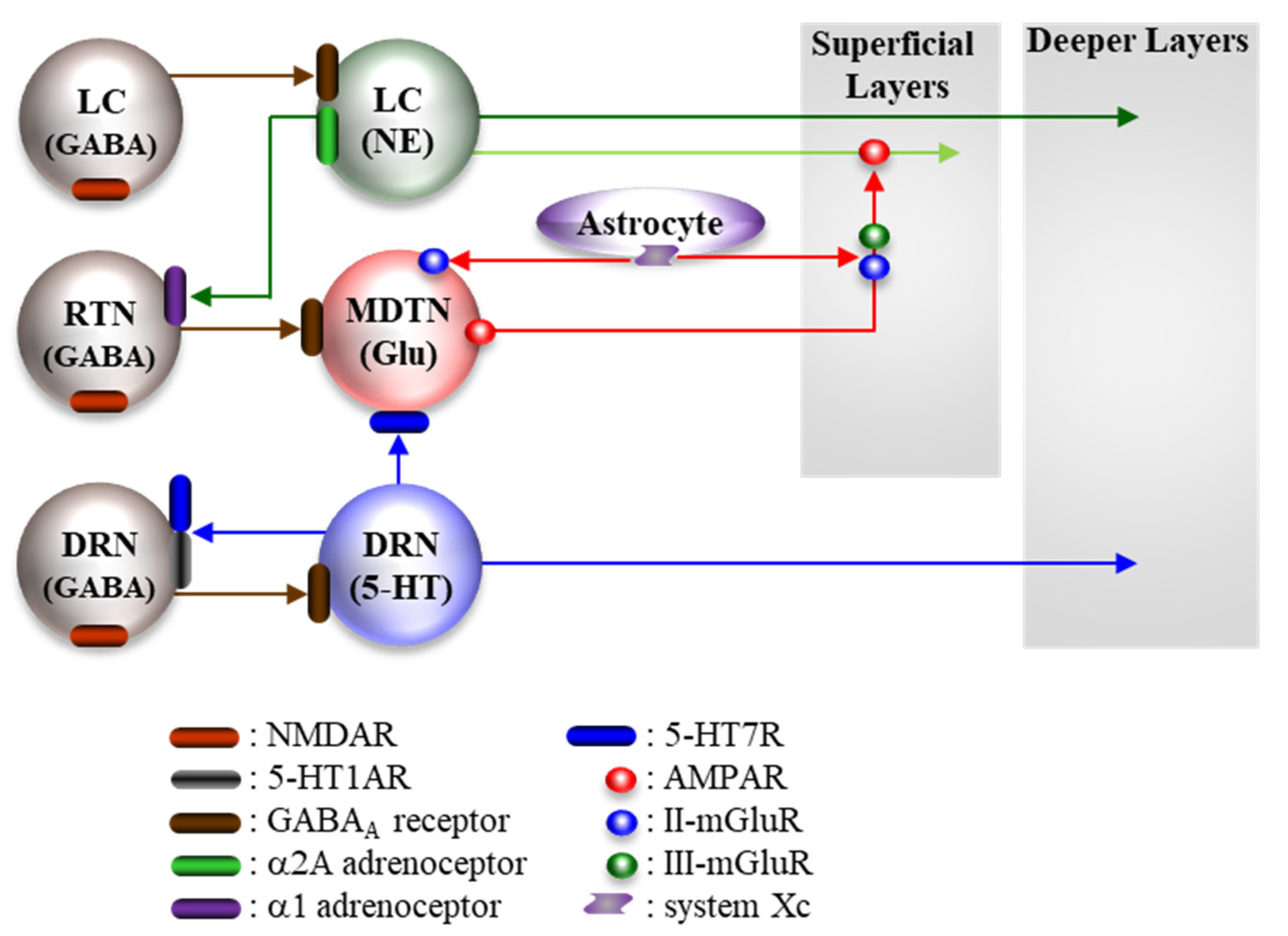
5.2. Pathophysiological Neural Circuits
5.2.1. Supressive Regulation of Enhanced Thalamocortical Glutamatergic Transmission
5.2.2. Stimulatory Regulation of Enhanced Thalamocortical Glutamatergic Transmission
6. Conclusions and Remaining Challenges
- Ideally, when a clear pharmacodynamic/pharmacokinetic distinction between the antidepressive and neuropsychotomimetic effects of NMDAR antagonists is achieved, then, according to the novel strategy, we can develop a new class of rapid-acting antidepressant for treatment of conventional monoaminergic antidepressant-resistant depression.
- Unfortunately, current findings suggest the induction mechanisms of NMDAR antagonists associated with antidepressive and neuropsychotomimetic effects are possibly identical. Therefore, it is important to identify the strategy of adjunctive therapies that gives antipsychotic effects without affecting the antidepressant effects of NMDAR antagonists.
- The high affinity dopamine D2 receptor partial agonistic action of ketamine possibly contributes to either its antidepressive or neuropsychotomimetic actions. Therefore, determination of the effects of adjuvant medication (typical and atypical antipsychotics) on the antidepressive and neuropsychotomimetic effects of ketamine is a rational strategy for the rapid-acting monoaminergic antidepressant-resistant depression therapy.
- If these above trials do not show beneficial outcomes, we should explore other neuromodulation therapies for prevention of the acute and chronic adverse effects of ketamine without affecting its antidepressive action.
- Preclinical findings suggest that distinct hippocampal and thalamic non-dopaminergic mechanisms play important roles in the ketamine-induced cognitive/memorial deficits. Thalamic nuclei that receive various inputs from cortical and subcortical regions integrate to give precise output to the frontal cortex. Therefore, conversion from tonic activation of thalamic activity induced by NMDAR inhibition to phasic activation/inhibition can lead to the development of cognitive promoting medication.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| II-mGluR | group II metabotropic glutamate receptors |
| III-mGluR | group III metabotropic glutamate receptors |
| 5-HT | serotonin |
| 5-HT2AR | serotonin receptor type 2A |
| 5-HT7R | serotonin receptor type 7 |
| ACTH | adrenocorticotropic hormone |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/glutamate receptor |
| BDNF | brain-derived neurotrophic factor |
| DRN | dorsal raphe nucleus |
| Erk | extracellular Signal-regulated Kinase |
| EMA | the European Medicines Agency |
| FDA | the Food and Drug Administration |
| fMRI | functional magnetic resonance imaging |
| GABA | γ-aminobutyrate |
| LC | locus coeruleus |
| MDTN | mediodorsal thalamic nucleus |
| MK801 | dizocilpine |
| mTOR | mammalian target of rapamycin |
| NMDAR | N-methyl-D-aspartate/glutamate receptor |
| OFC | orbitofrontal cortex |
| PPI | prepulse inhibition |
| RTN | reticular thalamic nucleus |
| SNRI | serotonin norepinephrine reuptake inhibitor |
| SSRI | selective serotonin reuptake inhibitor |
| VTA | ventral tegmental area |
References
- U.S. Food and Drug Administration. FDA Approves New Nasal Spray Medication for Treatment-Resistant Depression; Available only at a Certified Doctor’s Office or Clinic. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatment-resistant-depression-available-only-certified (accessed on 5 September 2020).
- European Medicines Agency. Esketamine Nasal Spray Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/spravato (accessed on 5 September 2020).
- Gaynes, B.N.; Warden, D.; Trivedi, M.H.; Wisniewski, S.R.; Fava, M.; Rush, A.J. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. 2009, 60, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.B.; Fedgchin, M.; Daly, E.J.; De Boer, P.; Cooper, K.; Lim, P.; Pinter, C.; Murrough, J.W.; Sanacora, G.; Shelton, R.C.; et al. A Double-Blind, Randomized, Placebo-Controlled, Dose-Frequency Study of Intravenous Ketamine in Patients With Treatment-Resistant Depression. Am. J. Psychiatry 2016, 173, 816–826. [Google Scholar] [CrossRef]
- DiazGranados, N.; Ibrahim, L.A.; Brutsche, N.E.; Ameli, R.; Henter, I.D.; Luckenbaugh, D.A.; Machado-Vieira, R.; Zarate, C.A., Jr. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J. Clin. Psychiatry 2010, 71, 1605–1611. [Google Scholar] [CrossRef]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Wilkinson, S.T.; Ballard, E.D.; Bloch, M.H.; Mathew, S.J.; Murrough, J.W.; Feder, A.; Sos, P.; Wang, G.; Zarate, C.A., Jr.; Sanacora, G. The Effect of a Single Dose of Intravenous Ketamine on Suicidal Ideation: A Systematic Review and Individual Participant Data Meta-Analysis. Am. J. Psychiatry 2018, 175, 150–158. [Google Scholar] [CrossRef]
- Lally, N.; Nugent, A.C.; Luckenbaugh, D.A.; Ameli, R.; Roiser, J.P.; Zarate, C.A. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl. Psychiatry 2014, 4, e469. [Google Scholar] [CrossRef]
- Moaddel, R.; Abdrakhmanova, G.; Kozak, J.; Jozwiak, K.; Toll, L.; Jimenez, L.; Rosenberg, A.; Tran, T.; Xiao, Y.; Zarate, C.A.; et al. Sub-anesthetic concentrations of (R, S)-ketamine metabolites inhibit acetylcholine-evoked currents in alpha7 nicotinic acetylcholine receptors. Eur. J. Pharmacol. 2013, 698, 228–234. [Google Scholar] [CrossRef]
- Ebert, B.; Mikkelsen, S.; Thorkildsen, C.; Borgbjerg, F.M. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur. J. Pharmacol. 1997, 333, 99–104. [Google Scholar] [CrossRef]
- White, P.F.; Schuttler, J.; Shafer, A.; Stanski, D.R.; Horai, Y.; Trevor, A.J. Comparative pharmacology of the ketamine isomers. Studies in volunteers. Br. J. Anaesth 1985, 57, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Negative schizophrenic symptomatology and the PCP (phencyclidine) model of schizophrenia. Hillside J. Clin. Psychiatry 1987, 9, 12–35. [Google Scholar] [PubMed]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [PubMed]
- Malhotra, A.K.; Pinals, D.A.; Adler, C.M.; Elman, I.; Clifton, A.; Pickar, D.; Breier, A. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Malhotra, A.K.; Pinals, D.A.; Weingartner, H.; Sirocco, K.; Missar, C.D.; Pickar, D.; Breier, A. NMDA receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 1996, 14, 301–307. [Google Scholar] [CrossRef]
- Martin, V.; Riffaud, A.; Marday, T.; Brouillard, C.; Franc, B.; Tassin, J.P.; Sevoz-Couche, C.; Mongeau, R.; Lanfumey, L. Response of Htr3a knockout mice to antidepressant treatment and chronic stress. Br. J. Pharm. 2017, 174, 2471–2483. [Google Scholar] [CrossRef]
- Conn, P.J.; Lindsley, C.W.; Jones, C.K. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharm. Sci. 2009, 30, 25–31. [Google Scholar] [CrossRef]
- Lisman, J.E.; Coyle, J.T.; Green, R.W.; Javitt, D.C.; Benes, F.M.; Heckers, S.; Grace, A.A. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008, 31, 234–242. [Google Scholar] [CrossRef]
- Aan Het Rot, M.; Zarate, C.A., Jr.; Charney, D.S.; Mathew, S.J. Ketamine for depression: Where do we go from here? Biol. Psychiatry 2012, 72, 537–547. [Google Scholar] [CrossRef]
- Leon, A.C.; Fiedorowicz, J.G.; Solomon, D.A.; Li, C.; Coryell, W.H.; Endicott, J.; Fawcett, J.; Keller, M.B. Risk of suicidal behavior with antidepressants in bipolar and unipolar disorders. J. Clin. Psychiatry 2014, 75, 720–727. [Google Scholar] [CrossRef]
- Beck, K.; Hindley, G.; Borgan, F.; Ginestet, C.; McCutcheon, R.; Brugger, S.; Driesen, N.; Ranganathan, M.; D’Souza, D.C.; Taylor, M.; et al. Association of Ketamine With Psychiatric Symptoms and Implications for Its Therapeutic Use and for Understanding Schizophrenia: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e204693. [Google Scholar] [CrossRef] [PubMed]
- Krystal, J.H.; Karper, L.P.; Seibyl, J.P.; Freeman, G.K.; Delaney, R.; Bremner, J.D.; Heninger, G.R.; Bowers, M.B., Jr.; Charney, D.S. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 1994, 51, 199–214. [Google Scholar] [CrossRef]
- Monyer, H.; Burnashev, N.; Laurie, D.J.; Sakmann, B.; Seeburg, P.H. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994, 12, 529–540. [Google Scholar] [CrossRef]
- Ulbrich, M.H.; Isacoff, E.Y. Rules of engagement for NMDA receptor subunits. Proc. Natl. Acad. Sci. USA 2008, 105, 14163–14168. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharm. 2012, 165, 1543–1555. [Google Scholar] [CrossRef]
- Gocho, Y.; Sakai, A.; Yanagawa, Y.; Suzuki, H.; Saitow, F. Electrophysiological and pharmacological properties of GABAergic cells in the dorsal raphe nucleus. J. Physiol. Sci. 2013, 63, 147–154. [Google Scholar] [CrossRef]
- Hu, H.; Jonas, P. A supercritical density of Na(+) channels ensures fast signaling in GABAergic interneuron axons. Nat. Neurosci. 2014, 17, 686–693. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K. Interaction between Mesocortical and Mesothalamic Catecholaminergic Transmissions Associated with NMDA Receptor in the Locus Coeruleus. Biomolecules 2020, 10, 990. [Google Scholar] [CrossRef]
- Okada, M.; Okubo, R.; Fukuyama, K. Vortioxetine Subchronically Activates Serotonergic Transmission via Desensitization of Serotonin 5-HT1A Receptor with 5-HT3 Receptor Inhibition in Rats. Int. J. Mol. Sci. 2019, 20, 6235. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Ueda, Y. Lurasidone inhibits NMDA/glutamate antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharm. 2019. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone sub-chronically activates serotonergic transmission via desensitization of 5-HT1A and 5-HT7 receptors in dorsal raphe nucleus. Pharmaceuticals 2019, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Nakano, T.; Ueda, Y. Pharmacological Discrimination of Effects of MK801 on Thalamocortical, Mesothalamic, and Mesocortical Transmissions. Biomolecules 2019, 9, 746. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc. Pharm. Res. Perspect 2019, 7, e00457. [Google Scholar] [CrossRef]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine Combines Astroglial System Xc(-) Activation with Glutamate/NMDA Receptor Inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine D2 and serotonin 5-HT1A receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef]
- Tanahashi, S.; Ueda, Y.; Nakajima, A.; Yamamura, S.; Nagase, H.; Okada, M. Novel delta1-receptor agonist KNT-127 increases the release of dopamine and L-glutamate in the striatum, nucleus accumbens and median pre-frontal cortex. Neuropharmacology 2012, 62, 2057–2067. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Nakagawa, M.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Tanii, H.; Shiroyama, T.; Okada, M. Effects of zotepine on extracellular levels of monoamine, GABA and glutamate in rat prefrontal cortex. Br. J. Pharm. 2009, 157, 656–665. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Kashimoto, K.; Nakagawa, M.; Kanehara, S.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Shiroyama, T.; et al. Effects of quetiapine on monoamine, GABA, and glutamate release in rat prefrontal cortex. Psychopharmacology 2009, 206, 243–258. [Google Scholar] [CrossRef]
- Stephenson, F.A. Structure and trafficking of NMDA and GABAA receptors. Biochem. Soc. Trans. 2006, 34, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Salussolia, C.L.; Prodromou, M.L.; Borker, P.; Wollmuth, L.P. Arrangement of subunits in functional NMDA receptors. J. Neurosci. J. Soc. Neurosci. 2011, 31, 11295–11304. [Google Scholar] [CrossRef] [PubMed]
- Perez-Otano, I.; Larsen, R.S.; Wesseling, J.F. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat. Reviews. Neurosci. 2016, 17, 623–635. [Google Scholar] [CrossRef]
- Pachernegg, S.; Strutz-Seebohm, N.; Hollmann, M. GluN3 subunit-containing NMDA receptors: Not just one-trick ponies. Trends Neurosci. 2012, 35, 240–249. [Google Scholar] [CrossRef]
- Gray, A.; Hyde, T.; Deep-Soboslay, A.; Kleinman, J.; Sodhi, M. Sex differences in glutamate receptor gene expression in major depression and suicide. Mol. Psychiatry 2015, 20, 1057–1068. [Google Scholar] [CrossRef]
- Lin, E.; Kuo, P.-H.; Liu, Y.-L.; Yu, Y.W.-Y.; Yang, A.C.; Tsai, S.-J. A deep learning approach for predicting antidepressant response in major depression using clinical and genetic biomarkers. Front. Psychiatry 2018, 9, 290. [Google Scholar] [CrossRef]
- Zhang, C.; Li, Z.; Wu, Z.; Chen, J.; Wang, Z.; Peng, D.; Hong, W.; Yuan, C.; Wang, Z.; Yu, S. A study of N-methyl-D-aspartate receptor gene (GRIN2B) variants as predictors of treatment-resistant major depression. Psychopharmacology 2014, 231, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Weder, N.; Zhang, H.; Jensen, K.; Yang, B.Z.; Simen, A.; Jackowski, A.; Lipschitz, D.; Douglas-Palumberi, H.; Ge, M.; Perepletchikova, F. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child. Adolesc. Psychiatry 2014, 53, 417–424. e415. [Google Scholar] [CrossRef]
- Kaut, O.; Schmitt, I.; Hofmann, A.; Hoffmann, P.; Schlaepfer, T.E.; Wüllner, U.; Hurlemann, R. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 331–341. [Google Scholar] [CrossRef]
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef]
- Calabrese, F.; Guidotti, G.; Molteni, R.; Racagni, G.; Mancini, M.; Riva, M.A. Stress-induced changes of hippocampal NMDA receptors: Modulation by duloxetine treatment. PLoS ONE 2012, 7, e37916. [Google Scholar] [CrossRef] [PubMed]
- Chandley, M.J.; Szebeni, A.; Szebeni, K.; Crawford, J.D.; Stockmeier, C.A.; Turecki, G.; Kostrzewa, R.M.; Ordway, G.A. Elevated gene expression of glutamate receptors in noradrenergic neurons from the locus coeruleus in major depression. Int. J. Neuropsychopharmacol. 2014, 17, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Karolewicz, B.; Stockmeier, C.A.; Ordway, G.A. Elevated levels of the NR2C subunit of the NMDA receptor in the locus coeruleus in depression. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2005, 30, 1557–1567. [Google Scholar] [CrossRef][Green Version]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Callado, L.F.; Meana, J.J.; Garzón-Niño, J. Schizophrenia and depression, two poles of endocannabinoid system deregulation. Transl. Psychiatry 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Feyissa, A.M.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 70–75. [Google Scholar] [CrossRef]
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 1888–1902. [Google Scholar] [CrossRef]
- Karolewicz, B.; Szebeni, K.; Gilmore, T.; Maciag, D.; Stockmeier, C.A.; Ordway, G.A. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. Int. J. Neuropsychopharmacol. 2009, 12, 143–153. [Google Scholar] [CrossRef]
- Weickert, C.S.; Fung, S.; Catts, V.; Schofield, P.; Allen, K.; Moore, L.; Newell, K.A.; Pellen, D.; Huang, X.-F.; Catts, S. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry 2013, 18, 1185–1192. [Google Scholar] [CrossRef]
- Akbarian, S.; Sucher, N.; Bradley, D.; Tafazzoli, A.; Trinh, D.; Hetrick, W.; Potkin, S.; Sandman, C.; Bunney, W.; Jones, E. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 1996, 16, 19–30. [Google Scholar] [CrossRef]
- Catts, V.S.; Derminio, D.S.; Hahn, C.-G.; Weickert, C.S. Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. npj Schizophrenia 2015, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Law, A.J.; Deakin, J. Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport 2001, 12, 2971–2974. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-M.; Sakai, K.; Roberts, R.C.; Conley, R.R.; Dean, B.; Tamminga, C.A. Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: Effects of schizophrenia. Am. J. Psychiatry 2000, 157, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, B.P. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: Evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 1998, 71, 2454–2464. [Google Scholar] [CrossRef]
- Banerjee, A.; Wang, H.-Y.; Borgmann-Winter, K.E.; MacDonald, M.L.; Kaprielian, H.; Stucky, A.; Kvasic, J.; Egbujo, C.; Ray, R.; Talbot, K. Src kinase as a mediator of convergent molecular abnormalities leading to NMDAR hypoactivity in schizophrenia. Mol. Psychiatry 2015, 20, 1091–1100. [Google Scholar] [CrossRef]
- Stone, J.M.; Erlandsson, K.; Arstad, E.; Squassante, L.; Teneggi, V.; Bressan, R.A.; Krystal, J.H.; Ell, P.J.; Pilowsky, L.S. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: A [(123)I] CNS-1261 SPET study. Psychopharmacology 2008, 197, 401–408. [Google Scholar] [CrossRef]
- Luby, E.D.; Cohen, B.D.; Rosenbaum, G.; Gottlieb, J.S.; Kelley, R. Study of a new schizophrenomimetic drug; sernyl. A.M.A. Arch. Neurol. Psychiatry 1959, 81, 363–369. [Google Scholar] [CrossRef]
- Domino, E.F.; Chodoff, P.; Corssen, G. Pharmacologic Effects of Ci-581, a New Dissociative Anesthetic, in Man. Clin. Pharmacol. Ther. 1965, 6, 279–291. [Google Scholar] [CrossRef]
- Lin, C.Y.; Liang, S.Y.; Chang, Y.C.; Ting, S.Y.; Kao, C.L.; Wu, Y.H.; Tsai, G.E.; Lane, H.Y. Adjunctive sarcosine plus benzoate improved cognitive function in chronic schizophrenia patients with constant clinical symptoms: A randomised, double-blind, placebo-controlled trial. World J. Biol. Psychiatry 2017, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.Y.; Lin, C.H.; Green, M.F.; Hellemann, G.; Huang, C.C.; Chen, P.W.; Tun, R.; Chang, Y.C.; Tsai, G.E. Add-on treatment of benzoate for schizophrenia: A randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry 2013, 70, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K.; Miyake, Y. Molecular cloning and chromosomal localization of a human gene encoding D-amino-acid oxidase. J. Biol. Chem. 1992, 267, 18631–18638. [Google Scholar] [PubMed]
- Sasabe, J.; Miyoshi, Y.; Suzuki, M.; Mita, M.; Konno, R.; Matsuoka, M.; Hamase, K.; Aiso, S. D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc. Natl. Acad. Sci. USA 2012, 109, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Murrough, J.W.; Soleimani, L.; DeWilde, K.E.; Collins, K.A.; Lapidus, K.A.; Iacoviello, B.M.; Lener, M.; Kautz, M.; Kim, J.; Stern, J.B.; et al. Ketamine for rapid reduction of suicidal ideation: A randomized controlled trial. Psychol. Med. 2015, 45, 3571–3580. [Google Scholar] [CrossRef]
- Grunebaum, M.F.; Galfalvy, H.C.; Choo, T.-H.; Keilp, J.G.; Moitra, V.K.; Parris, M.S.; Marver, J.E.; Burke, A.K.; Milak, M.S.; Sublette, M.E.; et al. Ketamine for Rapid Reduction of Suicidal Thoughts in Major Depression: A Midazolam-Controlled Randomized Clinical Trial. Am. J. Psychiatry 2018, 175, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Murrough, J.W.; Perez, A.M.; Pillemer, S.; Stern, J.; Parides, M.K.; aan het Rot, M.; Collins, K.A.; Mathew, S.J.; Charney, D.S.; Iosifescu, D.V. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol. Psychiatry 2013, 74, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Diazgranados, N.; Ibrahim, L.; Brutsche, N.E.; Newberg, A.; Kronstein, P.; Khalife, S.; Kammerer, W.A.; Quezado, Z.; Luckenbaugh, D.A.; Salvadore, G.; et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 2010, 67, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A., Jr.; Brutsche, N.E.; Ibrahim, L.; Franco-Chaves, J.; Diazgranados, N.; Cravchik, A.; Selter, J.; Marquardt, C.A.; Liberty, V.; Luckenbaugh, D.A. Replication of ketamine’s antidepressant efficacy in bipolar depression: A randomized controlled add-on trial. Biol. Psychiatry 2012, 71, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Murrough, J.W.; Iosifescu, D.V.; Chang, L.C.; Al Jurdi, R.K.; Green, C.E.; Perez, A.M.; Iqbal, S.; Pillemer, S.; Foulkes, A.; Shah, A.; et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: A two-site randomized controlled trial. Am. J. Psychiatry 2013, 170, 1134–1142. [Google Scholar] [CrossRef]
- Lapidus, K.A.; Levitch, C.F.; Perez, A.M.; Brallier, J.W.; Parides, M.K.; Soleimani, L.; Feder, A.; Iosifescu, D.V.; Charney, D.S.; Murrough, J.W. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol. Psychiatry 2014, 76, 970–976. [Google Scholar] [CrossRef]
- Hu, Y.D.; Xiang, Y.T.; Fang, J.X.; Zu, S.; Sha, S.; Shi, H.; Ungvari, G.S.; Correll, C.U.; Chiu, H.F.; Xue, Y.; et al. Single i.v. ketamine augmentation of newly initiated escitalopram for major depression: Results from a randomized, placebo-controlled 4-week study. Psychol. Med. 2016, 46, 623–635. [Google Scholar] [CrossRef]
- Li, C.-T.; Chen, M.-H.; Lin, W.-C.; Hong, C.-J.; Yang, B.-H.; Liu, R.-S.; Tu, P.-C.; Su, T.-P. The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: A randomized controlled study. Hum. Brain Mapp. 2016, 37, 1080–1090. [Google Scholar] [CrossRef]
- Su, T.-P.; Chen, M.-H.; Li, C.-T.; Lin, W.-C.; Hong, C.-J.; Gueorguieva, R.; Tu, P.-C.; Bai, Y.-M.; Cheng, C.-M.; Krystal, J.H. Dose-Related Effects of Adjunctive Ketamine in Taiwanese Patients with Treatment-Resistant Depression. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2017, 42, 2482–2492. [Google Scholar] [CrossRef] [PubMed]
- Grunebaum, M.F.; Ellis, S.P.; Keilp, J.G.; Moitra, V.K.; Cooper, T.B.; Marver, J.E.; Burke, A.K.; Milak, M.S.; Sublette, M.E.; Oquendo, M.A.; et al. Ketamine versus midazolam in bipolar depression with suicidal thoughts: A pilot midazolam-controlled randomized clinical trial. Bipolar Disord. 2017, 19, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Fava, M.; Freeman, M.P.; Flynn, M.; Judge, H.; Hoeppner, B.B.; Cusin, C.; Ionescu, D.F.; Mathew, S.J.; Chang, L.C.; Iosifescu, D.V.; et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol. Psychiatry 2018, 25, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.L.; Norris, S.; Talbot, J.; Birmingham, M.; Hatchard, T.; Ortiz, A.; Owoeye, O.; Batten, L.A.; Blier, P. Single, Repeated, and Maintenance Ketamine Infusions for Treatment-Resistant Depression: A Randomized Controlled Trial. Am. J. Psychiatry 2019, 176, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, D.F.; Bentley, K.H.; Eikermann, M.; Taylor, N.; Akeju, O.; Swee, M.B.; Pavone, K.J.; Petrie, S.R.; Dording, C.; Mischoulon, D.; et al. Repeat-dose ketamine augmentation for treatment-resistant depression with chronic suicidal ideation: A randomized, double blind, placebo controlled trial. J. Affect. Disord. 2019, 243, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.B.; Fedgchin, M.; Daly, E.; Xi, L.; Melman, C.; De Bruecker, G.; Tadic, A.; Sienaert, P.; Wiegand, F.; Manji, H.; et al. Intravenous Esketamine in Adult Treatment-Resistant Depression: A Double-Blind, Double-Randomization, Placebo-Controlled Study. Biol. Psychiatry 2016, 80, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Daly, E.J.; Singh, J.B.; Fedgchin, M.; Cooper, K.; Lim, P.; Shelton, R.C.; Thase, M.E.; Winokur, A.; Van Nueten, L.; Manji, H. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: A randomized clinical trial. JAMA Psychiatry 2018, 75, 139–148. [Google Scholar] [CrossRef]
- Canuso, C.M.; Singh, J.B.; Fedgchin, M.; Alphs, L.; Lane, R.; Lim, P.; Pinter, C.; Hough, D.; Sanacora, G.; Manji, H.; et al. Efficacy and Safety of Intranasal Esketamine for the Rapid Reduction of Symptoms of Depression and Suicidality in Patients at Imminent Risk for Suicide: Results of a Double-Blind, Randomized, Placebo-Controlled Study. Am. J. Psychiatry 2018, 175, 620–630. [Google Scholar] [CrossRef]
- Popova, V.; Daly, E.J.; Trivedi, M.; Cooper, K.; Lane, R.; Lim, P.; Mazzucco, C.; Hough, D.; Thase, M.E.; Shelton, R.C.; et al. Efficacy and Safety of Flexibly Dosed Esketamine Nasal Spray Combined With a Newly Initiated Oral Antidepressant in Treatment-Resistant Depression: A Randomized Double-Blind Active-Controlled Study. Am. J. Psychiatry 2019, 176, 428–438. [Google Scholar] [CrossRef]
- Fedgchin, M.; Trivedi, M.; Daly, E.J.; Melkote, R.; Lane, R.; Lim, P.; Vitagliano, D.; Blier, P.; Fava, M.; Liebowitz, M.; et al. Efficacy and Safety of Fixed-Dose Esketamine Nasal Spray Combined With a New Oral Antidepressant in Treatment-Resistant Depression: Results of a Randomized, Double-Blind, Active-Controlled Study (TRANSFORM-1). Int. J. Neuropsychopharmacol. 2019, 22, 616–630. [Google Scholar] [CrossRef]
- Correia-Melo, F.S.; Leal, G.C.; Vieira, F.; Jesus-Nunes, A.P.; Mello, R.P.; Magnavita, G.; Caliman-Fontes, A.T.; Echegaray, M.V.F.; Bandeira, I.D.; Silva, S.S.; et al. Efficacy and safety of adjunctive therapy using esketamine or racemic ketamine for adult treatment-resistant depression: A randomized, double-blind, non-inferiority study. J. Affect. Disord. 2020, 264, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Ochs-Ross, R.; Daly, E.J.; Zhang, Y.; Lane, R.; Lim, P.; Morrison, R.L.; Hough, D.; Manji, H.; Drevets, W.C.; Sanacora, G.; et al. Efficacy and Safety of Esketamine Nasal Spray Plus an Oral Antidepressant in Elderly Patients With Treatment-Resistant Depression—TRANSFORM-3. Am. J. Geriatr. Psychiatry 2020, 28, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Preskorn, S.H.; Baker, B.; Kolluri, S.; Menniti, F.S.; Krams, M.; Landen, J.W. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J. Clin. Psychopharmacol. 2008, 28, 631–637. [Google Scholar] [CrossRef]
- Ibrahim, L.; Diaz Granados, N.; Jolkovsky, L.; Brutsche, N.; Luckenbaugh, D.A.; Herring, W.J.; Potter, W.Z.; Zarate, C.A., Jr. A Randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J. Clin. Psychopharmacol. 2012, 32, 551–557. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A., Jr.; et al. Ketamine and Ketamine Metabolite Pharmacology: Insights into Therapeutic Mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef]
- Vollenweider, F.X.; Leenders, K.L.; Oye, I.; Hell, D.; Angst, J. Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET). Eur Neuropsychopharmacol 1997, 7, 25–38. [Google Scholar] [CrossRef]
- Short, B.; Fong, J.; Galvez, V.; Shelker, W.; Loo, C.K. Side-effects associated with ketamine use in depression: A systematic review. Lancet Psychiatry 2018, 5, 65–78. [Google Scholar] [CrossRef]
- Seeman, P.; Guan, H.C.; Hirbec, H. Dopamine D2High receptors stimulated by phencyclidines, lysergic acid diethylamide, salvinorin A, and modafinil. Synapse 2009, 63, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Kokkinou, M.; Ashok, A.H.; Howes, O.D. The effects of ketamine on dopaminergic function: Meta-analysis and review of the implications for neuropsychiatric disorders. Mol. Psychiatry 2018, 23, 59–69. [Google Scholar] [CrossRef]
- Seeman, P.; Ko, F.; Tallerico, T. Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol. Psychiatry 2005, 10, 877–883. [Google Scholar] [CrossRef]
- Kapur, S.; Seeman, P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol. Psychiatry 2002, 7, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Akhlaghi, N.; Payandemehr, P.; Yaseri, M.; Akhlaghi, A.A.; Abdolrazaghnejad, A. Premedication With Midazolam or Haloperidol to Prevent Recovery Agitation in Adults Undergoing Procedural Sedation With Ketamine: A Randomized Double-Blind Clinical Trial. Ann. Emerg. Med. 2019, 73, 462–469. [Google Scholar] [CrossRef]
- Chen, Y.M.; Lin, C.H.; Lane, H.Y. Survey of NMDA Receptor-related Biomarkers for Depression. Curr. Pharm. Des. 2020, 26, 228–235. [Google Scholar] [CrossRef]
- Singh, S.P.; Singh, V. Meta-analysis of the efficacy of adjunctive NMDA receptor modulators in chronic schizophrenia. CNS Drugs 2011, 25, 859–885. [Google Scholar] [CrossRef]
- Levin, R.; Dor-Abarbanel, A.E.; Edelman, S.; Durrant, A.R.; Hashimoto, K.; Javitt, D.C.; Heresco-Levy, U. Behavioral and cognitive effects of the N-methyl-D-aspartate receptor co-agonist D-serine in healthy humans: Initial findings. J. Psychiatr. Res. 2015, 61, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.X.; Fu, J.; Ma, S.R.; Peng, R.; Yu, J.B.; Cong, L.; Pan, L.B.; Zhang, Z.G.; Tian, H.; Che, C.T.; et al. Gut-brain axis metabolic pathway regulates antidepressant efficacy of albiflorin. Theranostics 2018, 8, 5945–5959. [Google Scholar] [CrossRef]
- Homayoun, H.; Stefani, M.R.; Adams, B.W.; Tamagan, G.D.; Moghaddam, B. Functional interaction between NMDA and mGlu5 receptors: Effects on working memory, instrumental learning, motor behaviors, and dopamine release. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2004, 29, 1259–1269. [Google Scholar] [CrossRef]
- Mouri, A.; Koseki, T.; Narusawa, S.; Niwa, M.; Mamiya, T.; Kano, S.-i.; Sawa, A.; Nabeshima, T. Mouse strain differences in phencyclidine-induced behavioural changes. Int. J. Neuropsychopharmacol. 2012, 15, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Domino, E.F. Genetic differences in the locomotor response to single and daily doses of phencyclidine in inbred mouse strains. Behav. Pharmacol. 1994, 5, 623–629. [Google Scholar] [CrossRef]
- Castañé, A.; Santana, N.; Artigas, F. PCP-based mice models of schizophrenia: Differential behavioral, neurochemical and cellular effects of acute and subchronic treatments. Psychopharmacology 2015, 232, 4085–4097. [Google Scholar] [CrossRef]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Tran, A.; Taylor, J.R.; Roth, R.H. Prefrontal cortical involvement in phencyclidine-induced activation of the mesolimbic dopamine system: Behavioral and neurochemical evidence. Psychopharmacology 1998, 138, 89–95. [Google Scholar] [CrossRef]
- Sams-Dodd, F. Effect of novel antipsychotic drugs on phencyclidine-induced stereotyped behaviour and social isolation in the rat social interaction test. Behav Pharm. 1997, 8, 196–215. [Google Scholar]
- Adell, A.; Jiménez-Sánchez, L.; López-Gil, X.; Romón, T. Is the acute NMDA receptor hypofunction a valid model of schizophrenia? Schizophr. Bull. 2012, 38, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Broberg, B.V.; Oranje, B.; Glenthoj, B.Y.; Fejgin, K.; Plath, N.; Bastlund, J.F. Assessment of auditory sensory processing in a neurodevelopmental animal model of schizophrenia--gating of auditory-evoked potentials and prepulse inhibition. Behav. Brain Res. 2010, 213, 142–147. [Google Scholar] [CrossRef]
- Bakshi, V.P.; Tricklebank, M.; Neijt, H.C.; Lehmann-Masten, V.; Geyer, M.A. Disruption of prepulse inhibition and increases in locomotor activity by competitive N-methyl-D-aspartate receptor antagonists in rats. J. Pharm. Exp. 1999, 288, 643–652. [Google Scholar]
- Thomson, D.M.; McVie, A.; Morris, B.J.; Pratt, J.A. Dissociation of acute and chronic intermittent phencyclidine-induced performance deficits in the 5-choice serial reaction time task: Influence of clozapine. Psychopharmacology 2011, 213, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Spielewoy, C.; Markou, A. Withdrawal from chronic phencyclidine treatment induces long-lasting depression in brain reward function. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2003, 28, 1106–1116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Neill, J.C.; Harte, M.K.; Haddad, P.M.; Lydall, E.S.; Dwyer, D.M. Acute and chronic effects of NMDA receptor antagonists in rodents, relevance to negative symptoms of schizophrenia: A translational link to humans. Eur. Neuropsychopharmacol. 2014, 24, 822–835. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Roth, R.H. The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 1999, 20, 201–225. [Google Scholar] [CrossRef]
- Egerton, A.; Reid, L.; McGregor, S.; Cochran, S.M.; Morris, B.J.; Pratt, J.A. Subchronic and chronic PCP treatment produces temporally distinct deficits in attentional set shifting and prepulse inhibition in rats. Psychopharmacology 2008, 198, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Maeng, S.; Zarate, C.A., Jr.; Du, J.; Schloesser, R.J.; McCammon, J.; Chen, G.; Manji, H.K. Cellular mechanisms underlying the antidepressant effects of ketamine: Role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 2008, 63, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, J.S.; Autio, H.; Vesa, L.; Antila, H.; Lindemann, L.; Hoener, M.C.; Skolnick, P.; Rantamaki, T.; Castren, E. The antidepressant-like effects of glutamatergic drugs ketamine and AMPA receptor potentiator LY 451646 are preserved in bdnf(+)/(-) heterozygous null mice. Neuropharmacology 2012, 62, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ren, Q.; Ma, M.; Chen, Q.X.; Hashimoto, K. Antidepressant Effects of (+)-MK-801 and (-)-MK-801 in the Social Defeat Stress Model. Int J. Neuropsychopharmacol 2016, 19, 1. [Google Scholar] [CrossRef]
- Garcia, L.S.; Comim, C.M.; Valvassori, S.S.; Reus, G.Z.; Stertz, L.; Kapczinski, F.; Gavioli, E.C.; Quevedo, J. Ketamine treatment reverses behavioral and physiological alterations induced by chronic mild stress in rats. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2009, 33, 450–455. [Google Scholar] [CrossRef]
- Reus, G.Z.; Abelaira, H.M.; dos Santos, M.A.; Carlessi, A.S.; Tomaz, D.B.; Neotti, M.V.; Liranco, J.L.; Gubert, C.; Barth, M.; Kapczinski, F.; et al. Ketamine and imipramine in the nucleus accumbens regulate histone deacetylation induced by maternal deprivation and are critical for associated behaviors. Behav. Brain Res. 2013, 256, 451–456. [Google Scholar] [CrossRef]
- Reus, G.Z.; Carlessi, A.S.; Titus, S.E.; Abelaira, H.M.; Ignacio, Z.M.; da Luz, J.R.; Matias, B.I.; Bruchchen, L.; Florentino, D.; Vieira, A.; et al. A single dose of S-ketamine induces long-term antidepressant effects and decreases oxidative stress in adulthood rats following maternal deprivation. Dev. Neurobiol. 2015, 75, 1268–1281. [Google Scholar] [CrossRef]
- Reus, G.Z.; Nacif, M.P.; Abelaira, H.M.; Tomaz, D.B.; dos Santos, M.A.; Carlessi, A.S.; da Luz, J.R.; Goncalves, R.C.; Vuolo, F.; Dal-Pizzol, F.; et al. Ketamine ameliorates depressive-like behaviors and immune alterations in adult rats following maternal deprivation. Neurosci. Lett. 2015, 584, 83–87. [Google Scholar] [CrossRef]
- Yang, C.; Shirayama, Y.; Zhang, J.C.; Ren, Q.; Yao, W.; Ma, M.; Dong, C.; Hashimoto, K. R-ketamine: A rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl. Psychiatry 2015, 5, e632. [Google Scholar] [CrossRef]
- Fukumoto, K.; Toki, H.; Iijima, M.; Hashihayata, T.; Yamaguchi, J.I.; Hashimoto, K.; Chaki, S. Antidepressant Potential of (R)-Ketamine in Rodent Models: Comparison with (S)-Ketamine. J. Pharm. Exp. 2017, 361, 9–16. [Google Scholar] [CrossRef]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef]
- Markou, A.; Chiamulera, C.; Geyer, M.A.; Tricklebank, M.; Steckler, T. Removing obstacles in neuroscience drug discovery: The future path for animal models. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2009, 34, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Levinstein, M.R.; Samuels, B.A. Mechanisms underlying the antidepressant response and treatment resistance. Front. Behav. Neurosci. 2014, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- Jayatissa, M.N.; Bisgaard, C.; Tingstrom, A.; Papp, M.; Wiborg, O. Hippocampal cytogenesis correlates to escitalopram-mediated recovery in a chronic mild stress rat model of depression. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2006, 31, 2395–2404. [Google Scholar] [CrossRef]
- Der-Avakian, A.; Mazei-Robison, M.S.; Kesby, J.P.; Nestler, E.J.; Markou, A. Enduring deficits in brain reward function after chronic social defeat in rats: Susceptibility, resilience, and antidepressant response. Biol. Psychiatry 2014, 76, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Araki, H.; Gomita, Y. Influence of ACTH on the effects of imipramine, desipramine and lithium on duration of immobility of rats in the forced swim test. Pharmacol. Biochem. Behav. 2002, 71, 63–69. [Google Scholar] [CrossRef]
- Sukoff Rizzo, S.J.; Neal, S.J.; Hughes, Z.A.; Beyna, M.; Rosenzweig-Lipson, S.; Moss, S.J.; Brandon, N.J. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Transl. Psychiatry 2012, 2, e199. [Google Scholar] [CrossRef]
- Belzung, C. Innovative drugs to treat depression: Did animal models fail to be predictive or did clinical trials fail to detect effects? Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2014, 39, 1041–1051. [Google Scholar] [CrossRef]
- Przegalinski, E.; Tatarczynska, E.; Deren-Wesolek, A.; Chojnacka-Wojcik, E. Antidepressant-like effects of a partial agonist at strychnine-insensitive glycine receptors and a competitive NMDA receptor antagonist. Neuropharmacology 1997, 36, 31–37. [Google Scholar] [CrossRef]
- Layer, R.T.; Popik, P.; Olds, T.; Skolnick, P. Antidepressant-like actions of the polyamine site NMDA antagonist, eliprodil (SL-82.0715). Pharmacol. Biochem. Behav. 1995, 52, 621–627. [Google Scholar] [CrossRef]
- Cryan, J.F.; Mombereau, C. In search of a depressed mouse: Utility of models for studying depression-related behavior in genetically modified mice. Mol. Psychiatry 2004, 9, 326–357. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.M.; Duman, R.S. Activation of mammalian target of rapamycin and synaptogenesis: Role in the actions of rapid-acting antidepressants. Biol. Psychiatry 2013, 73, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, L.; Campa, L.; Auberson, Y.P.; Adell, A. The role of GluN2A and GluN2B subunits on the effects of NMDA receptor antagonists in modeling schizophrenia and treating refractory depression. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2014, 39, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, L.W.; Ortiz, J.; Hamedani, A.G.; Nestler, E.J. Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: Common adaptations among cross-sensitizing agents. J. Neurosci. 1996, 16, 274–282. [Google Scholar] [CrossRef]
- Bartanusz, V.; Aubry, J.M.; Pagliusi, S.; Jezova, D.; Baffi, J.; Kiss, J.Z. Stress-induced changes in messenger RNA levels of N-methyl-D-aspartate and AMPA receptor subunits in selected regions of the rat hippocampus and hypothalamus. Neuroscience 1995, 66, 247–252. [Google Scholar] [CrossRef]
- Moghaddam, B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: Comparison to hippocampus and basal ganglia. J. Neurochem. 1993, 60, 1650–1657. [Google Scholar] [CrossRef]
- Pacheco, A.; Aguayo, F.I.; Aliaga, E.; Muñoz, M.; García-Rojo, G.; Olave, F.A.; Parra-Fiedler, N.A.; García-Pérez, A.; Tejos-Bravo, M.; Rojas, P.S. Chronic stress triggers expression of immediate early genes and differentially affects the expression of AMPA and NMDA subunits in dorsal and ventral hippocampus of rats. Front. Mol. Neurosci. 2017, 10, 244. [Google Scholar] [CrossRef]
- Sathyanesan, M.; Haiar, J.M.; Watt, M.J.; Newton, S.S. Restraint stress differentially regulates inflammation and glutamate receptor gene expression in the hippocampus of C57BL/6 and BALB/c mice. Stress 2017, 20, 197–204. [Google Scholar] [CrossRef]
- Masrour, F.F.; Peeri, M.; Azarbayjani, M.A.; Hosseini, M.-J. Voluntary exercise during adolescence mitigated negative the effects of maternal separation stress on the depressive-like behaviors of adult male rats: Role of NMDA receptors. Neurochem. Res. 2018, 43, 1067–1074. [Google Scholar] [CrossRef]
- Weiland, N.; Orchinik, M.; Tanapat, P. Chronic corticosterone treatment induces parallel changes in N-methyl-D-aspartate receptor subunit messenger RNA levels and antagonist binding sites in the hippocampus. Neuroscience 1997, 78, 653–662. [Google Scholar] [CrossRef]
- Dong, B.E.; Chen, H.; Sakata, K. BDNF deficiency and enriched environment treatment affect neurotransmitter gene expression differently across ages. J. Neurochem. 2020. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, S.Y.; Zhou, S.W.; Qian, G.S.; Peng, K.; Mo, Z.X.; Zhou, J.Y. Effects of rhynchophylline on GluN1 and GluN2B expressions in primary cultured hippocampal neurons. Fitoterapia 2014, 98, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Chaki, S. Requirement of AMPA receptor stimulation for the sustained antidepressant activity of ketamine and LY341495 during the forced swim test in rats. Behav. Brain Res. 2014, 271, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yamaki, V.N.; Wei, Z.; Zheng, Y.; Cai, X. Differential regulation of GluA1 expression by ketamine and memantine. Behav. Brain Res. 2017, 316, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Shinohara, R.; Fogaça, M.V.; Hare, B. Neurobiology of rapid-acting antidepressants: Convergent effects on GluA1-synaptic function. Mol. Psychiatry 2019, 24, 1816–1832. [Google Scholar] [CrossRef]
- Hashimoto, K. Role of the mTOR signaling pathway in the rapid antidepressant action of ketamine. Expert Rev. Neurother. 2011, 11, 33–36. [Google Scholar] [CrossRef]
- Yang, C.; Ren, Q.; Qu, Y.; Zhang, J.C.; Ma, M.; Dong, C.; Hashimoto, K. Mechanistic Target of Rapamycin-Independent Antidepressant Effects of (R)-Ketamine in a Social Defeat Stress Model. Biol. Psychiatry 2018, 83, 18–28. [Google Scholar] [CrossRef]
- Abe, N.; Borson, S.H.; Gambello, M.J.; Wang, F.; Cavalli, V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J. Biol. Chem. 2010, 285, 28034–28043. [Google Scholar] [CrossRef]
- Klann, E.; Antion, M.D.; Banko, J.L.; Hou, L. Synaptic plasticity and translation initiation. Learn. Mem. 2004, 11, 365–372. [Google Scholar] [CrossRef]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef]
- Li, Z.; Boules, M.; Williams, K.; Peris, J.; Richelson, E. The novel neurotensin analog NT69L blocks phencyclidine (PCP)-induced increases in locomotor activity and PCP-induced increases in monoamine and amino acids levels in the medial prefrontal cortex. Brain Res. 2010, 1311, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Amargos-Bosch, M.; Lopez-Gil, X.; Artigas, F.; Adell, A. Clozapine and olanzapine, but not haloperidol, suppress serotonin efflux in the medial prefrontal cortex elicited by phencyclidine and ketamine. Int. J. Neuropsychopharmacol. 2006, 9, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Ohoyama, K.; Yamamura, S.; Hamaguchi, T.; Nakagawa, M.; Motomura, E.; Shiroyama, T.; Tanii, H.; Okada, M. Effect of novel atypical antipsychotic, blonanserin, on extracellular neurotransmitter level in rat prefrontal cortex. Eur. J. Pharmacol. 2011, 653, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals 2020, 13, 117. [Google Scholar] [CrossRef]
- Ji, M.H.; Zhang, L.; Mao, M.-J.; Zhang, H.; Yang, J.J.; Qiu, L.L. Overinhibition mediated by parvalbumin interneurons might contribute to depression-like behavior and working memory impairment induced by lipopolysaccharide challenge. Behav. Brain Res. 2020, 383, 112509. [Google Scholar] [CrossRef]
- Page, C.E.; Shepard, R.; Heslin, K.; Coutellier, L. Prefrontal parvalbumin cells are sensitive to stress and mediate anxiety-related behaviors in female mice. Sci. Rep. 2019, 9, 19772. [Google Scholar] [CrossRef]
- Shepard, R.; Page, C.E.; Coutellier, L. Sensitivity of the prefrontal GABAergic system to chronic stress in male and female mice: Relevance for sex differences in stress-related disorders. Neuroscience 2016, 332, 1–12. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathomechanism of nocturnal paroxysmal dystonia in autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Biomed. Pharm. 2020, 126, 110070. [Google Scholar] [CrossRef]
- Yamamura, S.; Abe, M.; Nakagawa, M.; Ochi, S.; Ueno, S.; Okada, M. Different actions for acute and chronic administration of mirtazapine on serotonergic transmission associated with raphe nuclei and their innervation cortical regions. Neuropharmacology 2011, 60, 550–560. [Google Scholar] [CrossRef]
- Gerhard, D.M.; Pothula, S.; Liu, R.J.; Wu, M.; Li, X.Y.; Girgenti, M.J.; Taylor, S.R.; Duman, C.H.; Delpire, E.; Picciotto, M.; et al. GABA interneurons are the cellular trigger for ketamine’s rapid antidepressant actions. J. Clin. Investig. 2020, 130, 1336–1349. [Google Scholar] [CrossRef] [PubMed]
- Kumari, V.; Antonova, E.; Zachariah, E.; Galea, A.; Aasen, I.; Ettinger, U.; Mitterschiffthaler, M.T.; Sharma, T. Structural brain correlates of prepulse inhibition of the acoustic startle response in healthy humans. Neuroimage 2005, 26, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Hazlett, E.A.; Buchsbaum, M.S.; Zhang, J.; Newmark, R.E.; Glanton, C.F.; Zelmanova, Y.; Haznedar, M.M.; Chu, K.W.; Nenadic, I.; Kemether, E.M.; et al. Frontal-striatal-thalamic mediodorsal nucleus dysfunction in schizophrenia-spectrum patients during sensorimotor gating. Neuroimage 2008, 42, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Liebe, T.; Li, M.; Colic, L.; Munk, M.H.J.; Sweeney-Reed, C.M.; Woelfer, M.; Kretzschmar, M.A.; Steiner, J.; von During, F.; Behnisch, G.; et al. Ketamine influences the locus coeruleus norepinephrine network, with a dependency on norepinephrine transporter genotype—A placebo controlled fMRI study. Neuroimage Clin. 2018, 20, 715–723. [Google Scholar] [CrossRef]
- Sarkisyan, G.; Hedlund, P.B. The 5-HT7 receptor is involved in allocentric spatial memory information processing. Behav. Brain Res. 2009, 202, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharm. 2020, 177, 2143–2162. [Google Scholar] [CrossRef]
- Pergola, G.; Danet, L.; Pitel, A.L.; Carlesimo, G.A.; Segobin, S.; Pariente, J.; Suchan, B.; Mitchell, A.S.; Barbeau, E.J. The Regulatory Role of the Human Mediodorsal Thalamus. Trends. Cogn. Sci. 2018, 22, 1011–1025. [Google Scholar] [CrossRef]
- Golden, E.C.; Graff-Radford, J.; Jones, D.T.; Benarroch, E.E. Mediodorsal nucleus and its multiple cognitive functions. Neurology 2016, 87, 2161–2168. [Google Scholar] [CrossRef]
- McCormick, D.; Wang, Z. Serotonin and noradrenaline excite GABAergic neurones of the guinea-pig and cat nucleus reticularis thalami. J. Physiol. 1991, 442, 235–255. [Google Scholar] [CrossRef]
- Porrino, L.; Crane, A.; Goldman-Rakic, P. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J. Comp. Neurol. 1981, 198, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Russchen, F.; Amaral, D.G.; Price, J. The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis. J. Comp. Neurol. 1987, 256, 175–210. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Sherman, S.M. Thalamocortical Circuit Motifs: A General Framework. Neuron 2019, 103, 762–770. [Google Scholar] [CrossRef]
- Kuramoto, E.; Pan, S.; Furuta, T.; Tanaka, Y.R.; Iwai, H.; Yamanaka, A.; Ohno, S.; Kaneko, T.; Goto, T.; Hioki, H. Individual mediodorsal thalamic neurons project to multiple areas of the rat prefrontal cortex: A single neuron-tracing study using virus vectors. J. Comp. Neurol. 2017, 525, 166–185. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Fukuzawa, M.; Okubo, R.; Okada, M. Upregulated Connexin 43 Induced by Loss-of-Functional S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor Contributes to Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Pharmaceuticals 2020, 13, 58. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Okada, M. Upregulated and hyperactivated thalamic connexin 43 plays important roles in pathomechanisms of cognitive impairment and seizure of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant α4 subunit of nicotinic ACh receptor. Pharmaceuticals 2020, 13, 99. [Google Scholar] [CrossRef]
- Furtak, S.C.; Wei, S.M.; Agster, K.L.; Burwell, R.D. Functional neuroanatomy of the parahippocampal region in the rat: The perirhinal and postrhinal cortices. Hippocampus 2007, 17, 709–722. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Murata, M. A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43. Int. J. Mol. Sci. 2020, 21, 7019. [Google Scholar] [CrossRef]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of Astroglial Connexin is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells 2020, 9, 414. [Google Scholar] [CrossRef]
- Baker, D.A.; Madayag, A.; Kristiansen, L.V.; Meador-Woodruff, J.H.; Haroutunian, V.; Raju, I. Contribution of cystine-glutamate antiporters to the psychotomimetic effects of phencyclidine. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2008, 33, 1760–1772. [Google Scholar] [CrossRef]
- Zmudzka, E.; Salaciak, K.; Sapa, J.; Pytka, K. Serotonin receptors in depression and anxiety: Insights from animal studies. Life Sci. 2018, 210, 106–124. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, T.M.; Golkar, A.; Ekstrom, J.C.; Svenningsson, P.; Ogren, S.O. 5-HT7 receptor stimulation by 8-OH-DPAT counteracts the impairing effect of 5-HT(1A) receptor stimulation on contextual learning in mice. Eur. J. Pharmacol. 2008, 596, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Barbas, H.; Zikopoulos, B.; Timbie, C. Sensory pathways and emotional context for action in primate prefrontal cortex. Biol. Psychiatry 2011, 69, 1133–1139. [Google Scholar] [CrossRef]
- Schuetze, M.; Park, M.T.; Cho, I.Y.; MacMaster, F.P.; Chakravarty, M.M.; Bray, S.L. Morphological Alterations in the Thalamus, Striatum, and Pallidum in Autism Spectrum Disorder. Neuropsychopharmacol. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 2627–2637. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, A.S.; Korbo, S.; Uylings, H.B.; Pakkenberg, B. A stereological study of the mediodorsal thalamic nucleus in Down syndrome. Neuroscience 2014, 279, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Bonaventure, P.; Aluisio, L.; Shoblock, J.; Boggs, J.D.; Fraser, I.C.; Lord, B.; Lovenberg, T.W.; Galici, R. Pharmacological blockade of serotonin 5-HT(7) receptor reverses working memory deficits in rats by normalizing cortical glutamate neurotransmission. PLoS ONE 2011, 6, e20210. [Google Scholar] [CrossRef]
- Schmidt, S.; Furini, C.; Zinn, C.; Cavalcante, L.; Ferreira, F.; Behling, J.; Myskiw, J.; Izquierdo, I. Modulation of the consolidation and reconsolidation of fear memory by three different serotonin receptors in hippocampus. Neurobiol. Learn. Mem. 2017, 142, 48–54. [Google Scholar] [CrossRef]
- Devoto, P.; Flore, G. On the origin of cortical dopamine: Is it a co-transmitter in noradrenergic neurons? Curr. Neuropharmacol. 2006, 4, 115–125. [Google Scholar] [CrossRef]
- Devoto, P.; Flore, G.; Saba, P.; Fa, M.; Gessa, G.L. Stimulation of the locus coeruleus elicits noradrenaline and dopamine release in the medial prefrontal and parietal cortex. J. Neurochem. 2005, 92, 368–374. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Regimen | Diagnosis | Placebo (N) | Outcome Responder Ratio [Drug vs. Placebo] | Reference |
|---|---|---|---|---|---|
| Ketamine | |||||
| Double-blind | 0.5 mg/kg (40 min) single iv | Major and bipolar depression | Saline (9) | Reduced HDRS 240 min (initial) 72 h (sustain) | [8] |
| Double-blind | 0.5 mg/kg (40 min) single iv | Major depression | Propofol/fentanyl (70) | Reduced HDRS 24 h [71% vs. 0%] | [7] |
| Double-blind | 0.5 mg/kg (40 min) single iv | Treatment-resistant depression | Saline (18) | Reduced HDRS 110 min (initial) 7 days (sustain) [71% vs. 0%] | [7] |
| Double-blind (added on mood stabilizer) | 0.5 mg/kg (40 min) single iv (maintained Li or VPA) | Treatment-resistant bipolar depression | Saline (18) | Reduced MADRS 40 min (initial) 3 days (sustained) [71% vs. 6%] | [77] |
| Double-blind (added on mood stabilizer) | 0.5 mg/kg (40 min) single iv (maintained Li or VPA) | Treatment-resistant bipolar depression | Saline (15) | Reduced MADRS 40 min (initial) 3 days (sustained) [71% vs. 0%] | [78] |
| Double-blind | 0.5 mg/kg (40 min) single iv | Treatment-resistant depression | Midazolam (73) | Reduced MADRS 24 h (initial) 7 days (sustained) [64% vs. 28%] | [74,79] |
| Double-blind | 50 mg intranasal administration | Major depression | Saline (20) | Reduced MADRS 24 h (initial) 7 days (sustain) | [80] |
| Double-blind (added on SSRI) | 0.5 mg/kg (40 min) single iv | Major depression | Saline (30) | Reduced MADRS 2 h min (initial) [92% vs. 57%] | [81] |
| Double-blind | 0.5 mg/kg (40 min) 2~3 times iv over 15 days | Treatment-resistant depression | Saline (67) | Reduced MADRS 7 days (initial) 15 days (sustain) [69% vs. 9%] | [5] |
| Double-blind | 0.2, 0.5 mg/kg (40 min) single iv | Treatment-resistant depression | Saline (64) | Reduced HDRS 40 min (initial) [25% vs. 0%] | [82] |
| Double-blind | 0.2, 0.5 mg/kg (40 min) single iv | Treatment-resistant depression | Saline (95) | Reduced HDRS 40 min (initial) 28 days (sustain) [46% vs. 13%] | [83] |
| Double-blind | 0.5 mg/kg (40 min) single iv | Treatment-resistant bipolar depression | Midazolam (16) | Reduced HDRS 24 h (initial) [89% vs. 0%] | [84] |
| Double-blind | 0.5 mg/kg (40 min) single iv | Treatment-resistant depression | Midazolam (80) | Reduced HDRS 24 h (initial) [30% vs. 15%] | [75] |
| Double-blind | 0.1, 0.2, 0.5, 1.0 mg/kg (40 min) single iv | Treatment-resistant depression | Midazolam (99) | Reduced HDRS 24 h (initial) 21 days (sustain) [57% vs. 33%] | [85] |
| Double-blind | 0.5 mg/kg (40 min) 6 times iv over 14 days | Treatment-resistant depression | Midazolam (41) | Reduced MADRS 24 h (initial) 7 days (sustained) [59%] | [86] |
| Double-blind | 0.5 mg/kg (45 min) 6 times iv over 21 days | Treatment-resistant depression | Saline (26) | Reduced HDRS 21 days (sustain) [25% vs. 33%] | [87] |
| Esketamine | |||||
| Double-blind | 0.2 or 0.4 mg/kg single iv | Treatment-resistant depression | Saline (29) | Reduced MADRS 2 h (initial) 35 days (sustain) [64% vs. 0%] | [88] |
| Double-blind | 28, 56, 84 mg intranasal administration | Treatment-resistant depression | Simulated placebo of esketamine taste (denatonium benzoate) (126) | Reduced MADRS 2 h (initial) 74 days (sustain) [50% vs. 10%] | [89] |
| Double-blind | 84 mg intranasal administration | Treatment-resistant depression | Simulated placebo of esketamine taste (66) | Reduced MADRS 4 h (initial) 25 days (sustain) [50% vs. 10%] | [90] |
| Double-blind (added on SSRI or SNRI) | 56, 84 mg intranasal administration | Treatment-resistant depression | Simulated placebo of esketamine taste (197) | Reduced MADRS 24 h (initial) 74 days (sustain) [69.3% vs. 52%] | [91] |
| Double-blind (added on SSRI or SNRI) | 56, 84 mg intranasal administration (twice a week for 4 weeks) | Treatment-resistant depression | Simulated placebo of esketamine taste (346) | Reduced MADRS 24 h (initial) 28 days (sustain) [53.1% vs. 38.9%] | [92] |
| Double-blind | Esketamine (0.25 mg/kg, 40 min, single iv) | Treatment-resistant depression | Ketamine (0.5 mg/kg, 40 min, single iv) (63) | Reduced MADRS 24 h (initial)7 days [43.7% vs. 62.1%] | [93] |
| Double-blind (added on SSRI or SNRI) | 28, 56, 84 mg intranasal administration (twice a week for 4 weeks) | Treatment-resistant depression (>65 years old) | Simulated placebo of esketamine taste (denatonium benzoate) (137) | Reduced MADRS 28 days (sustain) [27.0% vs. 13.3%] | [94] |
| CP-101,606 | |||||
| Double-blind (added on paroxetine) | 0.75 mg/kg CP-101,606 (90 min) 2 times iv for 6.5 h | Paroxetine-resistant major depression | Saline (30) | Reduced HDRS 2 days (initial) 8 days (sustain) [60% vs. 20%] | [95] |
| MK-0657 | |||||
| Double-blind | 4 mg/day (po) increased 4, 8, 12 mg/day until 12 days | Treatment-resistant depression | Saline (5) | Reduced HDRS 5 days (initial) 12 days (sustain) | [96] |
| Model | Agent | Effect | Reference |
|---|---|---|---|
| Schizophrenia | |||
| locomotor activity stereotypical behaviour | MK801 Phencyclidine | hyperlocomotion | [109] [110,111,112] |
| prepulse inhibition (PPI) | MK801 Phencyclidine | disruptions | [117,118] |
| Depression | |||
| learned helplessness | Ketamine | rapid acting antidepressant effect | [4,124] |
| forced swimming | Ketamine MK801 Ro25-6981 CPP Imipramine fluoxetine NBQX | rapid acting antidepressant effect rapid acting antidepressant effect rapid acting antidepressant effect rapid acting antidepressant effect no antidepressant effects no antidepressant effects no antidepressant effects (supress antidepressant effects of ketamine, MK801 and Ro25-6981) | [4,124,125] [4,124,126] [124] [4,125] [4] [4] [124] |
| sucrose consumption (anhedonia test) (after chronic mild stress) | Ketamine | no antidepressant effects antidepressant/antianhedonic effect antidepressant effect | [4] [127] [4,127] |
| novelty-suppressed feeding (after chronic mild stress) | Ketamine | no antidepressant effects antidepressant effect | [4] [4] |
| fear conditioning | ketamine | No effect | [4] |
| passive avoidance tests | ketamine | not impair fear memory retention. | [124] |
| maternal deprivation | ketamine | antidepressant effect | [128,129,130] |
| TrkB knockout forced swimming novelty-suppressed feeding | Ketamine, MK801 ketamine | no antidepressant effects no antidepressant effects | [4] [4] |
| BDNF knockout Forced swimming | Ketamine MK801 | no antidepressant effects no antidepressant effects | [125] [4] |
| Arketamine/Esketamine | Arketamine | Esketamine | |
| learned helplessness | rapid acting antidepressant effect | no antidepressant effect | [131] |
| forced swimming | rapid acting antidepressant effect longer-lasting antidepressant effect than esketamine | rapid acting antidepressant effect | [132] |
| tail suspension | rapid acting antidepressant effect longer-lasting antidepressant effect than esketamine | rapid acting antidepressant effect | [132] |
| social defeat stress | rapid acting antidepressant effect longer-lasting antidepressant effect than esketamine | rapid acting antidepressant effect | [131] |
| repeated corticosterone | rapid acting antidepressant effect longer-lasting antidepressant effect than esketamine | rapid acting antidepressant effect | [132] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, M.; Kawano, Y.; Fukuyama, K.; Motomura, E.; Shiroyama, T. Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions. Int. J. Mol. Sci. 2020, 21, 7951. https://doi.org/10.3390/ijms21217951
Okada M, Kawano Y, Fukuyama K, Motomura E, Shiroyama T. Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions. International Journal of Molecular Sciences. 2020; 21(21):7951. https://doi.org/10.3390/ijms21217951
Chicago/Turabian StyleOkada, Motohiro, Yasuhiro Kawano, Kouji Fukuyama, Eishi Motomura, and Takashi Shiroyama. 2020. "Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions" International Journal of Molecular Sciences 21, no. 21: 7951. https://doi.org/10.3390/ijms21217951
APA StyleOkada, M., Kawano, Y., Fukuyama, K., Motomura, E., & Shiroyama, T. (2020). Candidate Strategies for Development of a Rapid-Acting Antidepressant Class That Does Not Result in Neuropsychiatric Adverse Effects: Prevention of Ketamine-Induced Neuropsychiatric Adverse Reactions. International Journal of Molecular Sciences, 21(21), 7951. https://doi.org/10.3390/ijms21217951