The Future of Direct Cardiac Reprogramming: Any GMT Cocktail Variety?



Abstract
1. Background
1.1. Cardiogenesis
- MEF2. MEF2 is a MAD-box containing transcription factor with a key role in heart morphogenesis and in the regulation of the CPC and CM gene program [4,18]. MEF2 is encoded by four genes, Mef2a, -b, -c, and -d. Mef2b and Mef2c are the first MEF2 isoforms expressed in the cardiac mesoderm at mouse E7.5, Mef2a and Mef2d are expressed in the linear heart tube between E8.0 and E8.5, and after E8.5, all four Mef2 genes are expressed throughout the developing heart [18]. Mef2c is required for activation of a subset of cardiac contractile protein genes, as well as for the development of cardiac structures derived from SHF [4]. In mice homozygous for a null mutation of Mef2c, the heart tube did not undergo looping morphogenesis, the future right ventricle did not form, and a subset of cardiac muscle genes was not expressed [19].
- HAND2. Hand1 and Hand2 encode basic helix-loop-helix transcription factors and are expressed in mesodermal and neural crest-derived structures of the developing heart. Hand2 is expressed in the outflow track, the epicardium, valve progenitors, and predominantly in the myocardial compartment of the right ventricle, while the related transcription factor Hand1 is predominantly expressed in the left ventricle [20,21]. Deletion of Hand2 results in severe hypoplasia of the right ventricle segment [22]. In fact, the absence of the right ventricular region of Mef2c mutant correlated with downregulation of the HAND2 [19]. HAND2 interacts with non-coding regions of many genes involved in cardiogenesis [21].
- GATA4. The Gata4 gene is expressed in CMs and their mesodermal precursors, as well as in the endocardium and the epicardium. GATA4 regulates expression of myocardium-related genes and is necessary for the proliferation of CMs, formation of the endocardial cushions, development of the right ventricle and septation of the outflow tract [23]. GATA4 binds and promotes deposition of H3K27ac, and subsequently, establish active chromatin regions, at multiple cardiac enhancers to stimulate transcription [24].
- BAF60c. Smarcd3 gene, encodes BAF60c, a cardiac-enriched subunit of the SWI/SNF-like BAF chromatin complex. BAF60c is expressed specifically in the heart and somites in the early mouse embryo. Smarcd3 silencing in mouse embryos causes defects in heart morphogenesis that reflect impaired expansion of the AHF, and results in abnormal cardiac and skeletal muscle differentiation [25]. Baf60c regulates a gene expression program that regulates the main functional properties of CMs, including genes encoding contractile proteins, modulators of sarcomere function, and cardiac metabolic genes. Interestingly, many of the genes deregulated in Baf60c null embryos are targets of the MYOCD, another important transcription factor in heart development [26], which can functionally interact with BAF60c [27].
- TBX5. Tbx5 gene is a T-box transcription factor, expressed early in development throughout the entire cardiac crescent. Lineage tracing of Tbx5 showed that this gene is expressed in the myocardium of the left ventricle, but not the right ventricle or outflow track, besides a population of the posterior SHF (contributing to the myocardium of the atria and the venous pole) [28]. TBX5 can have both positive and negative transcriptional activity depending on the transcription factors with which it interacts [29]. Interestingly, in 2009 Takeuchi et al. demonstrated the transdifferentiation of mouse mesoderm into beating CMs by the ectopic expression of GATA4, BAF60c, and TBX5. The authors described that BAF60c enabled binding of GATA4 to cardiac genes to initiate the cardiac expression program, whereas TBX5 repressed noncardiac mesodermal genes and promoted differentiation into CMs [30].
- NKX2.5. Nkx2.5 gene is a homeobox transcription factor essential for early heart formation. Nkx2.5 knockout mice die at E9.5-10.5 with severely underdeveloped heart [31]. NKX2.5 is expressed in the cardiac crescent stage and regulates CM differentiation [32]. Interestingly, NKX2.5 is expressed at lower levels in SHF progenitors than in FHF progenitors and CMs, and its expression level, combined with other factors, may trigger different outcomes. In SHF progenitors, NKX2.5 can promote proliferation and activate the expression of Fgf10 and Mef2c-AHF enhancer, together with FOXH1, whereas in the FHF, NKX2.5 reduces Fgf10 and Isl1 expression and induces differentiation [33].
- MESP1. Mesp1 is a basic helix-loop-helix transcription factor expressed in early mesoderm during gastrulation by the T-box transcription factor EOMES in response to low doses of NODAL/SMAD2/3 signaling [34]. MESP1 expressing cells migrate out from the primitive streak and are incorporated into the heart field to generate a single heart tube [35]. MESP1 acts as a master regulator of multipotent CPCs specification. It activates many genes that form the core cardiac transcriptional machinery and represses the expression of genes that control other early mesoderm and endoderm cell fates [36].
1.2. Regenerative Medicine to Treat Cardiac Diseases: Where Do We Stand?
1.3. The Current Progresses and Challenges to Cell Therapy For Heart Diseases
1.3.1. Cell Pre-Treatments
1.3.2. Genetically Modified Cells
1.3.3. Cells Encapsulated in Biomaterials
2. Direct Reprogramming for Heart Regeneration
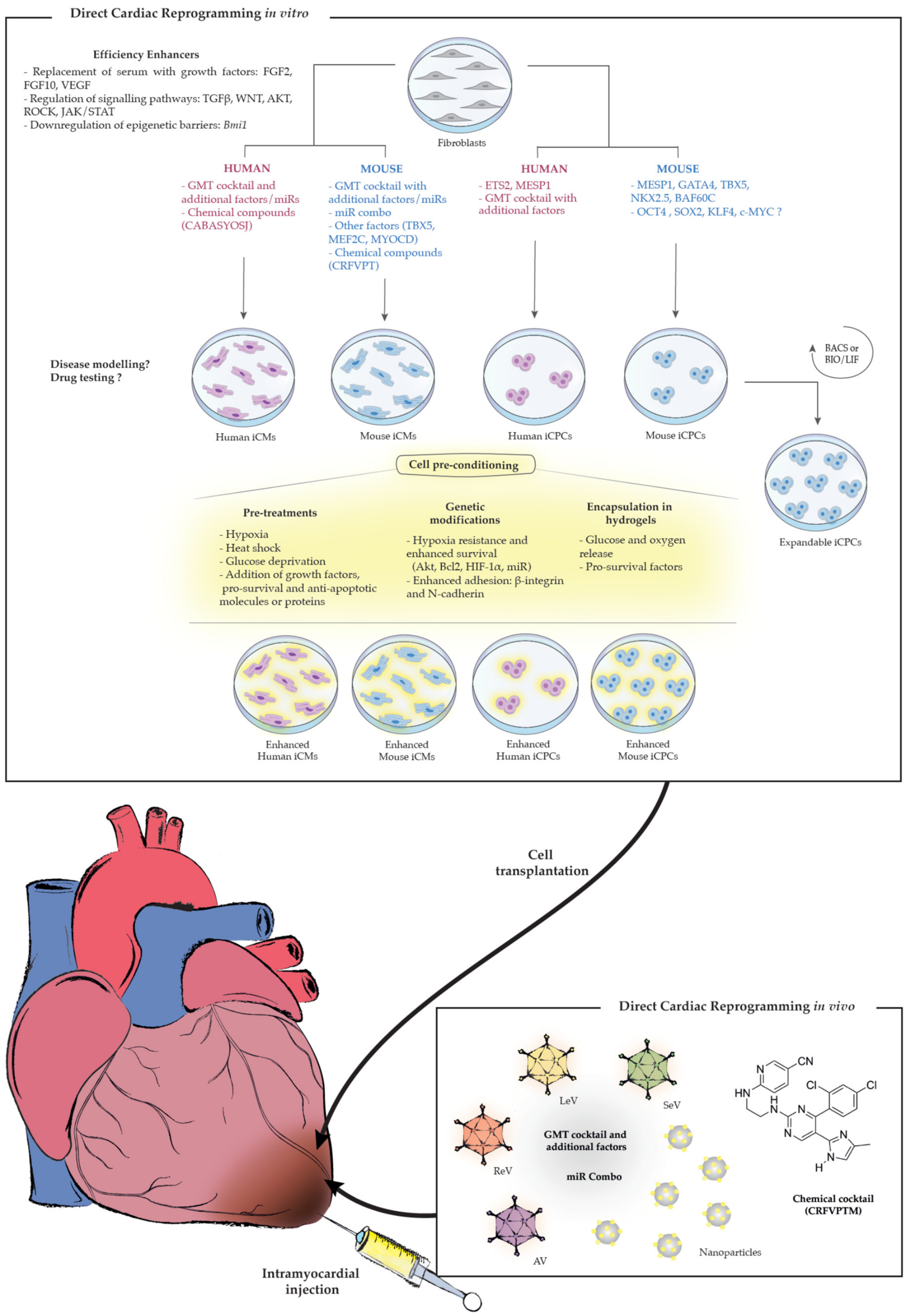
2.1. Direct Cardiac Reprogramming In Vitro
2.1.1. Direct Reprogramming into Mouse iCMs
- First Discovery: the GMT cocktail
- Modifications to the GMT cocktail
Stoichiometric Optimization of GMT Factors
Inclusion of Additional Transcription Factors: The Relevance of Hand2 Transcription Factor
Addition of miRs
Regulation of Signaling Pathways
Inhibition of Epigenetic Barriers
- Other cocktails different from GMT
2.1.2. Direct Reprogramming Into Human iCMs
- Modifications to the GMT cocktail
- Other cocktails different from GMT
2.1.3. Direct Reprogramming Into iCPCs
- First Discovery: the ETS2 and MESP1 combination
- Modifications to the GMT cocktail
- Other cocktails different from GMT: expandable mouse iCPCs
2.2. Direct Cardiac Reprogramming In Vivo
- The GMT cocktail
- Modifications to the GMT cocktail
- Other cocktails different from GMT
3. Future Directions and Challenges
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| iCMs | induced Cardiomyocytes |
| iCPCs | induced Cardiac Progenitor Cells |
| iPSCs | induced Pluripotent Stem Cells |
| AMI | Acute Myocardial Infarction |
| MHC | Myosin Heavy Chain |
| cTn | cardiac Troponin |
| GMT | GATA4, MEF2C, TBX5 |
| GHMT | GATA4, HAND2, MEF2C, TBX5 |
| FHF | First Heart Field |
| SHF | Second Heart Field |
| AHF | Anterior Heart Field |
| ReV | retrovirus |
| LeV | lentivirus |
| SeV | Sendai virus |
| AV | Adenovirus |
| miRs | microRNAs |
| CFs | cardiac fibroblasts |
| TTFs | tail-tip fibroblasts |
| MEFs | mouse embryonic fibroblasts |
| HDFs | human dermal fibroblasts |
| HFFs | human foreskin fibroblasts |
| hESC | human embryonic stem cell |
| AP | action potentials |
| CaT | calcium transients |
| SB | spontaneous beating |
| c-B | beating when co-cultured with murine CMs |
| HF | heart function |
| CS | cardiac tissue structure |
References
- Brade, T.; Pane, L.S.; Moretti, A.; Chien, K.R.; Laugwitz, K.L. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a013847. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G.; Buckingham, M.E.; Moorman, A.F. Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef]
- Vincent, S.D.; Buckingham, M.E. How to Make a Heart The Origin and Regulation of Cardiac Progenitor Cells. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2010; Volume 90, pp. 1–41. [Google Scholar]
- Dodou, E.; Verzi, M.P.; Anderson, J.P.; Xu, S.-M.; Black, B.L. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development 2004, 131, 3931–3942. [Google Scholar] [CrossRef] [PubMed]
- Soh, B.-S.; Buac, K.; Xu, H.; Li, E.; Ng, S.-Y.; Wu, H.; Chmielowiec, J.; Jiang, X.; Bu, L.; Li, R.A.; et al. N-cadherin prevents the premature differentiation of anterior heart field progenitors in the pharyngeal mesodermal microenvironment. Cell Res. 2014, 24, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.G. Chapter Two—The Second Heart Field. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2012; Volume 100, pp. 33–65. ISBN 9780123877864. [Google Scholar]
- Moretti, A.; Caron, L.; Nakano, A.; Lam, J.T.; Bernshausen, A.; Chen, Y.; Qyang, Y.; Bu, L.; Sasaki, M.; Martin-Puig, S.; et al. Multipotent Embryonic Isl1+ Progenitor Cells Lead to Cardiac, Smooth Muscle, and Endothelial Cell Diversification. Cell 2006, 127, 1151–1165. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.; Jiang, X.; Martin-Puig, S.; Caron, L.; Zhu, S.; Shao, Y.; Roberts, D.J.; Huang, P.L.; Domian, I.J.; Chien, K.R. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature 2009, 460, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Später, D.; Abramczuk, M.K.; Buac, K.; Zangi, L.; Stachel, M.W.; Clarke, J.; Sahara, M.; Ludwig, A.; Chien, K.R. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat. Cell Biol. 2013, 15, 1098–1106. [Google Scholar] [CrossRef]
- Ne, S.; Meilhac, M.; Lescroart, F.; Dric Blanpain, C.; Buckingham, M.E. Cardiac Cell Lineages that Form the Heart. Cold Spring Harb. Perspect. Med. 2014. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Später, D.; Hansson, E.M.; Zangi, L.; Chien, K.R. How to make a cardiomyocyte. Development 2014, 141, 4418–4431. [Google Scholar] [CrossRef]
- Woodcock, E.A.; Matkovich, S.J. Cardiomyocytes structure, function and associated pathologies. Int. J. Biochem. Cell Biol. 2005, 37, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin Heavy Chain Isoform Expression in the Failing and Nonfailing Human Heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef]
- Sharma, S.; Jackson, P.G.; Makan, J. Cardiac troponins. J. Clin. Pathol. 2004, 57, 1025–1026. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, D. Making or Breaking the Heart: From Lineage Determination to Morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef]
- Olson, E.N. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, C.; Naya, F. The Function of the MEF2 Family of Transcription Factors in Cardiac Development, Cardiogenomics, and Direct Reprogramming. J. Cardiovasc. Dev. Dis. 2016, 3, 26. [Google Scholar] [CrossRef]
- Lin, Q.; Schwarz, J.; Bucana, C.; Olson, E.N. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 1997, 276, 1404–1407. [Google Scholar] [CrossRef]
- Srivastava, D.; Cserjesi, P.; Olson, E.N. A subclass of bHLH proteins required for cardiac morphogenesis. Science 1995, 270, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Laurent, F.; Girdziusaite, A.; Gamart, J.; Barozzi, I.; Osterwalder, M.; Akiyama, J.A.; Lincoln, J.; Lopez-Rios, J.; Visel, A.; Zuniga, A.; et al. HAND2 Target Gene Regulatory Networks Control Atrioventricular Canal and Cardiac Valve Development. Cell Rep. 2017, 19, 1602–1613. [Google Scholar] [CrossRef]
- Srivastava, D.; Thomas, T.; Kirby, M.L.; Brown, D.; Olson, E.N. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat. Genet. 1997, 16, 154–160. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Ma, Q.; Juraszek, A.L.; Moses, K.; Schwartz, R.J.; Izumo, S.; Pu, W.T. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J. Clin. Investig. 2005, 115, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- He, A.; Gu, F.; Hu, Y.; Ma, Q.; Yi Ye, L.; Akiyama, J.A.; Visel, A.; Pennacchio, L.A.; Pu, W.T. Dynamic GATA4 enhancers shape the chromatin landscape central to heart development and disease. Nat. Commun. 2014, 5, 4907. [Google Scholar] [CrossRef]
- Lickert, H.; Takeuchi, J.K.; Von Both, I.; Walls, J.R.; McAuliffe, F.; Adamson, S.L.; Henkelman, R.M.; Wrana, J.L.; Rossant, J.; Bruneau, B.G. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 2004, 432, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Mokalled, M.H.; Carroll, K.J.; Cenik, B.K.; Chen, B.; Liu, N.; Olson, E.N.; Bassel-Duby, R. Myocardin-related transcription factors are required for cardiac development and function. Dev. Biol. 2015, 406, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hota, S.K.; Zhou, Y.Q.; Novak, S.; Miguel-Perez, D.; Christodoulou, D.; Seidman, C.E.; Seidman, J.G.; Gregorio, C.C.; Henkelman, R.M.; et al. Cardiac-enriched BAF chromatin-remodeling complex subunit Baf60c regulates gene expression programs essential for heart development and function. Biol. Open 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G.; Logan, M.; Davis, N.; Levi, T.; Tabin, C.J.; Seidman, J.G.; Seidman, C.E. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt- Oram syndrome. Dev. Biol. 1999, 211, 100–108. [Google Scholar] [CrossRef]
- Steimle, J.D.; Moskowitz, I.P. TBX5: A Key Regulator of Heart Development. In Current Topics in Developmental Biology; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 122, pp. 195–221. [Google Scholar]
- Takeuchi, J.K.; Bruneau, B.G. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 2009, 459, 708–711. [Google Scholar] [CrossRef]
- Lyons, I.; Parsons, L.M.; Hartley, L.; Li, R.; Andrews, J.E.; Robb, L.; Harvey, R.P. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995, 9, 1654–1666. [Google Scholar] [CrossRef]
- Stanley, E.G.; Biben, C.; Elefanty, A.; Barnett, L.; Koentgen, F.; Robb, L.; Harvey, R.P. Efficient Cre-mediated deletion in cardiac progenitor cells conferred by a 3’UTR-ires-Cre allele of the homeobox gene Nkx2-5. Int. J. Dev. Biol. 2002, 46, 431–439. [Google Scholar]
- Watanabe, Y.; Zaffran, S.; Kuroiwa, A.; Higuchi, H.; Ogura, T.; Harvey, R.P.; Kelly, R.G.; Buckingham, M. Fibroblast growth factor 10 gene regulation in the second heart field by Tbx1, Nkx2-5, and Islet1 reveals a genetic switch for down-regulation in the myocardium. Proc. Natl. Acad. Sci. USA 2012, 109, 18273–18280. [Google Scholar] [CrossRef]
- Costello, I.; Pimeisl, I.-M.; Dräger, S.; Bikoff, E.K.; Robertson, E.J.; Arnold, S.J. The T-box transcription factor Eomesodermin acts upstream of Mesp1 to specify cardiac mesoderm during mouse gastrulation. Nat. Cell Biol. 2011, 13, 1084–1091. [Google Scholar] [CrossRef]
- Saga, Y.; Miyagawa-Tomita, S.; Takagi, A.; Kitajima, S.; Miyazaki, J.I.; Inoue, T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 1999, 126, 3437–3447. [Google Scholar]
- Bondue, A.; Lapouge, G.; Paulissen, C.; Semeraro, C.; Iacovino, M.; Kyba, M.; Blanpain, C. Mesp1 acts as a master regulator of multipotent cardiovascular progenitor specification. Cell Stem Cell 2008, 3, 69–84. [Google Scholar] [CrossRef]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef]
- Laugwitz, K.L.; Moretti, A.; Lam, J.; Gruber, P.; Chen, Y.; Woodard, S.; Lin, L.Z.; Cai, C.L.; Lu, M.M.; Reth, M.; et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005, 433, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, F.; Mehrkens, D.; Friedrich, F.W.; Stubbendorff, M.; Hua, X.; Müller, J.C.; Schrepfer, S.; Evans, S.M.; Carrier, L.; Eschenhagen, T. Localization of Islet-1-positive cells in the healthy and infarcted adult murine heart. Circ. Res. 2012, 110, 1303–1310. [Google Scholar] [CrossRef]
- Messina, E.; De Angelis, L.; Frati, G.; Morrone, S.; Chimenti, S.; Fiordaliso, F.; Salio, M.; Battaglia, M.; Latronico, M.V.G.; Coletta, M.; et al. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ. Res. 2004, 95, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, K.; Quaini, F.; Tasca, G.; Torella, D.; Castaldo, C.; Nadal-Ginard, B.; Leri, A.; Kajstura, J.; Quaini, E.; Anversa, P. Intense myocyte formation from cardiac stem cells in human cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2003, 100, 10440–10445. [Google Scholar] [CrossRef] [PubMed]
- Sultana, N.; Zhang, L.; Yan, J.; Chen, J.; Cai, W.; Razzaque, S.; Jeong, D.; Sheng, W.; Bu, L.; Xu, M.; et al. Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat. Commun. 2015, 6, 8701. [Google Scholar] [CrossRef]
- Davis, D.R. Cardiac stem cells in the post-Anversa era. Eur. Heart J. 2019, 40, 1039–1041. [Google Scholar] [CrossRef]
- Carvajal-Vergara, X. Engineering Cardiac Stem Cells for the Treatment of the Damaged Heart. Adv. Tissue Eng. Regen. Med. Open Access 2017, 3. [Google Scholar] [CrossRef][Green Version]
- Jaźwińska, A.; Sallin, P. Regeneration versus scarring in vertebrate appendages and heart. J. Pathol. 2016, 238, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Murry, C.E. Heart regeneration. Nature 2011, 473, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, M.L.; Lee, R.T. Regeneration of the heart. EMBO Mol. Med. 2011, 3, 701–712. [Google Scholar] [CrossRef]
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef]
- Garbern, J.C.; Lee, R.T. Cardiac stem cell therapy and the promise of heart regeneration. Cell Stem Cell 2013, 12, 689–698. [Google Scholar] [CrossRef]
- Wu, R.; Hu, X.; Wang, J. Concise Review: Optimized Strategies for Stem Cell-Based Therapy in Myocardial Repair: Clinical Translatability and Potential Limitation. Stem Cells 2018. [Google Scholar] [CrossRef] [PubMed]
- Padda, J.; Sequiera, G.L.; Sareen, N.; Dhingra, S. Stem cell therapy for cardiac regeneration: Hits and misses 1. Can. J. Physiol. Pharmacol. 2015, 93, 835–841. [Google Scholar] [CrossRef]
- Gerbin, K.A.; Murry, C.E. The winding road to regenerating the human heart. Cardiovasc. Pathol. 2015, 24, 133–140. [Google Scholar] [CrossRef]
- Hashimoto, H.; Olson, E.N.; Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair HHS Public Access. Nat. Rev. Cardiol. 2018, 15, 585–600. [Google Scholar] [CrossRef]
- Madonna, R.; Van Laake, L.W.; Davidson, S.M.; Engel, F.B.; Hausenloy, D.J.; Lecour, S.; Leor, J.; Perrino, C.; Schulz, R.; Ytrehus, K.; et al. Position Paper of the European Society of Cardiology Working Group Cellular Biology of the Heart: Cell-based therapies for myocardial repair and regeneration in ischemic heart disease and heart failure. Eur. Heart J. 2016, 37, 1789–1798. [Google Scholar] [CrossRef]
- Cambria, E.; Pasqualini, F.S.; Wolint, P.; Günter, J.; Steiger, J.; Bopp, A.; Hoerstrup, S.P.; Emmert, M.Y. Translational cardiac stem cell therapy: Advancing from fi rst- generation to next-generation cell types. NPJ Regen. Med. 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Prowse, A.B.J.; Timmins, N.E.; Yau, T.M.; Li, R.-K.; Weisel, R.D.; Keller, G.; Zandstra, P.W. Transforming the promise of pluripotent stem cell-derived cardiomyocytes to a therapy: Challenges and solutions for clinical trials. Can. J. Cardiol. 2014, 30, 1335–1349. [Google Scholar] [CrossRef] [PubMed]
- Doppler, S.A.; Deutsch, M.-A.; Lange, R.; Krane, M. Direct Reprogramming-The Future of Cardiac Regeneration? Int. J. Mol. Sci. 2015, 16, 17368–17393. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.-J.N.; Song, K.; Olson, E.N. Heart repair by cardiac reprogramming. Nat. Med. 2013, 19, 413–415. [Google Scholar] [CrossRef]
- Sahara, M.; Santoro, F.; Chien, K.R. Programming and reprogramming a human heart cell. EMBO J. 2015, 34, 710–738. [Google Scholar] [CrossRef] [PubMed]
- Behfar, A.; Yamada, S.; Crespo-Diaz, R.; Nesbitt, J.J.; Rowe, L.A.; Perez-Terzic, C.; Gaussin, V.; Homsy, C.; Bartunek, J.; Terzic, A. Guided cardiopoiesis enhances therapeutic benefit of bone marrow human mesenchymal stem cells in chronic myocardial infarction. J. Am. Coll. Cardiol. 2010, 56, 721–734. [Google Scholar] [CrossRef]
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of Human Embryonic Stem Cell–Derived Cardiovascular Progenitors for Severe Ischemic Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438. [Google Scholar] [CrossRef]
- Inflammatory Microenvironment of Acute Myocardial Infarction Prevents Regeneration of Heart with Stem Cells Therapy. Cell. Physiol. Biochem. 2019, 53, 887–909. [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Simón, A.M.; Pogontke, C.; Castillo, M.I.; Abizanda, G.; Pelacho, B.; Sánchez-Domínguez, R.; Segovia, J.C.; Prósper, F.; Pérez-Pomares, J.M. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J. Am. Coll. Cardiol. 2015, 65, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.U.; Guo, Y.; Li, Q.-H.; Cao, P.; Al-Maqtari, T.; Vajravelu, B.N.; Du, J.; Book, M.J.; Zhu, X.; Nong, Y.; et al. c-kit+ Cardiac stem cells alleviate post-myocardial infarction left ventricular dysfunction despite poor engraftment and negligible retention in the recipient heart. PLoS ONE 2014, 9, e96725. [Google Scholar] [CrossRef]
- Limbourg, F.P.; Ringes-Lichtenberg, S.; Schaefer, A.; Jacoby, C.; Mehraein, Y.; Jäger, M.D.; Limbourg, A.; Fuchs, M.; Klein, G.; Ballmaier, M.; et al. Haematopoietic stem cells improve cardiac function after infarction without permanent cardiac engraftment. Eur. J. Heart Fail. 2005, 7, 722–729. [Google Scholar] [CrossRef]
- Liu, Y.-W.; Chen, B.; Yang, X.; Fugate, J.A.; Kalucki, F.A.; Futakuchi-Tsuchida, A.; Couture, L.; Vogel, K.W.; Astley, C.A.; Baldessari, A.; et al. Human embryonic stem cell-derived cardiomyocytes restore function in infarcted hearts of non-human primates. Nat. Biotechnol. 2018, 36. [Google Scholar] [CrossRef]
- Hu, X.; Xu, Y.; Zhong, Z.; Wu, Y.; Zhao, J.; Wang, Y.; Cheng, H.; Kong, M.; Zhang, F.; Chen, Q.; et al. A large-scale investigation of hypoxia-preconditioned allogeneic mesenchymal stem cells for myocardial repair in nonhuman primates: Paracrine activity without remuscularization. Circ. Res. 2016, 118, 970–983. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, W.; Meng, W.; Jegga, A.G.; Wang, Y.; Cai, W.; Kim, H.W.; Pasha, Z.; Wen, Z.; Rao, F.; et al. Heat shock improves sca-1+ stem cell survival and directs ischemic cardiomyocytes toward a prosurvival phenotype via exosomal transfer: A critical role for HSF1/miR-34a/HSP70 pathway. Stem Cells 2014, 32, 462–472. [Google Scholar] [CrossRef]
- Moya, A.; Larochette, N.; Paquet, J.; Deschepper, M.; Bensidhoum, M.; Izzo, V.; Kroemer, G.; Petite, H.; Logeart-Avramoglou, D. Quiescence Preconditioned Human Multipotent Stromal Cells Adopt a Metabolic Profile Favorable for Enhanced Survival under Ischemia. Stem Cells 2017, 35, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Chen, K.Y.; Naumova, A.V.; Muskheli, V.; Fugate, J.A.; Dupras, S.K.; Reinecke, H.; Xu, C.; Hassanipour, M.; Police, S.; et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 2007, 25, 1015–1024. [Google Scholar] [CrossRef]
- Lemcke, H.; Voronina, N.; Steinhoff, G.; David, R. Recent Progress in Stem Cell Modification for Cardiac Regeneration. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef]
- Huang, J.; Guo, J.; Beigi, F.; Hodgkinson, C.P.; Facundo, H.T.; Zhang, Z.; Espinoza-Derout, J.; Zhou, X.; Pratt, R.E.; Mirotsou, M.; et al. HASF is a stem cell paracrine factor that activates PKC epsilon mediated cytoprotection. J. Mol. Cell. Cardiol. 2014, 66, 157–164. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pereira, G.C.; Pasdois, P. The role of hexokinase in cardioprotection—Mechanism and potential for translation. Br. J. Pharmacol. 2015, 172, 2085–2100. [Google Scholar] [CrossRef]
- Li, X.; He, P.; Wang, X.L.; Zhang, S.; Devejian, N.; Bennett, E.; Cai, C. Sulfiredoxin-1 enhances cardiac progenitor cell survival against oxidative stress via the upregulation of the ERK/NRF2 signal pathway. Free Radic. Biol. Med. 2018, 123, 8–19. [Google Scholar] [CrossRef]
- Li, N.; Zhou, H.; Tang, Q. miR-133: A suppressor of cardiac remodeling? Front. Pharmacol. 2018, 9, 903. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Chen, W.; Xie, L.; Zhao, Z.A.; Yang, J.; Chen, Y.; Lei, W.; Shen, Z. MicroRNA-133 overexpression promotes the therapeutic efficacy of mesenchymal stem cells on acute myocardial infarction. Stem Cell Res. Ther. 2017, 8. [Google Scholar] [CrossRef]
- Li, J.; Rohailla, S.; Gelber, N.; Rutka, J.; Sabah, N.; Gladstone, R.A.; Wei, C.; Hu, P.; Kharbanda, R.K.; Redington, A.N. Microrna-144 is a circulating effector of remote ischemic preconditioning. Basic Res. Cardiol. 2014, 109, 423. [Google Scholar] [CrossRef]
- Guo, C.; Deng, Y.; Liu, J.; Qian, L. Cardiomyocyte-specific role of miR-24 in promoting cell survival. J. Cell. Mol. Med. 2015, 19, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Zhang, X.; Du, S.; Chen, D.; Che, R. Upregulation of miR-335 ameliorates myocardial ischemia reperfusion injury via targeting hypoxia inducible factor 1-alpha subunit inhibitor. Am. J. Transl. Res. 2018, 10, 4082–4094. [Google Scholar]
- Huang, F.; Zhu, X.; Hu, X.Q.; Fang, Z.F.; Tang, L.; Lu, X.L.; Zhou, S.H. Mesenchymal stem cells modified with miR-126 release angiogenic factors and activate Notch ligand Delta-like-4, enhancing ischemic angiogenesis and cell survival. Int. J. Mol. Med. 2013, 31, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Joladarashi, D.; Srikanth Garikipati, V.N.; Thandavarayan, R.A.; Verma, S.K.; Mackie, A.R.; Khan, M.; Gumpert, A.M.; Bhimaraj, A.; Youker, K.A.; Uribe, C.; et al. Enhanced cardiac regenerative ability of stem cells after ischemia-reperfusion injury: Role of human CD34+ cells deficient in microRNA-377. J. Am. Coll. Cardiol. 2015, 66, 2214–2226. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Guan, Q.; Dai, S.; Wei, W.; Zhang, Y. Integrin β1 increases stem cell survival and cardiac function after myocardial infarction. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Jiang, Z.; Qiao, L.; Guo, B.; Xiao, W.; Zhang, X.; Chang, L.; Li, Y. Integrin β-3 is required for the attachment, retention and therapeutic benefits of human cardiospheres in myocardial infarction. J. Cell. Mol. Med. 2018, 22, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; Zhao, M.; Fan, C.; Fast, V.G.; Valarmathi, M.T.; Zhu, W.; Zhang, J. N-cadherin overexpression enhances the reparative potency of human-induced pluripotent stem cell-derived cardiac myocytes in infarcted mouse hearts. Cardiovasc. Res. 2020, 116, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Swope, D.; Cheng, L.; Gao, E.; Li, J.; Radice, G.L. Loss of Cadherin-Binding Proteins-Catenin and Plakoglobin in the Heart Leads to Gap Junction Remodeling and Arrhythmogenesis. Mol. Cell. Biol. 2012, 32, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shemezis, J.R.; McQuinn, E.R.; Wang, J.; Sverdlov, M.; Chenn, A. AKT activation by N-cadherin regulates beta-catenin signaling and neuronal differentiation during cortical development. Neural Dev. 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.H.; Benson, A.P.; Li, P.; Holden, A.V. Regional localisation of left ventricular sheet structure: Integration with current models of cardiac fibre, sheet and band structure. Eur. J. Cardio-Thorac. Surg. 2007, 32, 231–249. [Google Scholar] [CrossRef]
- Nugraha, B.; Buono, M.F.; von Boehmer, L.; Hoerstrup, S.P.; Emmert, M.Y. Human Cardiac Organoids for Disease Modeling. Clin. Pharmacol. Ther. 2019, 105, 79–85. [Google Scholar] [CrossRef]
- Cyranoski, D. “Reprogrammed” stem cells approved to mend human hearts for the first time news /631/532. Nature 2018, 557, 619–620. [Google Scholar] [CrossRef]
- Alagarsamy, K.N.; Yan, W.; Srivastava, A.; Desiderio, V.; Dhingra, S. Application of injectable hydrogels for cardiac stem cell therapy and tissue engineering. Rev. Cardiovasc. Med. 2019, 20, 221–230. [Google Scholar] [PubMed]
- Salazar-Noratto, G.E.; Luo, G.; Denoeud, C.; Padrona, M.; Moya, A.; Bensidhoum, M.; Bizios, R.; Potier, E.; Logeart-Avramoglou, D.; Petite, H. Understanding and leveraging cell metabolism to enhance mesenchymal stem cell transplantation survival in tissue engineering and regenerative medicine applications. Stem Cells 2020, 38, 22–33. [Google Scholar] [CrossRef]
- 1592—Enzyme-controlled, Starch-based Hydrogels for Mesenchymal Stromal Cell Survival and Paracrine Functions | Morressier. Available online: https://www.morressier.com/article/1592--enzymecontrolled-starchbased-hydrogels-mesenchymal-stromal-cell-survival-paracrine-functions/5c8f909db5d368000a26af5f (accessed on 25 September 2020).
- Sart, S.; Ma, T.; Li, Y. Preconditioning Stem Cells for In Vivo Delivery. Biores. Open Access 2014, 3, 137–149. [Google Scholar] [CrossRef]
- Abdelwahid, E.; Kalvelyte, A.; Stulpinas, A.; De Carvalho, K.A.T.; Guarita-Souza, L.C.; Foldes, G. Stem cell death and survival in heart regeneration and repair. Apoptosis 2016, 21, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Robinton, D.A.; Daley, G.Q. The promise of induced pluripotent stem cells in research and therapy. Nature 2012, 481, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Sevilla, A.; Dsouza, S.L.; Ang, Y.-S.; Schaniel, C.; Lee, D.-F.; Yang, L.; Kaplan, A.D.; Adler, E.D.; Rozov, R.; et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 2010, 465. [Google Scholar] [CrossRef]
- Parrotta, E.I.; Lucchino, V.; Scaramuzzino, L.; Scalise, S.; Cuda, G. Modeling Cardiac Disease Mechanisms Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Progress, Promises and Challenges. Int. J. Mol. Sci. 2020, 21, 4354. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Domian, I.J.; Chiravuri, M.; van der Meer, P.; Feinberg, A.W.; Shi, X.; Shao, Y.; Wu, S.M.; Parker, K.K.; Chien, K.R. Generation of functional ventricular heart muscle from mouse ventricular progenitor cells. Science 2009, 326, 426–429. [Google Scholar] [CrossRef]
- Den Hartogh, S.C.; Schreurs, C.; Monshouwer-Kloots, J.J.; Davis, R.P.; Elliott, D.A.; Mummery, C.L.; Passier, R. Dual Reporter MESP1mCherry/w-NKX2-5eGFP/w hESCs Enable Studying Early Human Cardiac Differentiation. Stem Cells 2015, 33, 56–67. [Google Scholar] [CrossRef]
- Bondue, A.; Tännler, S.; Chiapparo, G.; Chabab, S.; Ramialison, M.; Paulissen, C.; Beck, B.; Harvey, R.; Blanpain, C. Defining the earliest step of cardiovascular progenitor specification during embryonic stem cell differentiation. J. Cell Biol. 2011, 192, 751–765. [Google Scholar] [CrossRef]
- Li, Y.; Lin, B.; Yang, L. Comparative transcriptomic analysis of multiple cardiovascular fates from embryonic stem cells predicts novel regulators in human cardiogenesis. Sci. Rep. 2015, 5, 9758. [Google Scholar] [CrossRef]
- Lalit, P.A.; Salick, M.R.; Nelson, D.O.; Squirrell, J.M.; Shafer, C.M.; Patel, N.G.; Saeed, I.; Schmuck, E.G.; Markandeya, Y.S.; Wong, R.; et al. Lineage Reprogramming of Fibroblasts into Proliferative Induced Cardiac Progenitor Cells by Defined Factors. Cell Stem Cell 2016, 18, 354–367. [Google Scholar] [CrossRef]
- Ieda, M.; Fu, J.-D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef]
- Chen, J.X.; Krane, M.; Deutsch, M.A.; Wang, L.; Rav-Acha, M.; Gregoire, S.; Engels, M.C.; Rajarajan, K.; Karra, R.; Abel, E.D.; et al. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Li, Y.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res. 2015, 116, 237–244. [Google Scholar] [CrossRef]
- Song, K.; Nam, Y.-J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef]
- Umei, T.C.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Isomi, M.; Haginiwa, S.; Kojima, H.; Kurotsu, S.; Tamura, F.; Osakabe, R.; et al. Single-construct polycistronic doxycycline-inducible vectors improve direct cardiac reprogramming and can be used to identify the critical timing of transgene expression. Int. J. Mol. Sci. 2017, 18, 1805. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Katoku-Kikyo, N.; Keirstead, S.A.; Kikyo, N. Accelerated direct reprogramming of fibroblasts into cardiomyocyte-like cells with the MyoD transactivation domain. Cardiovasc. Res. 2013, 100, 105–113. [Google Scholar] [CrossRef]
- Addis, R.C.; Ifkovits, J.L.; Pinto, F.; Kellam, L.D.; Esteso, P.; Rentschler, S.; Christoforou, N.; Epstein, J.A.; Gearhart, J.D. Optimization of direct fibroblast reprogramming to cardiomyocytes using calcium activity as a functional measure of success. J. Mol. Cell. Cardiol. 2013, 60, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Christoforou, N.; Chellappan, M.; Adler, A.F.; Kirkton, R.D.; Wu, T.; Addis, R.C.; Bursac, N.; Leong, K.W. Transcription Factors MYOCD, SRF, Mesp1 and SMARCD3 Enhance the Cardio-Inducing Effect of GATA4, TBX5, and MEF2C during Direct Cellular Reprogramming. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, A.D.; Kim, L.J.; Nam, Y.J. Ensuring expression of four core cardiogenic transcription factors enhances cardiac reprogramming. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, W.; Nam, Y.J. Stoichiometric optimization of Gata4, Hand2, Mef2c, and Tbx5 expression for contractile cardiomyocyte reprogramming. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, N.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Isomi, M.; Nakashima, H.; Akiyama, M.; Wada, R.; Inagawa, K.; et al. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J. 2014, 33, 1565–1581. [Google Scholar] [CrossRef] [PubMed]
- Ifkovits, J.L.; Addis, R.C.; Epstein, J.A.; Gearhart, J.D. Inhibition of TGFβ signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-Mediated In Vitro and In Vivo Direct Reprogramming of Cardiac Fibroblasts to CardiomyocytesNovelty and Significance. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef]
- Abad, M.; Hashimoto, H.; Zhou, H.; Morales, M.G.; Chen, B.; Bassel-Duby, R.; Olson, E.N. Notch Inhibition Enhances Cardiac Reprogramming by Increasing MEF2C Transcriptional Activity. Stem Cell Rep. 2017, 8, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Londono, P.; Cao, Y.; Sharpe, E.J.; Proenza, C.; O’Rourke, R.; Jones, K.L.; Jeong, M.Y.; Walker, L.A.; Buttrick, P.M.; et al. High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Zhou, H.; Dickson, M.E.; Kim, M.S.; Bassel-Duby, R.; Olson, E.N. Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 11864–11869. [Google Scholar] [CrossRef]
- Yamakawa, H.; Muraoka, N.; Miyamoto, K.; Sadahiro, T.; Isomi, M.; Haginiwa, S.; Kojima, H.; Umei, T.; Akiyama, M.; Kuishi, Y.; et al. Fibroblast Growth Factors and Vascular Endothelial Growth Factor Promote Cardiac Reprogramming under Defined Conditions. Stem Cell Rep. 2015, 5, 1128–1142. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.M.A.; Stone, N.R.; Berry, E.C.; Radzinsky, E.; Huang, Y.; Pratt, K.; Ang, Y.S.; Yu, P.; Wang, H.; Tang, S.; et al. Chemical enhancement of in vitro and in vivo direct cardiac reprogramming. Circulation 2017, 135, 978–995. [Google Scholar] [CrossRef]
- Muraoka, N.; Nara, K.; Tamura, F.; Kojima, H.; Yamakawa, H.; Sadahiro, T.; Miyamoto, K.; Isomi, M.; Haginiwa, S.; Tani, H.; et al. Role of cyclooxygenase-2-mediated prostaglandin E2-prostaglandin E receptor 4 signaling in cardiac reprogramming. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, L.; Vaseghi, H.R.; Liu, Z.; Lu, R.; Alimohamadi, S.; Yin, C.; Fu, J.D.; Wang, G.G.; Liu, J.; et al. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 2016, 18, 382–395. [Google Scholar] [CrossRef]
- Protze, S.; Khattak, S.; Poulet, C.; Lindemann, D.; Tanaka, E.M.; Ravens, U. A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. J. Mol. Cell. Cardiol. 2012, 53, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Finch, E.A.; Zhang, L.; Zhang, H.; Hodgkinson, C.P.; Pratt, R.E.; Rosenberg, P.B.; Mirotsou, M.; Dzau, V.J. MicroRNA induced cardiac reprogramming in vivo evidence for mature cardiac myocytes and improved cardiac function. Circ. Res. 2014, 116, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Huang, C.; Xu, X.; Gu, H.; Ye, Y.; Jiang, C.; Qiu, Z.; Xie, X. Direct reprogramming of mouse fibroblasts into cardiomyocytes with chemical cocktails. Cell Res. 2015, 25, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Wada, R.; Muraoka, N.; Inagawa, K.; Yamakawa, H.; Miyamoto, K.; Sadahiro, T.; Umei, T.; Kaneda, R.; Suzuki, T.; Kamiya, K.; et al. Induction of human cardiomyocyte-like cells from fibroblasts by defined factors. Proc. Natl. Acad. Sci. USA 2013, 110, 12667–12672. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.D.; Stone, N.R.; Liu, L.; Spencer, C.I.; Qian, L.; Hayashi, Y.; Delgado-Olguin, P.; Ding, S.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of human fibroblasts toward a cardiomyocyte-like state. Stem Cell Rep. 2013, 1, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Mathison, M.; Patel, V.; Sanagasetti, D.; Gibson, B.W.; Yang, J.; Rosengart, T.K. MiR-590 Promotes Transdifferentiation of Porcine and Human Fibroblasts Toward a Cardiomyocyte-Like Fate by Directly Repressing Specificity Protein 1. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Christoforou, N.; Chakraborty, S.; Kirkton, R.D.; Adler, A.F.; Addis, R.C.; Leong, K.W. Core transcription factors, MicroRNAs, and small molecules drive transdifferentiation of human fibroblasts towards the cardiac cell lineage. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Nam, Y.-J.; Song, K.; Luo, X.; Daniel, E.; Lambeth, K.; West, K.; Hill, J.A.; DiMaio, J.M.; Baker, L.A.; Bassel-Duby, R.; et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc. Natl. Acad. Sci. USA 2013, 110, 5588–5593. [Google Scholar] [CrossRef]
- Cao, N.; Huang, Y.; Zheng, J.; Spencer, C.I.; Zhang, Y.; Fu, J.-D.; Nie, B.; Xie, M.; Zhang, M.; Wang, H.; et al. Conversion of human fibroblasts into functional cardiomyocytes by small molecules. Science 2016, 352, aaf1502. [Google Scholar] [CrossRef]
- Islas, J.F.; Liu, Y.; Weng, K.-C.; Robertson, M.J.; Zhang, S.; Prejusa, A.; Harger, J.; Tikhomirova, D.; Chopra, M.; Iyer, D.; et al. Transcription factors ETS2 and MESP1 transdifferentiate human dermal fibroblasts into cardiac progenitors. Proc. Natl. Acad. Sci. USA 2012, 109, 13016–13021. [Google Scholar] [CrossRef] [PubMed]
- Pratico, E.D.; Feger, B.J.; Watson, M.J.; Sullenger, B.A.; Bowles, D.E.; Milano, C.A.; Nair, S.K. RNA-Mediated Reprogramming of Primary Adult Human Dermal Fibroblasts into c-kit+ Cardiac Progenitor Cells. Stem Cells Dev. 2015, 24, 2622–2633. [Google Scholar] [CrossRef]
- Li, X.-H.; Li, Q.; Jiang, L.; Deng, C.; Liu, Z.; Fu, Y.; Zhang, M.; Tan, H.; Feng, Y.; Shan, Z.; et al. Generation of Functional Human Cardiac Progenitor Cells by High-Efficiency Protein Transduction. Stem Cells Transl. Med. 2015, 4, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, X.; Zhao, L.; Zuo, S.; Chen, X.; Zhang, L.; Lin, Z.; Zhao, X.; Qin, Y.; Zhou, X.; et al. Lineage reprogramming of fibroblasts into induced cardiac progenitor cells by CRISPR/Cas9-based transcriptional activators. Acta Pharm. Sin. B 2020, 10, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, N.; Huang, Y.; Spencer, C.I.; Fu, J.-D.D.; Yu, C.; Liu, K.; Nie, B.; Xu, T.; Li, K.; et al. Expandable Cardiovascular Progenitor Cells Reprogrammed from Fibroblasts. Cell Stem Cell 2016, 18, 368–381. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Verheul, C.; Sommer, A.G.; Brumbaugh, J.; Schwarz, B.A.; Lipchina, I.; Huebner, A.J.; Mostoslavsky, G.; Hochedlinger, K. Lineage conversion induced by pluripotency factors involves transient passage through an iPSC stage. Nat. Biotechnol. 2015, 33, 761–768. [Google Scholar] [CrossRef]
- Maza, I.; Caspi, I.; Zviran, A.; Chomsky, E.; Rais, Y.; Viukov, S.; Geula, S.; Buenrostro, J.D.; Weinberger, L.; Krupalnik, V.; et al. Transient acquisition of pluripotency during somatic cell transdifferentiation with iPSC reprogramming factors. Nat. Biotechnol. 2015, 33, 769–774. [Google Scholar] [CrossRef]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef]
- Inagawa, K.; Miyamoto, K.; Yamakawa, H.; Muraoka, N.; Sadahiro, T.; Umei, T.; Wada, R.; Katsumata, Y.; Kaneda, R.; Nakade, K.; et al. Induction of cardiomyocyte-like cells in infarct hearts by gene transfer of Gata4, Mef2c, and Tbx5. Circ. Res. 2012, 111, 1147–1156. [Google Scholar] [CrossRef]
- Ma, H.; Wang, L.; Yin, C.; Liu, J.; Qian, L. In vivo cardiac reprogramming using an optimal single polycistronic construct. Cardiovasc. Res. 2015, 108, 217–219. [Google Scholar] [CrossRef]
- Mathison, M.; Gersch, R.P.; Nasser, A.; Lilo, S.; Korman, M.; Fourman, M.; Hackett, N.; Shroyer, K.; Yang, J.; Ma, Y.; et al. In vivo cardiac cellular reprogramming efficacy is enhanced by angiogenic preconditioning of the infarcted myocardium with vascular endothelial growth factor. J. Am. Heart Assoc. 2012, 1, e005652. [Google Scholar] [CrossRef] [PubMed]
- Mathison, M.; Singh, V.P.; Gersch, R.P.; Ramirez, M.O.; Cooney, A.; Kaminsky, S.M.; Chiuchiolo, M.J.; Nasser, A.; Yang, J.; Crystal, R.G.; et al. “triplet” polycistronic vectors encoding Gata4, Mef2c, and Tbx5 enhances postinfarct ventricular functional improvement compared with singlet vectors. J. Thorac. Cardiovasc. Surg. 2014, 148, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Mathison, M.; Singh, V.P.; Chiuchiolo, M.J.; Sanagasetti, D.; Mao, Y.; Patel, V.B.; Yang, J.; Kaminsky, S.M.; Crystal, R.G.; Rosengart, T.K. In situ reprogramming to transdifferentiate fibroblasts into cardiomyocytes using adenoviral vectors: Implications for clinical myocardial regeneration. J. Thorac. Cardiovasc. Surg. 2017, 153, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Akiyama, M.; Tamura, F.; Isomi, M.; Yamakawa, H.; Sadahiro, T.; Muraoka, N.; Kojima, H.; Haginiwa, S.; Kurotsu, S.; et al. Direct In Vivo Reprogramming with Sendai Virus Vectors Improves Cardiac Function after Myocardial Infarction. Cell Stem Cell 2018, 22, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Lee, E.; Kim, J.; Kwon, Y.W.; Kwon, Y.; Kim, J. Efficient in vivo direct conversion of fibroblasts into cardiomyocytes using a nanoparticle-based gene carrier. Biomaterials 2019, 192, 500–509. [Google Scholar] [CrossRef]
- Huang, C.; Tu, W.; Fu, Y.; Wang, J.; Xie, X. Chemical-induced cardiac reprogramming in vivo. Cell Res. 2018, 28, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857. [Google Scholar] [CrossRef]
- Lian, X.; Zhang, J.; Azarin, S.; Zhu, K.; Hazeltine, L. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013, 8, 162–175. [Google Scholar] [CrossRef]
- Itskovitz-Eldor, J.; Schuldiner, M.; Karsenti, D.; Eden, A.; Yanuka, O.; Amit, M.; Soreq, H.; Benvenisty, N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000, 6, 88–95. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Z.; Tian, H.; Chen, X. Production and clinical development of nanoparticles for gene delivery. Mol. Ther. Methods Clin. Dev. 2016, 3, 16023. [Google Scholar] [CrossRef]
- Traxler, L.; Edenhofer, F.; Mertens, J. Next-generation disease modeling with direct conversion: A new path to old neurons. FEBS Lett. 2019, 593, 3316–3337. [Google Scholar] [CrossRef]


{kind=link}
| Cell Origin | Reprogramming Cocktails | Efficiency | Functionality | References |
|---|---|---|---|---|
| Direct cardiac reprogramming into iCMs in vitro | ||||
| GMT and modifications to GMT cocktail | ||||
| Mouse | GATA4, MEF2C, TBX5 | 4-6% αMHC-GFP+/cTnT+ iCMs from CFs | AP,CaT, SB | [105] |
| MEF2C, GATA4, TBX5 | ~10% αMHC-GFP+ and ~4.8% cTnT+ iCMs from CFs | AP,CaT, SB | [107] | |
| GATA4, MEF2C, TBX5, HAND2 | 9.2% and 6.8% αMHC+/TnT+ iCMs from TTFs and CFs, respectively | CaT, SB | [108] | |
| GATA4, MEF2C, TBX5, HAND2 | ~1.5% cTnT+ in pDox-GMT; 13% cTnT+ in pMX–GMT/pDox–Hand2 iCMs, from MEFs | CaT, SB | [109] | |
| GATA4, MEF2C, TBX5, HAND2 | ~70–80% of cells expressing GMT(H) were Titin-eGFP+ or α-actinin+ iCMs from MEFs | CaT, SB | [113] | |
| MEF2C, GATA4, TBX5, HAND2 | ~25% Titin-eGFP+/α-actinin+ iCMs from MEFs | CaT, SB | [114] | |
| GATA4, MYOD-MEF2C, TBX5, HAND2 | 10-20% cTnT+ iCMs from embryonic head fibroblasts | CaT, SB | [110] | |
| GATA4, MEF2C, TBX5, HAND2, NKX2.5 | 1.6% cTnT-GCaMP5+ iCMs from MEFs | CaT, SB | [111] | |
| GATA4, MEF2C, TBX5, MYOCD, SRF, (MESP1, BAF60C) | 2.4% αMHC-GFP+ iCMs from MEFs | CaT, no SB | [112] | |
| GATA4, MEF2C, TBX5, (miR-133 or MESP1, MYOCD) | 9.5% αMHC-GFP+/ cTnT+ and 19.9% α-actinin+ iCMs from MEFs | CaT, SB | [115] | |
| GATA4, MEF2C, TBX5, HAND2, NKX2.5, SB431542 | 17% cTnT-GCaMP5+ iCMs from MEFs; 9.27% cTnT-GCaMP5+ iCMs from CFs | CaT, SB | [116] | |
| GATA4, MEF2C, TBX5, HAND2, DAPT | ~38% cTnT+ and ~35% α-actinin+ iCMs from MEFs | CaT, SB | [118] | |
| GATA4, MEF2C, TBX5, HAND2, miR-1, miR-133, A83-01, Y-27632 | 60% cTnT+ and 60% α-actinin+ iCMs from MEFs | AP, CaT, SB | [119] | |
| GATA4, MEF2C, TBX5, HAND2, AKT1 | 23.3% αMHC-GFP+/cTnT+ iCMs from MEFs; 50% beating iCMs from MEFs at Day 21 | CaT, SB | [120] | |
| GATA4, MEF2C, TBX5, (HAND2 or MESP1, MYOCD), FGF2, FGF10, VEGF | ~13% αMHC-GFP+ and ~2% cTnT+ iCMs from MEFs | CaT, SB | [121] | |
| GATA4, MEF2C, TBX5, SB431542, XAV939 | ~30% αMHC-GFP+ iCMs from CFs | AP,CaT, SB | [122] | |
| GATA4, MEF2C, TBX5, HAND2, Diclofenac | ~5% cTnT+/ αMHC+ iCMs from postnatal TTFs | CaT, SB | [123] | |
| GATA4, MEF2C, TBX5, (HAND2), Bmi1 shRNA | 22% αMHC+/TnT+ iCMs from CFs | CaT, SB | [124] | |
| Human | GATA4, MEF2C, TBX5, MESP1, MYOCD | 5.9% cTnT+ and 5.5% α-actinin+ iCMs from HCFs | AP, CaT, c-B | [129] |
| GATA4, MEF2C, TBX5, ESRGG, MESP1, MYOCD, ZFPM2 | 13% αMHC-mCherry+/cTnT+ iCMs from hESC-derived fibroblasts | AP, CaT, no SB | [130] | |
| GATA4, MEF2C, TBX5, MESP1, MYOCD, miR-133 | 27.8% cTnT+ and 8% α-actinin+ iCMs from HCFs | CaT, no SB | [115] | |
| GATA4, MEF2C, TBX5, MYOCD, NKX2.5, mir-1, miR-133, JAK1i, GSK3βi or NRG | ~3.8% cTnT+ iCMs from HDFs | CaT, no SB | [132] | |
| Human, rat, porcine | GATA4, MEF2C, TBX5, (HAND2, MYOCD or miR-590) | ~40% αMHC-GFP+ and ~5-6% cTnT+ iCMs from adult HCFs | No SB in human iCMs | [131] |
| Other cocktails different from GMT | ||||
| Mouse | TBX5, MEF2C, MYOCD | ~11% cTnT+ iCMs from CFs | AP | [125] |
| miR-1, miR-133, miR-208, miR-499a, JI1 | ~28% αMHC-CFP+ iCMs from CFs | AP, CaT, SB | [117] | |
| CHIR99021, RepSox, Forskolin, VPA, Parnate, TTNPB | 14.5% α-actinin+ and 9% α-MHC+ iCMs from MEFs | AP, CaT, SB | [127] | |
| Human | GATA4, HAND2, TBX5, MYOCD, miR-1, miR-133 | ~35% cTnT+ and ~42% tropomyosin+ iCMs from HFFs | CaT, SB | [133] |
| CHIR99021, A83-01, BIX01294, AS8351, SC1, Y27632, OAC2, SU16F, JNJ10198409 | 7% cTnT+ iCMs from HFFs | AP, CaT, SB | [134] | |
| Direct reprogramming into iCPCs in vitro | ||||
| Mouse | MESP1, TBX5, GATA4, NKX2.5, BAF60C, BIO, LIF | > 90% Nkx2.5-YFP+, Gata4+ and Irx4+ iCPCs from adult CFs | Expandable; Tri-lineage dif.; In vivo in AMI | [104] |
| OCT4, SOX2, KLF4, C-MYC, BMP4, Activin A, CHIR99021, SU5402 | 70% Flk1+/Pdgfrα+ iCPCs from MEFs | Expandable; Tri-lineage dif.; In vivo in AMI | [139] | |
| Human | ETS2, MESP1, Activin A, BMP2 | 9.3% NKX2.5-tdTomato+ iCPCs from HDFs | Not expandable; Unipotent (CM) | [135] |
| GATA4, MEF2C, TBX5, HAND2 | 4.9% c-Kit+ iCPCs from adult HDFs | Not expandable; Unipotent (CM) | [136] | |
| GATA4, MEF2C, TBX5, HAND2, BMP4, Activin A, bFGF | 81% Flk1+ and 83% Isl1+ iCPCs from HDFs | Not expandable; Tri-lineage dif.; In vivo in AMI | [137] | |
| GATA4, MEF2C, TBX5, HAND2 | ~72% of GATA4+ cells were NKX2.5+; ~85% of HAND2+ cells were ISL1+, from HFFs | Not expandable; Tri-lineage dif. | [138] | |
| Direct reprogramming into iCMs in vivo | ||||
| GMT cocktail | ||||
| Mouse | GATA4, MEF2C, TBX5, (ReV vector), Thymosin β4 (intramyocardial) | Periostin-Cre: R26R-lacZ mice: 35% β-Gal+ and α-actinin+ iCMs | Improvement in HF and CS | [142] |
| TBX5, MEF2C, GATA4 (ReV vector) | 1% α-actinin+ iCMs derived from GMT transduced cells | Improvement in HF and CS | [143] | |
| MEF2C, GATA4, TBX5 (ReV vector) | Periostin-Cre: R26R-lacZ mice: ~80 β-Gal+/α-actinin+ iCMs per section | Improvement in HF and CS | [144] | |
| GATA4, MEF2C, TBX5 (SeV vector) | TCF21iCre/R26-tdTomato mice: ∼1.5% tdTomato+/cTnT+ iCMs | Improvement in HF and CS | [148] | |
| GATA4, MEF2C, TBX5 (Nanoparticles) | In vitro: 22% αMHC-eGFP+ iCMs from MEFs; In vivo: ND | Improvement in HF and CS | [149] | |
| Rat | GATA4, MEF2C, TBX5 (AV vector) | In vitro: ~6.5% cTnT+ iCMs from rat CFs; In vivo: ND | Improvement in HF and CS | [147] |
| GATA4, TBX5, MEF2C (LeV vector), VEGF (AV vector) | ND | Improvement in HF and CS | [145,146] | |
| Modifications to GMT cocktail | ||||
| Mouse | GATA4, MEF2C, TBX5, HAND2 (ReV vector) | Fsp1-Cre x R26LacZ mice: ~6.5% β-Gal+ iCMs; TCF21-iCre x R26tdTomato mice: ~2.4% tdTomato+ iCMs | Improvement in HF and CS | [108] |
| Other cocktails different from GMT | ||||
| Mouse | miR-1, miR-133, miR-208, miR-499a (LeV vector) | Fsp1-Cre: R26R-tdTomato mice: 12% tdTomato+/cTnT+ iCMs | Improvement in HF and CS | [126] |
| GATA4, MEF2C, TBX5 (ReV vector) SB431542, XAV939 (intraperitoneal) | ROSA-YFP/Periostin-Cre mice: 150-200 YFP+/cTnT+ iCMs per section | Improvement in HF and CS | [122] | |
| CHIR99021, RepSox, Forskolin, TTNPB, Rolipram (oral) VPA, Parnate (intraperitoneal) | Fsp1-Cre: R26RtdTomato: 0.78% tdTomato+/α-actinin+ iCMs | Improvement in HF and CS | [150] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Muneta, L.; Miranda-Arrubla, J.; Carvajal-Vergara, X. The Future of Direct Cardiac Reprogramming: Any GMT Cocktail Variety? Int. J. Mol. Sci. 2020, 21, 7950. https://doi.org/10.3390/ijms21217950
López-Muneta L, Miranda-Arrubla J, Carvajal-Vergara X. The Future of Direct Cardiac Reprogramming: Any GMT Cocktail Variety? International Journal of Molecular Sciences. 2020; 21(21):7950. https://doi.org/10.3390/ijms21217950
Chicago/Turabian StyleLópez-Muneta, Leyre, Josu Miranda-Arrubla, and Xonia Carvajal-Vergara. 2020. "The Future of Direct Cardiac Reprogramming: Any GMT Cocktail Variety?" International Journal of Molecular Sciences 21, no. 21: 7950. https://doi.org/10.3390/ijms21217950
APA StyleLópez-Muneta, L., Miranda-Arrubla, J., & Carvajal-Vergara, X. (2020). The Future of Direct Cardiac Reprogramming: Any GMT Cocktail Variety? International Journal of Molecular Sciences, 21(21), 7950. https://doi.org/10.3390/ijms21217950