Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Organic Cation Transporters (OCTs) in the Liver
3. OCTs in the Kidney
4. OCTs in the Intestines
5. OCTs in Other Tissues
5.1. Central Nervous System
5.2. Inner Ear
5.3. Cardiovascular System
5.4. Skeletal Muscle
5.5. Reproductive Organs
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| OCT | Organic cation transporter |
| OCTN | Organic cation transporter novel |
| SLC | Solute carrier |
| TMA | Trimethylamine |
| TMAO | Trimethylamine N-oxide |
References
- Bioparadigms SLC TABLES. Available online: http://slc.bioparadigms.org/ (accessed on 5 April 2020).
- Cesar-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef]
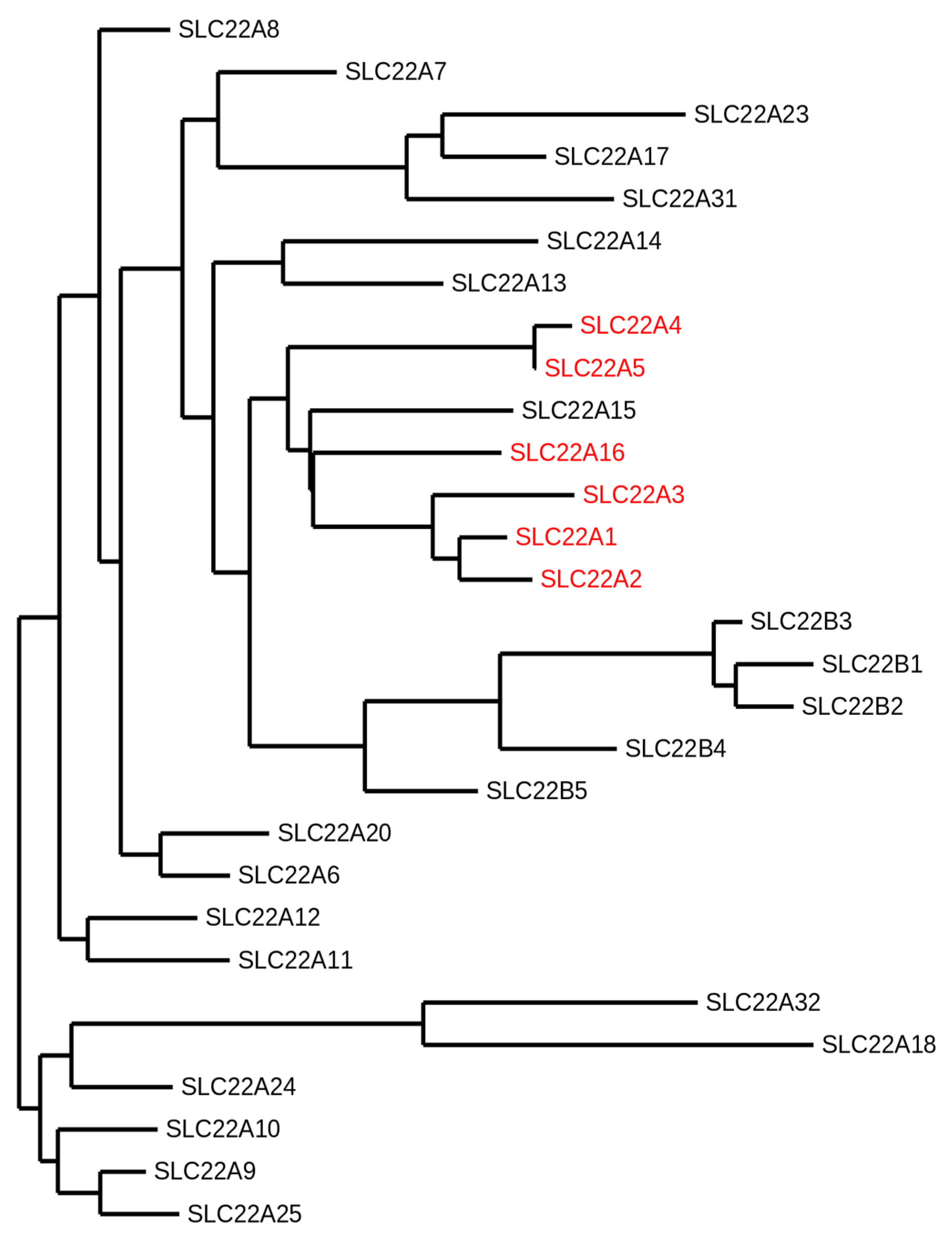
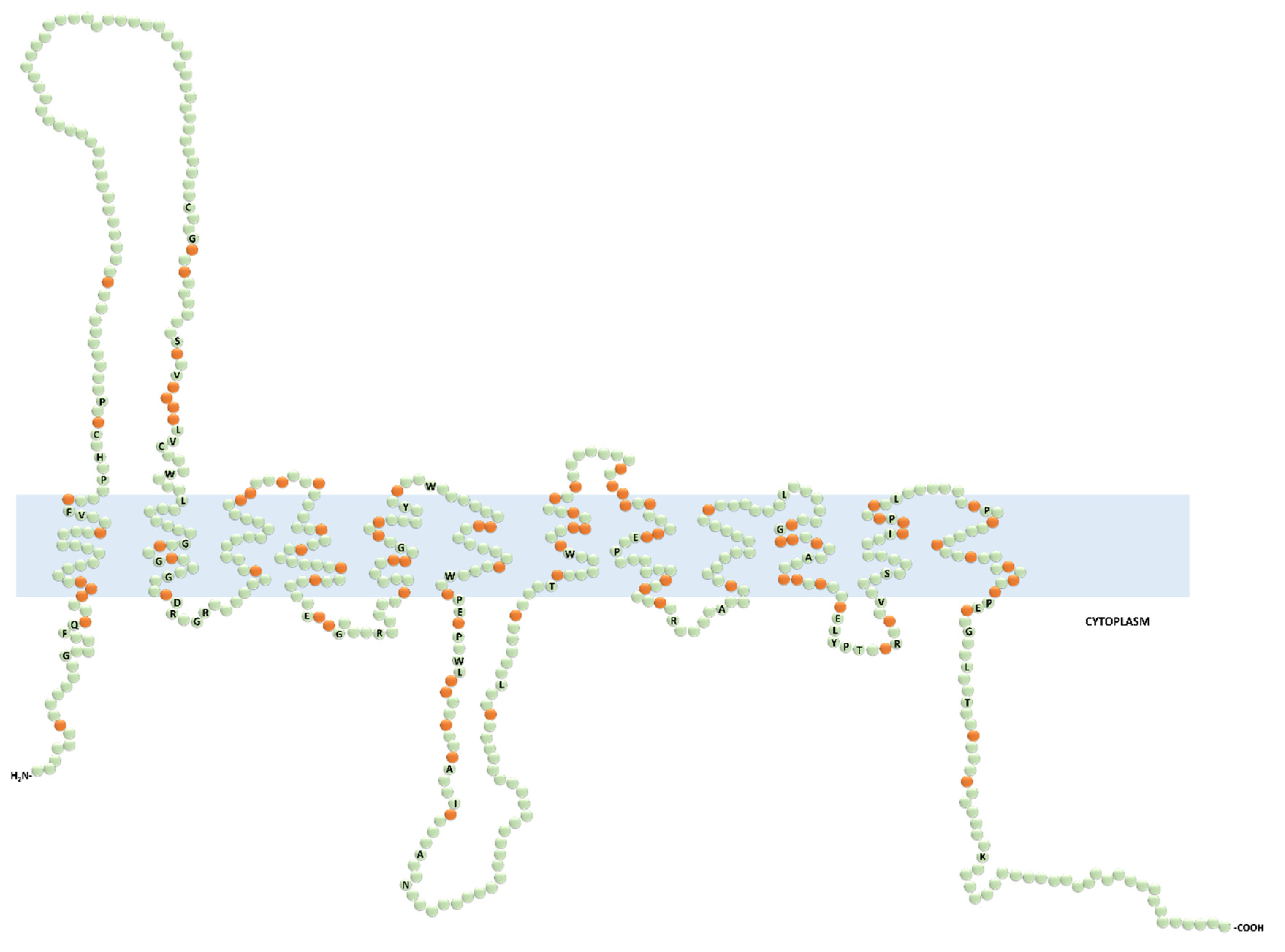
- Koepsell, H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Asp. Med. 2013, 34, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Nigam, K.B.; Date, R.C.; Bush, K.T.; Springer, S.A.; Saier, M.H., Jr.; Wu, W.; Nigam, S.K. Evolutionary Analysis and Classification of OATs, OCTs, OCTNs, and Other SLC22 Transporters: Structure-Function Implications and Analysis of Sequence Motifs. PLoS ONE 2015, 10, e0140569. [Google Scholar] [CrossRef] [PubMed]
- Eraly, S.A.; Nigam, S.K. Novel human cDNAs homologous to Drosophila Orct and mammalian carnitine transporters. Biochem. Biophys. Res. Commun. 2002, 297, 1159–1166. [Google Scholar] [CrossRef]
- Okada, R.; Koshizuka, K.; Yamada, Y.; Moriya, S.; Kikkawa, N.; Kinoshita, T.; Hanazawa, T.; Seki, N. Regulation of Oncogenic Targets by miR-99a-3p (Passenger Strand of miR-99a-Duplex) in Head and Neck Squamous Cell Carcinoma. Cells 2019, 8, 1535. [Google Scholar] [CrossRef]
- Zhu, G.; Qian, M.; Lu, L.; Chen, Y.; Zhang, X.; Wu, Q.; Liu, Y.; Bian, Z.; Yang, Y.; Guo, S.; et al. O-GlcNAcylation of YY1 stimulates tumorigenesis in colorectal cancer cells by targeting SLC22A15 and AANAT. Carcinogenesis 2019, 40, 1121–1131. [Google Scholar] [CrossRef]
- Drake, K.A.; Torgerson, D.G.; Gignoux, C.R.; Galanter, J.M.; Roth, L.A.; Huntsman, S.; Eng, C.; Oh, S.S.; Yee, S.W.; Lin, L.; et al. A genome-wide association study of bronchodilator response in Latinos implicates rare variants. J. Allergy Clin. Immunol. 2014, 133, 370–378. [Google Scholar] [CrossRef]
- Phylogeny.fr: Robust Phylogenetic Analysis for the Non-Specialist. Available online: www.phylogeny.fr/index.cgi (accessed on 3 March 2020).
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465-9. [Google Scholar] [CrossRef]
- Dereeper, A.; Audic, S.; Claverie, J.M.; Blanc, G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMS Evol. Biol. 2010, 10, 8. [Google Scholar] [CrossRef]
- Koepsell, H. Substrate recognition and translocation by polyspecific organic cation transporters. Biol. Chem. 2011, 392, 95–101. [Google Scholar] [CrossRef]
- Schmitt, B.M.; Gorbunov, D.; Schlachtbauer, P.; Egenberger, B.; Gorboulev, V.; Wischmeyer, E.; Muller, T.; Koepsell, H. Charge-to-substrate ratio during organic cation uptake by rat OCT2 is voltage dependent and altered by exchange of glutamate 448 with glutamine. Am. J. Physiol. Ren. Physiol. 2009, 296, F709–F722. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharm. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef] [PubMed]
- Pelis, R.M.; Wright, S.H. SLC22, SLC44, and SLC47 transporters--organic anion and cation transporters: Molecular and cellular properties. Curr. Top. Membr. 2014, 73, 233–261. [Google Scholar] [PubMed]
- Visentin, M.; Gai, Z.; Torozi, A.; Hiller, C.; Kullak-Ublick, G.A. Colistin is substrate of the carnitine/organic cation transporter 2 (OCTN2, SLC22A5). Drug Metab. Dispos. 2017, 45, 1240–1244. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Visentin, M.; Hiller, C.; Krajnc, E.; Li, T.; Zhen, J.; Kullak-Ublick, G.A. Organic Cation Transporter 2 Overexpression May Confer an Increased Risk of Gentamicin-Induced Nephrotoxicity. Antimicrob. Agents Chemother. 2016, 60, 5573–5580. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Nakanishi, T.; Shibue, Y.; Kobayashi, D.; Moseley, R.H.; Shirasaka, Y.; Tamai, I. Hepatic uptake of gamma-butyrobetaine, a precursor of carnitine biosynthesis, in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G681–G686. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J. Biol. Chem. 1998, 273, 20378–20382. [Google Scholar] [CrossRef]
- Koepsell, H. Multiple binding sites in organic cation transporters require sophisticated procedures to identify interactions of novel drugs. Biol. Chem. 2019, 400, 195–207. [Google Scholar] [CrossRef]
- Visentin, M.; Torozi, A.; Gai, Z.; Hausler, S.; Li, C.; Hiller, C.; Schraml, P.H.; Moch, H.; Kullak-Ublick, G.A. Fluorocholine Transport Mediated by the Organic Cation Transporter 2 (OCT2, SLC22A2): Implication for Imaging of Kidney Tumors. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1129–1136. [Google Scholar] [CrossRef]
- Hormann, S.; Gai, Z.; Kullak-Ublick, G.A.; Visentin, M. Plasma Membrane Cholesterol Regulates the Allosteric Binding of 1-Methyl-4-Phenylpyridinium to Organic Cation Transporter 2 (SLC22A2). J. Pharmacol. Exp. Ther. 2020, 372, 46–53. [Google Scholar] [CrossRef]
- Nies, A.T.; Koepsell, H.; Winter, S.; Burk, O.; Klein, K.; Kerb, R.; Zanger, U.M.; Keppler, D.; Schwab, M.; Schaeffeler, E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 2009, 50, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dresser, M.J.; Gray, A.T.; Yost, S.C.; Terashita, S.; Giacomini, K.M. Cloning and functional expression of a human liver organic cation transporter. Mol. Pharmacol. 1997, 51, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Sakurai, Y.; Saito, H.; Masuda, S.; Urakami, Y.; Goto, M.; Fukatsu, A.; Ogawa, O.; Inui, K. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol. 2002, 13, 866–874. [Google Scholar] [PubMed]
- Enomoto, A.; Wempe, M.F.; Tsuchida, H.; Shin, H.J.; Cha, S.H.; Anzai, N.; Goto, A.; Sakamoto, A.; Niwa, T.; Kanai, Y.; et al. Molecular identification of a novel carnitine transporter specific to human testis. Insights into the mechanism of carnitine recognition. J. Biol. Chem. 2002, 277, 36262–36271. [Google Scholar] [CrossRef]
- Sagwal, S.K.; Pasqual-Melo, G.; Bodnar, Y.; Gandhirajan, R.K.; Bekeschus, S. Combination of chemotherapy and physical plasma elicits melanoma cell death via upregulation of SLC22A16. Cell Death Dis. 2018, 9, 1179. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Y.; Yue, X. SLC22A16 upregulation is an independent unfavorable prognostic indicator in gastric cancer. Future Oncol. 2018, 14, 2139–2148. [Google Scholar] [CrossRef]
- Tecza, K.; Pamula-Pilat, J.; Lanuszewska, J.; Butkiewicz, D.; Grzybowska, E. Pharmacogenetics of toxicity of 5-fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget 2018, 9, 9114–9136. [Google Scholar] [CrossRef]
- Faraji, A.; Dehghan Manshadi, H.R.; Mobaraki, M.; Zare, M.; Houshmand, M. Association of ABCB1 and SLC22A16 Gene Polymorphisms with Incidence of Doxorubicin-Induced Febrile Neutropenia: A Survey of Iranian Breast Cancer Patients. PLoS ONE 2016, 11, e0168519. [Google Scholar] [CrossRef]
- Wu, Y.; Hurren, R.; MacLean, N.; Gronda, M.; Jitkova, Y.; Sukhai, M.A.; Minden, M.D.; Schimmer, A.D. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis 2015, 20, 1099–1108. [Google Scholar] [CrossRef]
- Bray, J.; Sludden, J.; Griffin, M.J.; Cole, M.; Verrill, M.; Jamieson, D.; Boddy, A.V. Influence of pharmacogenetics on response and toxicity in breast cancer patients treated with doxorubicin and cyclophosphamide. Br. J. Cancer 2010, 102, 1003–1009. [Google Scholar] [CrossRef]
- Aouida, M.; Poulin, R.; Ramotar, D. The human carnitine transporter SLC22A16 mediates high affinity uptake of the anticancer polyamine analogue bleomycin-A5. J. Biol. Chem. 2010, 285, 6275–6284. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Ito, K.; Akahira, J.; Sato, N.; Onogawa, T.; Moriya, T.; Unno, M.; Abe, T.; Niikura, H.; Takano, T.; et al. Expression of organic cation transporter SLC22A16 in human epithelial ovarian cancer: A possible role of the adriamycin importer. Int. J. Gynecol. Pathol. 2007, 26, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Wong, Z.W.; Jada, S.R.; Xiang, X.; Chen Shu, X.; Ang, P.C.; Figg, W.D.; Lee, E.J.; Chowbay, B. Novel SLC22A16 polymorphisms and influence on doxorubicin pharmacokinetics in Asian breast cancer patients. Pharmacogenomics 2007, 8, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Okabe, M.; Unno, M.; Harigae, H.; Kaku, M.; Okitsu, Y.; Sasaki, T.; Mizoi, T.; Shiiba, K.; Takanaga, H.; Terasaki, T.; et al. Characterization of the organic cation transporter SLC22A16: A doxorubicin importer. Biochem. Biophys. Res. Commun. 2005, 333, 754–762. [Google Scholar] [CrossRef]
- Gong, S.; Lu, X.; Xu, Y.; Swiderski, C.F.; Jordan, C.T.; Moscow, J.A. Identification of OCT6 as a novel organic cation transporter preferentially expressed in hematopoietic cells and leukemias. Exp. Hematol. 2002, 30, 1162–1169. [Google Scholar] [CrossRef]
- Sato, N.; Ito, K.; Onogawa, T.; Akahira, J.; Unno, M.; Abe, T.; Niikura, H.; Yaegashi, N. Expression of organic cation transporter SLC22A16 in human endometria. Int. J. Gynecol. Pathol. 2007, 26, 53–60. [Google Scholar] [CrossRef]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstadt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar] [CrossRef]
- Koepsell, H.; Schmitt, B.M.; Gorboulev, V. Organic cation transporters. Rev. Physiol. Biochem. Pharmacol. 2003, 150, 36–90. [Google Scholar]
- Neuhoff, S.; Ungell, A.L.; Zamora, I.; Artursson, P. pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: Implications for drug-drug interactions. Pharm. Res. 2003, 20, 1141–1148. [Google Scholar] [CrossRef]
- Tamai, I.; China, K.; Sai, Y.; Kobayashi, D.; Nezu, J.; Kawahara, E.; Tsuji, A. Na(+)-coupled transport of L-carnitine via high-affinity carnitine transporter OCTN2 and its subcellular localization in kidney. Biochim. et Biophys. Acta 2001, 1512, 273–284. [Google Scholar] [CrossRef]
- Meier, Y.; Eloranta, J.J.; Darimont, J.; Ismair, M.G.; Hiller, C.; Fried, M.; Kullak-Ublick, G.A.; Vavricka, S.R. Regional distribution of solute carrier mRNA expression along the human intestinal tract. Drug Metab. Dispos. Biol. Fate Chem. 2007, 35, 590–594. [Google Scholar] [CrossRef]
- Sugiura, T.; Kato, Y.; Wakayama, T.; Silver, D.L.; Kubo, Y.; Iseki, S.; Tsuji, A. PDZK1 regulates two intestinal solute carriers (Slc15a1 and Slc22a5) in mice. Drug Metab. Dispos. Biol. Fate Chem. 2008, 36, 1181–1188. [Google Scholar] [CrossRef]
- McCloud, E.; Ma, T.Y.; Grant, K.E.; Mathis, R.K.; Said, H.M. Uptake of L-carnitine by a human intestinal epithelial cell line, Caco-2. Gastroenterology 1996, 111, 1534–1540. [Google Scholar] [CrossRef]
- Schulze, U.; Brast, S.; Grabner, A.; Albiker, C.; Snieder, B.; Holle, S.; Schlatter, E.; Schroter, R.; Pavenstadt, H.; Herrmann, E.; et al. Tetraspanin CD63 controls basolateral sorting of organic cation transporter 2 in renal proximal tubules. Faseb. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 1421–1433. [Google Scholar] [CrossRef]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef]
- Muth, T.R.; Caplan, M.J. Transport protein trafficking in polarized cells. Annu. Rev. Cell Dev. Biol. 2003, 19, 333–366. [Google Scholar] [CrossRef]
- Koizumi, T.; Nikaido, H.; Hayakawa, J.; Nonomura, A.; Yoneda, T. Infantile disease with microvesicular fatty infiltration of viscera spontaneously occurring in the C3H-H-2(0) strain of mouse with similarities to Reye’s syndrome. Lab Anim. 1988, 22, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Sugiura, M.; Sugiura, T.; Wakayama, T.; Kubo, Y.; Kobayashi, D.; Sai, Y.; Tamai, I.; Iseki, S.; Tsuji, A. Organic cation/carnitine transporter OCTN2 (Slc22a5) is responsible for carnitine transport across apical membranes of small intestinal epithelial cells in mouse. Mol. Pharmacol. 2006, 70, 829–837. [Google Scholar] [CrossRef]
- Bohmer, T.; Eiklid, K.; Jonsen, J. Carnitine uptake into human heart cells in culture. Biochim. et Biophys. Acta 1977, 465, 627–633. [Google Scholar] [CrossRef]
- Yokogawa, K.; Yonekawa, M.; Tamai, I.; Ohashi, R.; Tatsumi, Y.; Higashi, Y.; Nomura, M.; Hashimoto, N.; Nikaido, H.; Hayakawa, J.; et al. Loss of wild-type carrier-mediated L-carnitine transport activity in hepatocytes of juvenile visceral steatosis mice. Hepatology 1999, 30, 997–1001. [Google Scholar] [CrossRef]
- Grundemann, D.; Gorboulev, V.; Gambaryan, S.; Veyhl, M.; Koepsell, H. Drug excretion mediated by a new prototype of polyspecific transporter. Nature 1994, 372, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Saborowski, M.; Kullak-Ublick, G.A.; Eloranta, J.J. The human organic cation transporter-1 gene is transactivated by hepatocyte nuclear factor-4alpha. J. Pharmacol. Exp. Ther. 2006, 317, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Suzuki, H.; Hirohashi, T.; Ishikawa, T.; Meier, P.J.; Hirose, K.; Akizawa, T.; Yoshioka, M.; Sugiyama, Y. Characterization of inducible nature of MRP3 in rat liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G438–G446. [Google Scholar] [CrossRef]
- Denk, G.U.; Soroka, C.J.; Mennone, A.; Koepsell, H.; Beuers, U.; Boyer, J.L. Down-regulation of the organic cation transporter 1 of rat liver in obstructive cholestasis. Hepatology 2004, 39, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.E.; Hong, S.S.; Choi, M.K.; Maeng, H.J.; Kim, D.D.; Chung, S.J.; Shim, C.K. Reduced antidiabetic effect of metformin and down-regulation of hepatic Oct1 in rats with ethynylestradiol-induced cholestasis. Pharm. Res. 2009, 26, 549–559. [Google Scholar] [CrossRef]
- Visentin, M.; van Rosmalen, B.V.; Hiller, C.; Bieze, M.; Hofstetter, L.; Verheij, J.; Kullak-Ublick, G.A.; Koepsell, H.; Phoa, S.S.; Tamai, I.; et al. Impact of Organic Cation Transporters (OCT-SLC22A) on Differential Diagnosis of Intrahepatic Lesions. Drug Metab. Dispos. 2017, 45, 166–173. [Google Scholar] [CrossRef]
- Heise, M.; Lautem, A.; Knapstein, J.; Schattenberg, J.M.; Hoppe-Lotichius, M.; Foltys, D.; Weiler, N.; Zimmermann, A.; Schad, A.; Grundemann, D.; et al. Downregulation of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) in human hepatocellular carcinoma and their prognostic significance. BMC Cancer 2012, 12, 109. [Google Scholar] [CrossRef]
- Schaeffeler, E.; Hellerbrand, C.; Nies, A.T.; Winter, S.; Kruck, S.; Hofmann, U.; van der Kuip, H.; Zanger, U.M.; Koepsell, H.; Schwab, M. DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med. 2011, 3, 82. [Google Scholar] [CrossRef]
- Ciarimboli, G.; Koepsell, H.; Iordanova, M.; Gorboulev, V.; Durner, B.; Lang, D.; Edemir, B.; Schroter, R.; Van Le, T.; Schlatter, E. Individual PKC-phosphorylation sites in organic cation transporter 1 determine substrate selectivity and transport regulation. J. Am. Soc. Nephrol. 2005, 16, 1562–1570. [Google Scholar] [CrossRef]
- Keller, T.; Gorboulev, V.; Mueller, T.D.; Dotsch, V.; Bernhard, F.; Koepsell, H. Rat Organic Cation Transporter 1 Contains Three Binding Sites for Substrate 1-Methyl-4-phenylpyridinium per Monomer. Mol. Pharmacol. 2019, 95, 169–182. [Google Scholar] [CrossRef]
- Shu, Y.; Sheardown, S.A.; Brown, C.; Owen, R.P.; Zhang, S.; Castro, R.A.; Ianculescu, A.G.; Yue, L.; Lo, J.C.; Burchard, E.G.; et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J. Clin. Investig. 2007, 117, 1422–1431. [Google Scholar] [CrossRef]
- Manzetti, S.; Zhang, J.; van der Spoel, D. Thiamin function, metabolism, uptake, and transport. Biochemistry 2014, 53, 821–835. [Google Scholar] [CrossRef]
- Chen, L.; Shu, Y.; Liang, X.; Chen, E.C.; Yee, S.W.; Zur, A.A.; Li, S.; Xu, L.; Keshari, K.R.; Lin, M.J.; et al. OCT1 is a high-capacity thiamine transporter that regulates hepatic steatosis and is a target of metformin. Proc. Natl. Acad. Sci. USA 2014, 111, 9983–9988. [Google Scholar] [CrossRef]
- Nies, A.T.; Schwab, M. Organic cation transporter pharmacogenomics and drug-drug interaction. Expert Rev. Clin. Pharmacol. 2010, 3, 707–711. [Google Scholar] [CrossRef]
- Tzvetkov, M.V.; Matthaei, J.; Pojar, S.; Faltraco, F.; Vogler, S.; Prukop, T.; Seitz, T.; Brockmoller, J. Increased Systemic Exposure and Stronger Cardiovascular and Metabolic Adverse Reactions to Fenoterol in Individuals with Heritable OCT1 Deficiency. Clin. Pharmacol. Ther. 2018, 103, 868–878. [Google Scholar] [CrossRef]
- Sundelin, E.; Gormsen, L.C.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Christensen, M.; Brosen, K.; Frokiaer, J.; Jessen, N. Genetic Polymorphisms in Organic Cation Transporter 1 Attenuates Hepatic Metformin Exposure in Humans. Clin. Pharmacol. Ther. 2017, 102, 841–848. [Google Scholar] [CrossRef]
- Tzvetkov, M.V. OCT1 pharmacogenetics in pain management: Is a clinical application within reach? Pharmacogenomics 2017, 18, 1515–1523. [Google Scholar] [CrossRef]
- Zolk, O. Disposition of metformin: Variability due to polymorphisms of organic cation transporters. Ann. Med. 2012, 44, 119–129. [Google Scholar] [CrossRef]
- Lanvers-Kaminsky, C.; Sprowl, J.A.; Malath, I.; Deuster, D.; Eveslage, M.; Schlatter, E.; Mathijssen, R.H.; Boos, J.; Jurgens, H.; Am Zehnhoff-Dinnesen, A.G.; et al. Human OCT2 variant c.808G>T confers protection effect against cisplatin-induced ototoxicity. Pharmacogenomics 2015, 16, 323–332. [Google Scholar] [CrossRef]
- Grundemann, D.; Schechinger, B.; Rappold, G.A.; Schomig, E. Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat. Neurosci. 1998, 1, 349–351. [Google Scholar] [CrossRef]
- Vollmar, J.; Kim, Y.O.; Marquardt, J.U.; Becker, D.; Galle, P.R.; Schuppan, D.; Zimmermann, T. Deletion of organic cation transporter Oct3 promotes hepatic fibrosis via upregulation of TGFbeta. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G195–G202. [Google Scholar] [CrossRef]
- Kunzmann, S.; Schmidt-Weber, C.; Zingg, J.M.; Azzi, A.; Kramer, B.W.; Blaser, K.; Akdis, C.A.; Speer, C.P. Connective tissue growth factor expression is regulated by histamine in lung fibroblasts: Potential role of histamine in airway remodeling. J. Allergy Clin. Immunol. 2007, 119, 1398–1407. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D.; Mahmoudi, A.; Mougenot, N.; Varoquaux, O.; Le Nahour, G.; Fouret, P.; Lechat, P. Catecholamine effects on cardiac remodelling, oxidative stress and fibrosis in experimental heart failure. Redox Rep. 2002, 7, 145–151. [Google Scholar] [CrossRef]
- Vaz, F.M.; Wanders, R.J. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361 Pt 3, 417–429. [Google Scholar] [CrossRef]
- Almannai, M.; Alfadhel, M.; El-Hattab, A.W. Carnitine Inborn Errors of Metabolism. Molecules 2019, 24, 3251. [Google Scholar] [CrossRef]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. et Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Yokogawa, K.; Higashi, Y.; Tamai, I.; Nomura, M.; Hashimoto, N.; Nikaido, H.; Hayakawa, J.; Miyamoto, K.; Tsuji, A. Decreased tissue distribution of L-carnitine in juvenile visceral steatosis mice. J. Pharmacol. Exp. Ther. 1999, 289, 224–230. [Google Scholar]
- Nezu, J.; Tamai, I.; Oku, A.; Ohashi, R.; Yabuuchi, H.; Hashimoto, N.; Nikaido, H.; Sai, Y.; Koizumi, A.; Shoji, Y.; et al. Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat. Genet. 1999, 21, 91–94. [Google Scholar] [CrossRef]
- Ringseis, R.; Luci, S.; Spielmann, J.; Kluge, H.; Fischer, M.; Geissler, S.; Wen, G.; Hirche, F.; Eder, K. Clofibrate treatment up-regulates novel organic cation transporter (OCTN)-2 in tissues of pigs as a model of non-proliferating species. Eur. J. Pharmacol. 2008, 583, 11–17. [Google Scholar] [CrossRef]
- Ringseis, R.; Posel, S.; Hirche, F.; Eder, K. Treatment with pharmacological peroxisome proliferator-activated receptor alpha agonist clofibrate causes upregulation of organic cation transporter 2 in liver and small intestine of rats. Pharmacol. Res. Off. J. Ital. Pharmacol. Soc. 2007, 56, 175–183. [Google Scholar]
- Maeda, T.; Wakasawa, T.; Funabashi, M.; Fukushi, A.; Fujita, M.; Motojima, K.; Tamai, I. Regulation of Octn2 transporter (SLC22A5) by peroxisome proliferator activated receptor alpha. Biol. Pharm. Bull. 2008, 31, 1230–1236. [Google Scholar] [CrossRef]
- Hirai, T.; Fukui, Y.; Motojima, K. PPARalpha agonists positively and negatively regulate the expression of several nutrient/drug transporters in mouse small intestine. Biol. Pharm. Bull. 2007, 30, 2185–2190. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Konig, B.; Stangl, G.I.; Eder, K. PPAR alpha mediates transcriptional upregulation of novel organic cation transporters-2 and -3 and enzymes involved in hepatic carnitine synthesis. Exp. Biol. Med. (Maywood) 2008, 233, 356–365. [Google Scholar] [CrossRef]
- van Vlies, N.; Ferdinandusse, S.; Turkenburg, M.; Wanders, R.J.; Vaz, F.M. PPAR alpha-activation results in enhanced carnitine biosynthesis and OCTN2-mediated hepatic carnitine accumulation. Biochim. et Biophys. Acta 2007, 1767, 1134–1142. [Google Scholar] [CrossRef]
- Kiens, B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol. Rev. 2006, 86, 205–243. [Google Scholar] [CrossRef]
- Stephens, F.B.; Constantin-Teodosiu, D.; Laithwaite, D.; Simpson, E.J.; Greenhaff, P.L. Insulin stimulates L-carnitine accumulation in human skeletal muscle. Faseb. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 377–379. [Google Scholar] [CrossRef]
- Tang, Y.; Masuo, Y.; Sakai, Y.; Wakayama, T.; Sugiura, T.; Harada, R.; Futatsugi, A.; Komura, T.; Nakamichi, N.; Sekiguchi, H.; et al. Localization of Xenobiotic Transporter OCTN1/SLC22A4 in Hepatic Stellate Cells and Its Protective Role in Liver Fibrosis. J. Pharm. Sci. 2016, 105, 1779–1789. [Google Scholar] [CrossRef]
- McBride, B.F.; Yang, T.; Liu, K.; Urban, T.J.; Giacomini, K.M.; Kim, R.B.; Roden, D.M. The organic cation transporter, OCTN1, expressed in the human heart, potentiates antagonism of the HERG potassium channel. J. Cardiovasc. Pharmacol. 2009, 54, 63–71. [Google Scholar] [CrossRef]
- Tamai, I.; Yabuuchi, H.; Nezu, J.; Sai, Y.; Oku, A.; Shimane, M.; Tsuji, A. Cloning and characterization of a novel human pH-dependent organic cation transporter, OCTN1. FEBS Lett. 1997, 419, 107–111. [Google Scholar] [CrossRef]
- Motohashi, H.; Nakao, Y.; Masuda, S.; Katsura, T.; Kamba, T.; Ogawa, O.; Inui, K. Precise comparison of protein localization among OCT, OAT, and MATE in human kidney. J. Pharm. Sci. 2013, 102, 3302–3308. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, W.; Prasad, P.D.; Seth, P.; Rajan, D.P.; Leibach, F.H.; Chen, J.; Conway, S.J.; Ganapathy, V. Functional characteristics and tissue distribution pattern of organic cation transporter 2 (OCTN2), an organic cation/carnitine transporter. J. Pharmacol. Exp. Ther. 1999, 290, 1482–1492. [Google Scholar] [PubMed]
- Jonker, J.W.; Wagenaar, E.; Van Eijl, S.; Schinkel, A.H. Deficiency in the organic cation transporters 1 and 2 (Oct1/Oct2 [Slc22a1/Slc22a2]) in mice abolishes renal secretion of organic cations. Mol. Cell Biol. 2003, 23, 7902–7908. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Bleasby, K.; Chan, G.H.; Nunes, I.; Evers, R. The Complexities of Interpreting Reversible Elevated Serum Creatinine Levels in Drug Development: Does a Correlation with Inhibition of Renal Transporters Exist? Drug Metab. Dispos. 2016, 44, 1498–1509. [Google Scholar] [CrossRef]
- Levey, A.S.; Perrone, R.D.; Madias, N.E. Serum creatinine and renal function. Annu. Rev. Med. 1988, 39, 465–490. [Google Scholar] [CrossRef] [PubMed]
- Lepist, E.I.; Zhang, X.; Hao, J.; Huang, J.; Kosaka, A.; Birkus, G.; Murray, B.P.; Bannister, R.; Cihlar, T.; Huang, Y.; et al. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014, 86, 350–357. [Google Scholar] [CrossRef]
- Perrone, R.D.; Madias, N.E.; Levey, A.S. Serum creatinine as an index of renal function: New insights into old concepts. Clin. Chem. 1992, 38, 1933–1953. [Google Scholar] [CrossRef] [PubMed]
- Urakami, Y.; Kimura, N.; Okuda, M.; Inui, K. Creatinine transport by basolateral organic cation transporter hOCT2 in the human kidney. Pharm. Res. 2004, 21, 976–981. [Google Scholar] [CrossRef]
- Scotcher, D.; Arya, V.; Yang, X.; Zhao, P.; Zhang, L.; Huang, S.M.; Rostami-Hodjegan, A.; Galetin, A. Mechanistic Models as Framework for Understanding Biomarker Disposition: Prediction of Creatinine-Drug Interactions. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 282–293. [Google Scholar] [CrossRef]
- Gutierrez, F.; Fulladosa, X.; Barril, G.; Domingo, P. Renal tubular transporter-mediated interactions of HIV drugs: Implications for patient management. Aids. Rev. 2014, 16, 199–212. [Google Scholar]
- Nakada, T.; Kudo, T.; Kume, T.; Kusuhara, H.; Ito, K. Estimation of changes in serum creatinine and creatinine clearance caused by renal transporter inhibition in healthy subjects. Drug Metab. Pharm. 2019, 34, 233–238. [Google Scholar] [CrossRef]
- EMA Guideline on the Investigation of Drug Interactions. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf (accessed on 19 August 2020).
- FDA, U. In Vitro Metabolism- and Transporter- Mediated Drug-Drug Interaction Studies Guidance for Industry. 2019. Available online: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (accessed on 19 August 2020).
- Somogyi, A.; Stockley, C.; Keal, J.; Rolan, P.; Bochner, F. Reduction of metformin renal tubular secretion by cimetidine in man. Br. J. Clin. Pharm. 1987, 23, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.C.; Liang, X.; Yee, S.W.; Geier, E.G.; Stocker, S.L.; Chen, L.; Giacomini, K.M. Targeted disruption of organic cation transporter 3 attenuates the pharmacologic response to metformin. Mol. Pharm. 2015, 88, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Yin, O.Q.; Tomlinson, B.; Chow, M.S. OCT2 polymorphisms and in-vivo renal functional consequence: Studies with metformin and cimetidine. Pharm. Genom. 2008, 18, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Kusuhara, H.; Yokochi, M.; Toyoshima, J.; Inoue, K.; Yuasa, H.; Sugiyama, Y. Competitive inhibition of the luminal efflux by multidrug and toxin extrusions, but not basolateral uptake by organic cation transporter 2, is the likely mechanism underlying the pharmacokinetic drug-drug interactions caused by cimetidine in the kidney. J. Pharmacol. Exp. Ther. 2012, 340, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Terada, T.; Ueba, M.; Sato, T.; Masuda, S.; Katsura, T.; Inui, K. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J. Pharmacol. Exp. Ther. 2009, 329, 185–191. [Google Scholar] [CrossRef]
- Wright, S.H. Molecular and cellular physiology of organic cation transporter 2. Am. J. Physiol. Ren. Physiol. 2019, 317, F1669–F1679. [Google Scholar] [CrossRef]
- Sandoval, P.J.; Morales, M.; Secomb, T.W.; Wright, S.H. Kinetic basis of metformin-MPP interactions with organic cation transporter OCT2. Am. J. Physiol. Ren. Physiol. 2019, 317, F720–F734. [Google Scholar] [CrossRef]
- Yonezawa, A.; Inui, K. Organic cation transporter OCT/SLC22A and H(+)/organic cation antiporter MATE/SLC47A are key molecules for nephrotoxicity of platinum agents. Biochem. Pharm. 2011, 81, 563–568. [Google Scholar] [CrossRef]
- Manohar, S.; Leung, N. Cisplatin nephrotoxicity: A review of the literature. J. Nephrol. 2018, 31, 15–25. [Google Scholar] [CrossRef]
- Ciarimboli, G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014, 34, 547–550. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G. Membrane transporters as mediators of Cisplatin effects and side effects. Science (Cairo) 2012, 2012, 473829. [Google Scholar] [CrossRef]
- Ciarimboli, G.; Ludwig, T.; Lang, D.; Pavenstadt, H.; Koepsell, H.; Piechota, H.J.; Haier, J.; Jaehde, U.; Zisowsky, J.; Schlatter, E. Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am. J. Pathol. 2005, 167, 1477–1484. [Google Scholar] [CrossRef]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharm. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef]
- Harrach, S.; Ciarimboli, G. Role of transporters in the distribution of platinum-based drugs. Front. Pharmacol. 2015, 6, 85. [Google Scholar] [CrossRef]
- Sleijfer, D.T.; Offerman, J.J.; Mulder, N.H.; Verweij, M.; van der Hem, G.K.; Schraffordt Koops, H.S.; Meijer, S. The protective potential of the combination of verapamil and cimetidine on cisplatin-induced nephrotoxicity in man. Cancer 1987, 60, 2823–2828. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, W. Ameliorative effects of SLC22A2 gene polymorphism 808 G/T and cimetidine on cisplatin-induced nephrotoxicity in Chinese cancer patients. Food Chem. Toxicol. 2012, 50, 2289–2293. [Google Scholar] [CrossRef]
- Katsuda, H.; Yamashita, M.; Katsura, H.; Yu, J.; Waki, Y.; Nagata, N.; Sai, Y.; Miyamoto, K. Protecting cisplatin-induced nephrotoxicity with cimetidine does not affect antitumor activity. Biol. Pharm. Bull. 2010, 33, 1867–1871. [Google Scholar] [CrossRef]
- Fox, E.; Levin, K.; Zhu, Y.; Segers, B.; Balamuth, N.; Womer, R.; Bagatell, R.; Balis, F. Pantoprazole, an Inhibitor of the Organic Cation Transporter 2, Does Not Ameliorate Cisplatin-Related Ototoxicity or Nephrotoxicity in Children and Adolescents with Newly Diagnosed Osteosarcoma Treated with Methotrexate, Doxorubicin, and Cisplatin. Oncologist 2018, 23, 762–e79. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, J.; Yuan, Z.; Jiang, Z.; Shu, T.; Xu, D.; He, J.; Zhang, L.; Huang, X. Key role of organic cation transporter 2 for the nephrotoxicity effect of triptolide in rheumatoid arthritis. Int. Immunopharmacol. 2019, 77, 105959. [Google Scholar] [CrossRef]
- Qi, X.; Zhu, L.; Yang, B.; Luo, H.; Xu, W.; He, X.; Huang, K. Mitigation of cell apoptosis induced by ochratoxin A (OTA) is possibly through organic cation transport 2 (OCT2) knockout. Food Chem. Toxicol. 2018, 121, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Uwai, Y.; Ida, H.; Tsuji, Y.; Katsura, T.; Inui, K. Renal transport of adefovir, cidofovir, and tenofovir by SLC22A family members (hOAT1, hOAT3, and hOCT2). Pharm. Res. 2007, 24, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Holle, S.K.; Vollenbrocker, B.; Hagos, Y.; Reuter, S.; Burckhardt, G.; Bierer, S.; Herrmann, E.; Pavenstadt, H.; Rossi, R.; et al. New clues for nephrotoxicity induced by ifosfamide: Preferential renal uptake via the human organic cation transporter 2. Mol. Pharm. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Cho, H.Y.; Yoo, H.D.; Kim, S.M.; Lee, Y.B. Influences of organic cation transporter polymorphisms on the population pharmacokinetics of metformin in healthy subjects. AAPS J. 2013, 15, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Te Brinke, H.; Ruiter, J.P.N.; Haasjes, J.; Oostheim, W.; van Lenthe, H.; IJlst, L.; Ebberink, M.S.; Wanders, R.J.A.; Vaz, F.M.; et al. A mutation creating an upstream translation initiation codon in SLC22A5 5’UTR is a frequent cause of primary carnitine deficiency. Hum. Mutat. 2019, 40, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Frigeni, M.; Balakrishnan, B.; Yin, X.; Calderon, F.R.O.; Mao, R.; Pasquali, M.; Longo, N. Functional and molecular studies in primary carnitine deficiency. Hum. Mutat. 2017, 38, 1684–1699. [Google Scholar] [CrossRef]
- Li, F.Y.; El-Hattab, A.W.; Bawle, E.V.; Boles, R.G.; Schmitt, E.S.; Scaglia, F.; Wong, L.J. Molecular spectrum of SLC22A5 (OCTN2) gene mutations detected in 143 subjects evaluated for systemic carnitine deficiency. Hum. Mutat. 2010, 31, E1632–E1651. [Google Scholar] [CrossRef]
- Kishimoto, S.; Suda, K.; Yoshimoto, H.; Teramachi, Y.; Nishino, H.; Koteda, Y.; Itoh, S.; Kudo, Y.; Iemura, M.; Matsuishi, T. Thirty-year follow-up of carnitine supplementation in two siblings with hypertrophic cardiomyopathy caused by primary systemic carnitine deficiency. Int. J. Cardiol. 2012, 159, e14–e15. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Huener, G.; Baykal, T.; Demirkol, M.; Duran, M.; Wanders, R.; Nezu, J.; Mayatepek, E. Silent and symptomatic primary carnitine deficiency within the same family due to identical mutations in the organic cation/carnitine transporter OCTN2. J. Inherit. Metab. Dis. 2003, 26, 613–615. [Google Scholar] [CrossRef]
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012, 33, 118–123. [Google Scholar] [CrossRef]
- Vasiljevski, E.R.; Summers, M.A.; Little, D.G.; Schindeler, A. Lipid storage myopathies: Current treatments and future directions. Prog. Lipid Res. 2018, 72, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.S.; Kim, G.; Moon, K.S.; Kim, Y.B.; Oh, J.H.; Kim, H.S.; Jeong, J.; Shin, J.G.; Kim, D.H. Characterization of urinary metabolites as biomarkers of colistin-induced nephrotoxicity in rats by a liquid chromatography/mass spectrometry-based metabolomics approach. Toxicol. Lett. 2016, 248, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Tune, B.M. Effects of L-carnitine on the renal tubular transport of cephaloridine. Biochem. Pharmacol. 1995, 50, 562–564. [Google Scholar] [CrossRef]
- Tune, B.M.; Hsu, C.Y. Toxicity of cephaloridine to carnitine transport and fatty acid metabolism in rabbit renal cortical mitochondria: Structure-activity relationships. J. Pharmacol. Exp. Ther. 1994, 270, 873–880. [Google Scholar]
- Gai, Z.; Samodelov, S.L.; Kullak-Ublick, G.A.; Visentin, M. Molecular Mechanisms of Colistin-Induced Nephrotoxicity. Molecules 2019, 24, 653. [Google Scholar] [CrossRef]
- Ganapathy, M.E.; Huang, W.; Rajan, D.P.; Carter, A.L.; Sugawara, M.; Iseki, K.; Leibach, F.H.; Ganapathy, V. beta-lactam antibiotics as substrates for OCTN2, an organic cation/carnitine transporter. J. Biol. Chem. 2000, 275, 1699–1707. [Google Scholar] [CrossRef]
- Peltekova, V.D.; Wintle, R.F.; Rubin, L.A.; Amos, C.I.; Huang, Q.; Gu, X.; Newman, B.; Van Oene, M.; Cescon, D.; Greenberg, G.; et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat. Genet. 2004, 36, 471–475. [Google Scholar] [CrossRef]
- Kekuda, R.; Prasad, P.D.; Wu, X.; Wang, H.; Fei, Y.J.; Leibach, F.H.; Ganapathy, V. Cloning and functional characterization of a potential-sensitive, polyspecific organic cation transporter (OCT3) most abundantly expressed in placenta. J. Biol. Chem. 1998, 273, 15971–15979. [Google Scholar] [CrossRef]
- Grundemann, D.; Liebich, G.; Kiefer, N.; Koster, S.; Schomig, E. Selective substrates for non-neuronal monoamine transporters. Mol. Pharmacol. 1999, 56, 1–10. [Google Scholar] [CrossRef]
- Sinclair, C.J.; Chi, K.D.; Subramanian, V.; Ward, K.L.; Green, R.M. Functional expression of a high affinity mammalian hepatic choline/organic cation transporter. J. Lipid Res. 2000, 41, 1841–1848. [Google Scholar]
- Busch, A.E.; Quester, S.; Ulzheimer, J.C.; Waldegger, S.; Gorboulev, V.; Arndt, P.; Lang, F.; Koepsell, H. Electrogenic properties and substrate specificity of the polyspecific rat cation transporter rOCT1. J. Biol. Chem. 1996, 271, 32599–32604. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kato, S.; Shimizu, T.; Wakayama, T.; Nakamichi, N.; Kubo, Y.; Iwata, D.; Suzuki, K.; Soga, T.; Asano, M.; et al. Functional expression of carnitine/organic cation transporter OCTN1/SLC22A4 in mouse small intestine and liver. Drug Metab. Dispos. 2010, 38, 1665–1672. [Google Scholar] [CrossRef]
- Kato, Y.; Kubo, Y.; Iwata, D.; Kato, S.; Sudo, T.; Sugiura, T.; Kagaya, T.; Wakayama, T.; Hirayama, A.; Sugimoto, M.; et al. Gene knockout and metabolome analysis of carnitine/organic cation transporter OCTN1. Pharm. Res. 2010, 27, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Kato, Y.; Sai, Y.; Tsuji, A. Functional characterization of human organic cation transporter OCTN1 single nucleotide polymorphisms in the Japanese population. J. Pharm. Sci. 2004, 93, 2920–2926. [Google Scholar] [CrossRef]
- Newman, B.; Gu, X.J.; Wintle, R.; Cescon, D.; Yazdanpanah, M.; Liu, X.D.; Peltekova, V.; Van Oene, M.; Amos, C.I.; Siminovitch, K.A. A risk haplotype in the solute carrier family 22A4/22A5 gene cluster influences phenotypic expression of Crohn’s disease. Gastroenterology 2005, 128, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, O.; Latiano, A.; Valvano, R.; D’Inca, R.; Vecchi, M.; Sturniolo, G.C.; Saibeni, S.; Peyvandi, F.; Bossa, F.; Zagaria, C.; et al. Variants of OCTN1-2 cation transporter genes are associated with both Crohn’s disease and ulcerative colitis. Aliment. Pharmacol. Ther. 2006, 23, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Babusukumar, U.; Wang, T.; McGuire, E.; Broeckel, U.; Kugathasan, S. Contribution of OCTN variants within the IBD5 locus to pediatric onset Crohn’s disease. Am. J. Gastroenterol. 2006, 101, 1354–1361. [Google Scholar] [CrossRef]
- Russell, R.K.; Drummond, H.E.; Nimmo, E.R.; Anderson, N.H.; Noble, C.L.; Wilson, D.C.; Gillett, P.M.; McGrogan, P.; Hassan, K.; Weaver, L.T.; et al. Analysis of the influence of OCTN1/2 variants within the IBD5 locus on disease susceptibility and growth indices in early onset inflammatory bowel disease. Gut 2006, 55, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Waller, S.; Tremelling, M.; Bredin, F.; Godfrey, L.; Howson, J.; Parkes, M. Evidence for association of OCTN genes and IBD5 with ulcerative colitis. Gut 2006, 55, 809–814. [Google Scholar] [CrossRef][Green Version]
- Silverberg, M.S.; Duerr, R.H.; Brant, S.R.; Bromfield, G.; Datta, L.W.; Jani, N.; Kane, S.V.; Rotter, J.I.; Philip Schumm, L.; Hillary Steinhart, A.; et al. Refined genomic localization and ethnic differences observed for the IBD5 association with Crohn’s disease. Eur. J. Hum. Genet. 2007, 15, 328–335. [Google Scholar] [CrossRef]
- Tarasova, L.; Kalnina, I.; Geldnere, K.; Bumbure, A.; Ritenberga, R.; Nikitina-Zake, L.; Fridmanis, D.; Vaivade, I.; Pirags, V.; Klovins, J. Association of genetic variation in the organic cation transporters OCT1, OCT2 and multidrug and toxin extrusion 1 transporter protein genes with the gastrointestinal side effects and lower BMI in metformin-treated type 2 diabetes patients. Pharm. Genom. 2012, 22, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Jonker, J.W.; Kato, Y.; Kusuhara, H.; Schinkel, A.H.; Sugiyama, Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J. Pharmacol. Exp. Ther. 2002, 302, 510–515. [Google Scholar] [CrossRef]
- Dawed, A.Y.; Zhou, K.; van Leeuwen, N.; Mahajan, A.; Robertson, N.; Koivula, R.; Elders, P.J.M.; Rauh, S.P.; Jones, A.G.; Holl, R.W.; et al. Variation in the Plasma Membrane Monoamine Transporter (PMAT) (Encoded by SLC29A4) and Organic Cation Transporter 1 (OCT1) (Encoded by SLC22A1) and Gastrointestinal Intolerance to Metformin in Type 2 Diabetes: An IMI DIRECT Study. Diabetes Care 2019, 42, 1027–1033. [Google Scholar] [CrossRef]
- Nakamichi, N.; Shima, H.; Asano, S.; Ishimoto, T.; Sugiura, T.; Matsubara, K.; Kusuhara, H.; Sugiyama, Y.; Sai, Y.; Miyamoto, K.; et al. Involvement of carnitine/organic cation transporter OCTN1/SLC22A4 in gastrointestinal absorption of metformin. J. Pharm. Sci. 2013, 102, 3407–3417. [Google Scholar] [CrossRef]
- Busch, A.E.; Karbach, U.; Miska, D.; Gorboulev, V.; Akhoundova, A.; Volk, C.; Arndt, P.; Ulzheimer, J.C.; Sonders, M.S.; Baumann, C.; et al. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol. Pharmacol. 1998, 54, 342–352. [Google Scholar] [CrossRef]
- Haag, C.; Berkels, R.; Grundemann, D.; Lazar, A.; Taubert, D.; Schomig, E. The localisation of the extraneuronal monoamine transporter (EMT) in rat brain. J. Neurochem. 2004, 88, 291–297. [Google Scholar] [CrossRef]
- Vialou, V.; Balasse, L.; Callebert, J.; Launay, J.M.; Giros, B.; Gautron, S. Altered aminergic neurotransmission in the brain of organic cation transporter 3-deficient mice. J. Neurochem. 2008, 106, 1471–1482. [Google Scholar] [CrossRef]
- Gasser, P.J.; Lowry, C.A.; Orchinik, M. Corticosterone-sensitive monoamine transport in the rat dorsomedial hypothalamus: Potential role for organic cation transporter 3 in stress-induced modulation of monoaminergic neurotransmission. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 8758–8766. [Google Scholar] [CrossRef]
- Cui, M.; Aras, R.; Christian, W.V.; Rappold, P.M.; Hatwar, M.; Panza, J.; Jackson-Lewis, V.; Javitch, J.A.; Ballatori, N.; Przedborski, S.; et al. The organic cation transporter-3 is a pivotal modulator of neurodegeneration in the nigrostriatal dopaminergic pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 8043–8048. [Google Scholar] [CrossRef]
- Bacq, A.; Balasse, L.; Biala, G.; Guiard, B.; Gardier, A.M.; Schinkel, A.; Louis, F.; Vialou, V.; Martres, M.P.; Chevarin, C.; et al. Organic cation transporter 2 controls brain norepinephrine and serotonin clearance and antidepressant response. Mol. Psychiatry 2012, 17, 926–939. [Google Scholar] [CrossRef]
- Courousse, T.; Bacq, A.; Belzung, C.; Guiard, B.; Balasse, L.; Louis, F.; Le Guisquet, A.M.; Gardier, A.M.; Schinkel, A.H.; Giros, B.; et al. Brain organic cation transporter 2 controls response and vulnerability to stress and GSK3beta signaling. Mol. Psychiatry 2015, 20, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Sprowl, J.A.; Ciarimboli, G.; Lancaster, C.S.; Giovinazzo, H.; Gibson, A.A.; Du, G.; Janke, L.J.; Cavaletti, G.; Shields, A.F.; Sparreboom, A. Oxaliplatin-induced neurotoxicity is dependent on the organic cation transporter OCT2. Proc. Natl. Acad. Sci. USA 2013, 110, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.M.; Leblanc, A.F.; Uddin, M.E.; Kim, J.Y.; Chen, M.; Eisenmann, E.D.; Gibson, A.A.; Li, Y.; Hong, K.W.; DiGiacomo, D.; et al. Neuronal uptake transporters contribute to oxaliplatin neurotoxicity in mice. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chaves, C.; Campanelli, F.; Chapy, H.; Gomez-Zepeda, D.; Glacial, F.; Smirnova, M.; Taghi, M.; Pallud, J.; Perriere, N.; Decleves, X.; et al. An Interspecies Molecular and Functional Study of Organic Cation Transporters at the Blood-Brain Barrier: From Rodents to Humans. Pharmaceutics 2020, 12, 308. [Google Scholar] [CrossRef]
- Laurell, G. Pharmacological intervention in the field of ototoxicity. HNO 2019, 67, 434–439. [Google Scholar] [CrossRef]
- Hellberg, V.; Gahm, C.; Liu, W.; Ehrsson, H.; Rask-Andersen, H.; Laurell, G. Immunohistochemical localization of OCT2 in the cochlea of various species. Laryngoscope 2015, 125, E320–E325. [Google Scholar] [CrossRef]
- Ben Said, M.; Grati, M.; Ishimoto, T.; Zou, B.; Chakchouk, I.; Ma, Q.; Yao, Q.; Hammami, B.; Yan, D.; Mittal, R.; et al. A mutation in SLC22A4 encoding an organic cation transporter expressed in the cochlea strial endothelium causes human recessive non-syndromic hearing loss DFNB60. Hum. Genet. 2016, 135, 513–524. [Google Scholar] [CrossRef]
- Grundemann, D. The ergothioneine transporter controls and indicates ergothioneine activity—A review. Prev. Med. 2012, 54, S71–S74. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and Characterization of Trimethylamine-N-oxide Uptake and Efflux Transporters. Mol. Pharm. 2017, 14, 310–318. [Google Scholar] [CrossRef]
- Miyake, T.; Mizuno, T.; Mochizuki, T.; Kimura, M.; Matsuki, S.; Irie, S.; Ieiri, I.; Maeda, K.; Kusuhara, H. Involvement of Organic Cation Transporters in the Kinetics of Trimethylamine N-oxide. J. Pharm. Sci. 2017, 106, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Helsley, R.N.; Brown, A.L.; Buffa, J.A.; Choucair, I.; Nemet, I.; Gogonea, C.B.; Gogonea, V.; Wang, Z.; Garcia-Garcia, J.C.; et al. Small molecule inhibition of gut microbial choline trimethylamine lyase activity alters host cholesterol and bile acid metabolism. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1474–H1486. [Google Scholar] [CrossRef]
- Miller, M.J.; Bostwick, B.L.; Kennedy, A.D.; Donti, T.R.; Sun, Q.; Sutton, V.R.; Elsea, S.H. Chronic Oral L-Carnitine Supplementation Drives Marked Plasma TMAO Elevations in Patients with Organic Acidemias Despite Dietary Meat Restrictions. JIMD Rep. 2016, 30, 39–44. [Google Scholar] [PubMed]
- Vallance, H.D.; Koochin, A.; Branov, J.; Rosen-Heath, A.; Bosdet, T.; Wang, Z.; Hazen, S.L.; Horvath, G. Marked elevation in plasma trimethylamine-N-oxide (TMAO) in patients with mitochondrial disorders treated with oral l-carnitine. Mol. Genet. Metab. Rep. 2018, 15, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Samulak, J.J.; Sawicka, A.K.; Hartmane, D.; Grinberga, S.; Pugovics, O.; Lysiak-Szydlowska, W.; Olek, R.A. L-Carnitine Supplementation Increases Trimethylamine-N-Oxide but not Markers of Atherosclerosis in Healthy Aged Women. Ann. Nutr. Metab. 2019, 74, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Yamagishi, S.; Sakai, K.; Kaida, Y.; Yokoro, M.; Ueda, S.; Wada, Y.; Takeuchi, M.; Shimizu, M.; Yamazaki, H.; et al. Oral L-carnitine supplementation increases trimethylamine-N-oxide but reduces markers of vascular injury in hemodialysis patients. J. Cardiovasc. Pharmacol. 2015, 65, 289–295. [Google Scholar] [CrossRef]
- Bordoni, L.; Sawicka, A.K.; Szarmach, A.; Winklewski, P.J.; Olek, R.A.; Gabbianelli, R. A Pilot Study on the Effects of l-Carnitine and Trimethylamine-N-Oxide on Platelet Mitochondrial DNA Methylation and CVD Biomarkers in Aged Women. Int. J. Mol. Sci. 2020, 21, 1047. [Google Scholar] [CrossRef]
- Olek, R.A.; Samulak, J.J.; Sawicka, A.K.; Hartmane, D.; Grinberga, S.; Pugovics, O.; Lysiak-Szydlowska, W. Increased Trimethylamine N-Oxide Is Not Associated with Oxidative Stress Markers in Healthy Aged Women. Oxid. Med. Cell Longev. 2019, 2019, 6247169. [Google Scholar] [CrossRef]
- Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients 2020, 12, 2982. [Google Scholar] [CrossRef]
- Heianza, Y.; Ma, W.; Manson, J.E.; Rexrode, K.M.; Qi, L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J. Am. Heart Assoc. 2017, 6, e004947. [Google Scholar] [CrossRef]
- Mueller, D.M.; Allenspach, M.; Othman, A.; Saely, C.H.; Muendlein, A.; Vonbank, A.; Drexel, H.; von Eckardstein, A. Plasma levels of trimethylamine-N-oxide are confounded by impaired kidney function and poor metabolic control. Atherosclerosis 2015, 243, 638–644. [Google Scholar] [CrossRef]
- Jia, J.; Dou, P.; Gao, M.; Kong, X.; Li, C.; Liu, Z.; Huang, T. Assessment of Causal Direction Between Gut Microbiota-Dependent Metabolites and Cardiometabolic Health: A Bidirectional Mendelian Randomization Analysis. Diabetes 2019, 68, 1747–1755. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1104–1115. [Google Scholar] [CrossRef]
- Aldana-Hernandez, P.; Leonard, K.A.; Zhao, Y.Y.; Curtis, J.M.; Field, C.J.; Jacobs, R.L. Dietary Choline or Trimethylamine N-oxide Supplementation Does Not Influence Atherosclerosis Development in Ldlr-/- and Apoe-/- Male Mice. J. Nutr. 2020, 150, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Pawlikowski, B.; Schlessinger, A.; More, S.S.; Stryke, D.; Johns, S.J.; Portman, M.A.; Chen, E.; Ferrin, T.E.; Sali, A.; et al. Role of organic cation transporter 3 (SLC22A3) and its missense variants in the pharmacologic action of metformin. Pharm. Genom. 2010, 20, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. OCTN cation transporters in health and disease: Role as drug targets and assay development. J. Biomol. Screen 2013, 18, 851–867. [Google Scholar] [CrossRef]
- Pochini, L.; Galluccio, M.; Scalise, M.; Console, L.; Indiveri, C. OCTN: A Small Transporter Subfamily with Great Relevance to Human Pathophysiology, Drug Discovery, and Diagnostics. Slas. Discov. 2019, 24, 89–110. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samodelov, S.L.; Kullak-Ublick, G.A.; Gai, Z.; Visentin, M. Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology. Int. J. Mol. Sci. 2020, 21, 7890. https://doi.org/10.3390/ijms21217890
Samodelov SL, Kullak-Ublick GA, Gai Z, Visentin M. Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology. International Journal of Molecular Sciences. 2020; 21(21):7890. https://doi.org/10.3390/ijms21217890
Chicago/Turabian StyleSamodelov, Sophia L., Gerd A. Kullak-Ublick, Zhibo Gai, and Michele Visentin. 2020. "Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology" International Journal of Molecular Sciences 21, no. 21: 7890. https://doi.org/10.3390/ijms21217890
APA StyleSamodelov, S. L., Kullak-Ublick, G. A., Gai, Z., & Visentin, M. (2020). Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology. International Journal of Molecular Sciences, 21(21), 7890. https://doi.org/10.3390/ijms21217890