Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
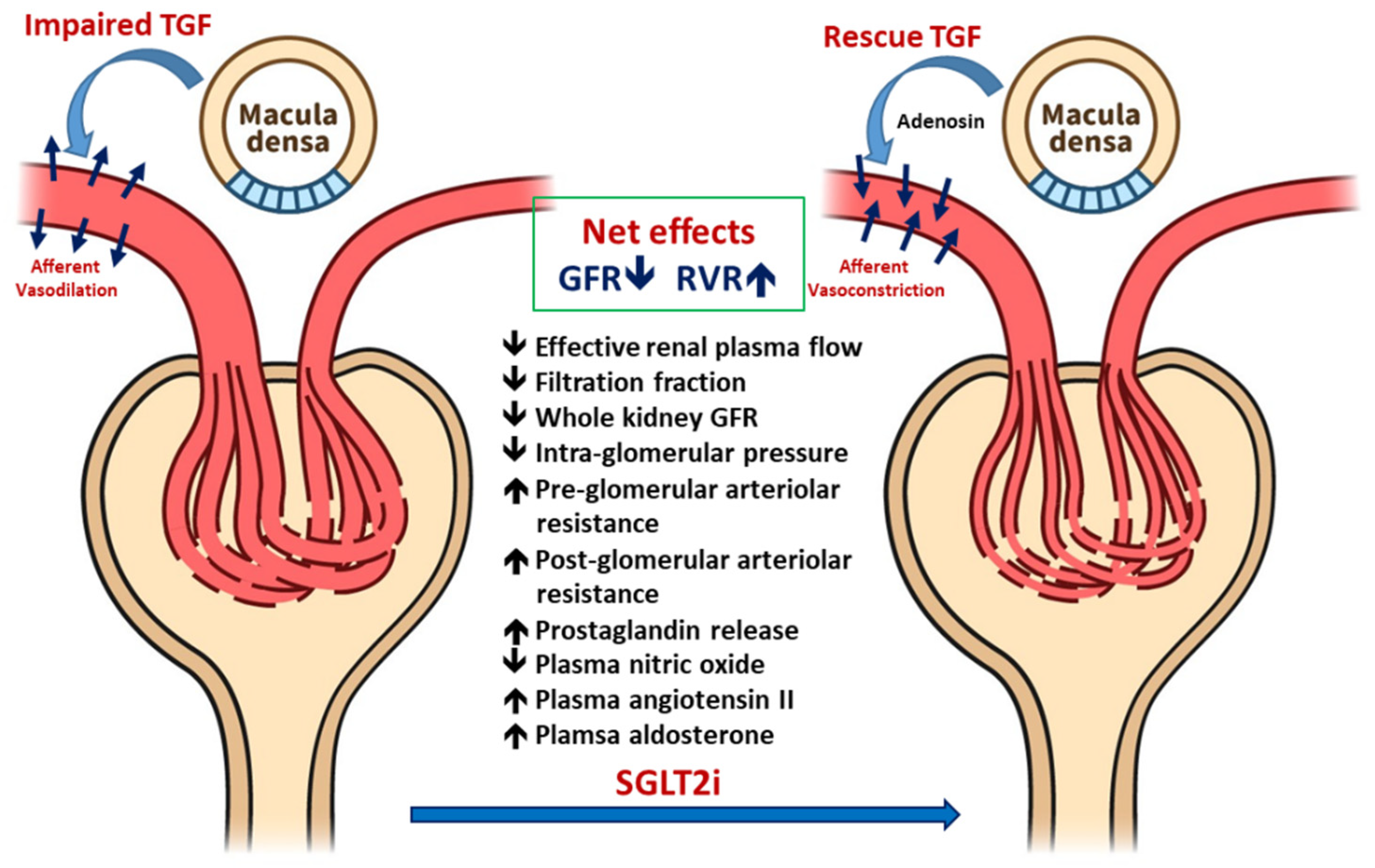
2. Mechanism of Glomerular Hyperfiltration in Diabetes
3. Hemodynamic Factors Affected by SGLT2i
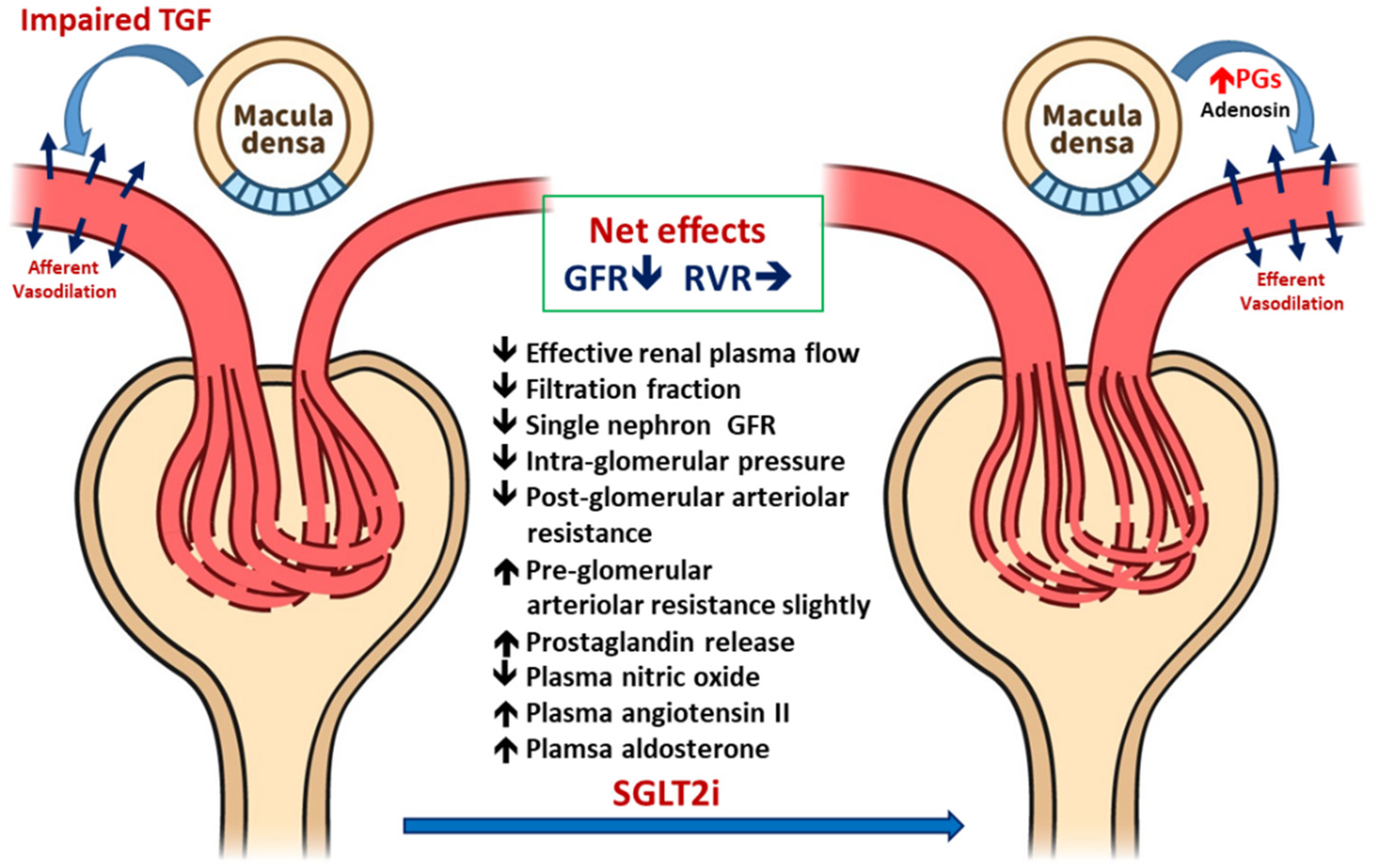
3.1. Improved Glomerular Hyperfiltration and Renal Hemodynamics
3.1.1. Renal Hemodynamic Effects of SGLT2i in Patients with T1DM
3.1.2. Renal Hemodynamic Effect of SGLT2i in Patients with T2DM
3.2. Effects of SGLT2i on Systemic and Intrarenal RAS
3.2.1. Effects on Systemic RAS
3.2.2. Effects on Intrarenal RAS
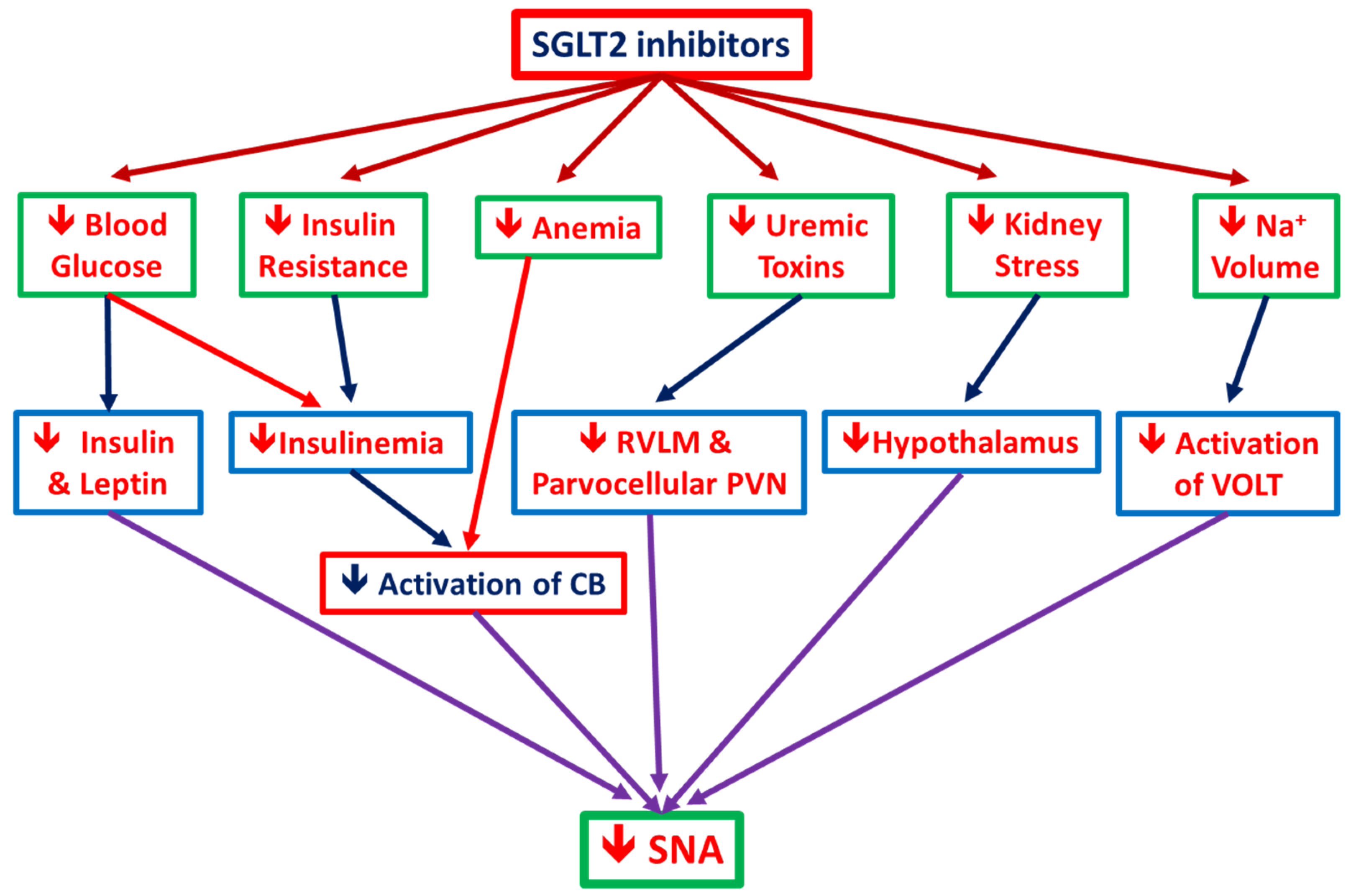
3.3. Ameliorate the Chronic Activation of the Sympathetic Nervous System
3.3.1. SGLT2i Reduces SNS Activity: Animal Data
3.3.2. SGLT2i Reduces SNS Activity: Human Data
3.3.3. SGLT2i Reduces SNS Activity (SNA): The postulated mechanisms
4. Metabolic Factors Influenced by SGLT2i
4.1. Pancreatic α Cell Secretion of Glucagon (Hypoglycemia Prevention)
4.2. Uric Acid-Lowering Effect of SGLT2i
4.3. Effects of SGLT2i on Insulin Sensitivity and β-Cell Function
4.4. Effects of SGLT2i on Serum Electrolytes and Renal Epithelial Transporter Activity
4.5. Effects of SGLT2 Inhibition on Kidney Pathological Findings
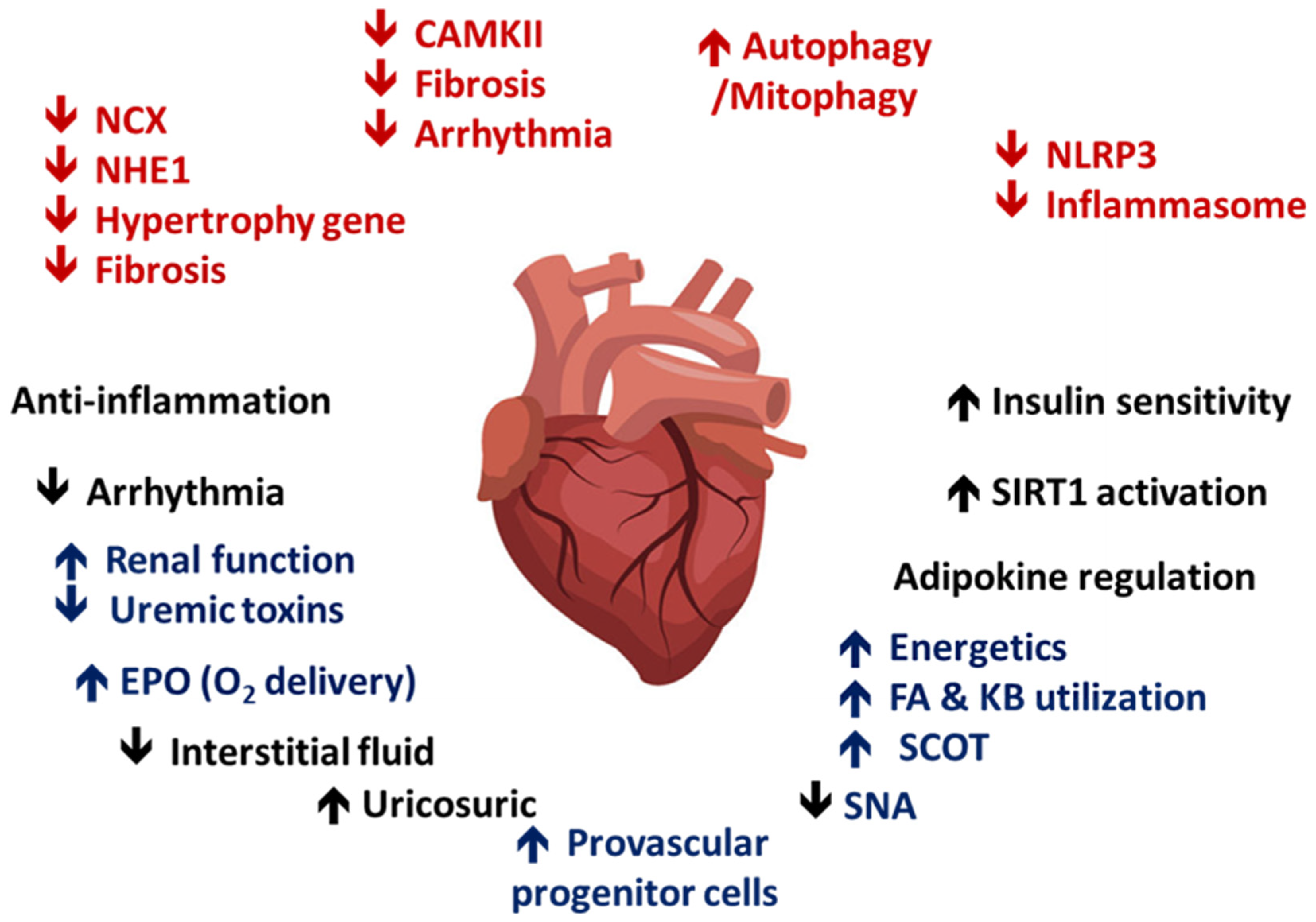
4.6. SGLT2i Contributes to Cardiac and Renal Metabolism
4.7. Anti-Inflammatory Effects of SGLT2i
4.8. SGLT2i Reduces Renal Fibrosis and Enhances EPO Production
4.9. SGLT2i in Acute Kidney Injury
4.10. Effects of SGLT2i on Bone Metabolism
4.11. SGLT2i on Non-Osmotic Sodium Storage and Interstitial Fluid Dynamics
4.12. Uremic Toxin-Lowering Effect of Relative Nonspecific SGLT2i
5. Effects of SGLT2i on Clinical Parameters and Outcomes in Patients with T2DM
5.1. The Protection of SGLT2i in Cardiac and Kidney Outcomes
5.2. The Concerns for SGLT2i-Induced Ketoacidosis: Volume Depletion and Insulinopenia
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | Angiotensin converting enzyme |
| AKI | Acute kidney injury |
| AQP-2 | Aquaporin 2 |
| AV3V | Anteroventral third ventricle |
| BP | Blood pressure |
| CB | Carotid Body |
| CKD | Chronic kidney disease |
| DKA | Diabetic ketoacidosis |
| DM | Diabetes mellitus |
| DPP4 | Dipeptidyl peptidase-IV |
| ECM | Extracellular matrix |
| EPO | Erythropoietin |
| ESRD | End stage renal disease |
| GFR | Glomerular filtration rate |
| GLUT2 | Glucose transporter 2 |
| GLUT-9 | Glucose transporter 9 |
| HF | Heart failure |
| HIF | Hypoxia inducible factor |
| HOMA | Homeostasis model assessment |
| IML | Intermediolateral nucleus of spinal cord |
| IS | Indoxyl sulfate |
| NHE3 | Na+/H+ exchanger isoform 3 |
| NKCC-2 | Na-K-Cl cotransporter 2 |
| OVLT | Organum vasculosum laminae terminalis |
| PAC | Plasma aldosterone concentration |
| PAG | Periaqueductal gray |
| PCS | P-cresol sulfate |
| PRA | Plasma renin activity |
| PT | Proximal tubules |
| PVN | Paraventricular nucleus of hypothalamus |
| RAS | Renin-angiotensin system |
| RCT | Randomized clinical trials |
| ROS | Reactive oxygen species) |
| RVLM | Rostral ventrolateral medulla |
| SGLT-2 | Sodium–glucose cotransporter 2 |
| SGLT2i | Sodium–glucose cotransporter 2 inhibitor |
| SIRT-1 | Sirtulin-1 |
| SNA | Sympathetic nerve activity |
| SNS | Sympathetic nerve system |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TGF | Tubular-glomerular feedback |
| TGFβ | Transforming growth factor β |
| TNFα | Tumor necrosis factor α |
| VEGF | Vascular endothelial growth factor |
| VHL-E3 | Von Hippel-Lindau ubiquitin ligase-E3 |
| URAT1 | Urate transporter 1 |
References
- Mather, A.; Pollock, C. Glucose handling by the kidney. Kidney Int. 2011, 79, S1–S6. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetology 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Renal function in diabetic disease models: The tubular system in the pathophysiology of the diabetic kidney. Annu. Rev. Physiol. 2012, 74, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Rose, M.; Gerasimova, M.; Satriano, J.; Platt, K.A.; Koepsell, H.; Cunard, R.; Sharma, K.; Thomson, S.C.; Rieg, T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. Physiol. 2013, 304, F156–F167. [Google Scholar] [CrossRef] [PubMed]
- Tonneijck, L.; Muskiet, M.H.A.; Smits, M.M.; Van Bommel, E.J.; Heerspink, H.J.L.; Van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.O.; Steadman, R. Diabetic nephropathy: The central role of renal proximal tubular cells in tubulointerstitial injury. Histol. Histopathol. 2002, 17, 247–252. [Google Scholar]
- Vervoort, G.; Veldman, B.; Berden, J.H.M.; Smits, P.; Wetzels, J.F.M. Glomerular hyperfiltration in type 1 diabetes mellitus results from primary changes in proximal tubular sodium handling without changes in volume expansion. Eur. J. Clin. Investig. 2005, 35, 330–336. [Google Scholar] [CrossRef]
- Pollock, C.A.; Lawrence, J.R.; Field, M.J. Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am. J. Physiol. Physiol. 1991, 260, F946–F952. [Google Scholar] [CrossRef]
- Curthoys, N.P.; Moe, O.W. Proximal Tubule Function and Response to Acidosis. Clin. J. Am. Soc. Nephrol. 2013, 9, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Schwark, J.-R.; Richter, K.; Hropot, M. Role of Na+/H+ exchanger NHE3 in nephron function: Micropuncture studies with S3226, an inhibitor of NHE3. Am. J. Physiol. Physiol. 2000, 278, F375–F379. [Google Scholar] [CrossRef]
- Vallon, V.; Komers, R. Pathophysiology of the Diabetic Kidney. In Comprehensive Physiology; Wiley: Hoboken, NJ, USA, 2011; Volume 1, pp. 1175–1232. [Google Scholar]
- Umino, H.; Hasegawa, K.; Minakuchi, H.; Muraoka, H.; Kawaguchi, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. High Basolateral Glucose Increases Sodium-Glucose Cotransporter 2 and Reduces Sirtuin-1 in Renal Tubules through Glucose Transporter-2 Detection. Sci. Rep. 2018, 8, 6791. [Google Scholar] [CrossRef]
- Lee, E.M.; Pollock, C.A.; Drumm, K.; Barden, J.A.; Poronnik, P. Effects of pathophysiological concentrations of albumin on NHE3 activity and cell proliferation in primary cultures of human proximal tubule cells. Am. J. Physiol. Physiol. 2003, 285, F748–F757. [Google Scholar] [CrossRef][Green Version]
- Pessoa, T.D.; Campos, L.C.G.; Carraro-Lacroix, L.; Girardi, A.C.C.; Malnic, G. Functional Role of Glucose Metabolism, Osmotic Stress, and Sodium-Glucose Cotransporter Isoform-Mediated Transport on Na+/H+ Exchanger Isoform 3 Activity in the Renal Proximal Tubule. Clin. J. Am. Soc. Nephrol. 2014, 25, 2028–2039. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Patel, R.; Kim, Y.C.; Nespoux, J.; Freeman, B.; et al. Effect of renal tubule-specific knockdown of the Na+/H+ exchanger NHE3 in Akita diabetic mice. Am. J. Physiol. Renal Physiol. 2019, 317, 419–434. [Google Scholar] [CrossRef]
- Dos Santos, D.S.; Polidoro, J.Z.; Borges-Júnior, F.A.; Girardi, A.C.C. Cardioprotection conferred by sodium-glucose cotransporter 2 inhibitors: A renal proximal tubule perspective. Am. J. Physiol. Physiol. 2020, 318, C328–C336. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Kim, S.; Son, M.; Kim, M.; Koh, E.S.; Shin, S.J.; Ko, S.-H.; Kim, H.-S. Empagliflozin Contributes to Polyuria via Regulation of Sodium Transporters and Water Channels in Diabetic Rat Kidneys. Front. Physiol. 2019, 10, 271. [Google Scholar] [CrossRef] [PubMed]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.H.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; Von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Maione, M.; Lai, V.; Lee, A.; Fagan, N.M.; Woerle, H.J.; Johansen, O.E.; Broedl, U.C.; et al. Renal Hemodynamic Effect of Sodium-Glucose Cotransporter 2 Inhibition in Patients With Type 1 Diabetes Mellitus. Circulation 2014, 129, 587–597. [Google Scholar] [CrossRef]
- Bidani, A.K.; Picken, M.; Hacioglu, R.; Williamson, G.; Griffin, K.A. Spontaneously reduced blood pressure load in the rat streptozotocin-induced diabetes model: Potential pathogenetic relevance. Am. J. Physiol. Physiol. 2006, 292, F647–F654. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Epstein, M.; Loutzenhiser, R.; Forster, H. Impaired myogenic responsiveness of the afferent arteriole in streptozotocin-induced diabetic rats: Role of eicosanoid derangements. J. Am. Soc. Nephrol. 1992, 2, 1578–1586. [Google Scholar] [PubMed]
- Vallon, V.; Blantz, R.C.; Thomson, S. Homeostatic efficiency of tubuloglomerular feedback is reduced in established diabetes mellitus in rats. Am. J. Physiol. Physiol. 1995, 269, F876–F883. [Google Scholar] [CrossRef]
- Vallon, V. Salt-Sensitivity of Proximal Reabsorption Alters Macula Densa Salt and Explains the Paradoxical Effect of Dietary Salt on Glomerular Filtration Rate in Diabetes Mellitus. J. Am. Soc. Nephrol. 2002, 13, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; Bjornstad, P. Role of sodium-glucose cotransporter 2 inhibition to mitigate diabetic kidney disease risk in type 1 diabetes. Nephrol. Dial. Transplant. 2020, 35, i24–i32. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, M.J.; Saunders, A.J.; Viberti, G.; Keen, H. Effect of Blood Glucose Control on Increased Glomerular Filtration Rate and Kidney Size in Insulin-Dependent Diabetes. N. Engl. J. Med. 1985, 312, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Van Bommel, E.J.; Muskiet, M.H.; Van Baar, M.J.; Tonneijck, L.; Smits, M.M.; Emanuel, A.L.; Bozovic, A.; Danser, A.J.; Geurts, F.; Hoorn, E.J.; et al. The renal hemodynamic effects of the SGLT2 inhibitor dapagliflozin are caused by post-glomerular vasodilatation rather than pre-glomerular vasoconstriction in metformin-treated patients with type 2 diabetes in the randomized, double-blind RED trial. Kidney Int. 2020, 97, 202–212. [Google Scholar] [CrossRef]
- Vallon, V.; Mühlbauer, B.; Osswald, H. Adenosine and Kidney Function. Physiol. Rev. 2006, 86, 901–940. [Google Scholar] [CrossRef]
- Bjornstad, P.; Nelson, R.G.; Pavkov, M.E. Do sodium-glucose cotransporter-2 inhibitors affect renal hemodynamics by different mechanisms in type 1 and type 2 diabetes? Kidney Int. 2020, 97, 31–33. [Google Scholar] [CrossRef]
- Yamazaki, D.; Hitomi, H.; Nishiyama, A. Hypertension with diabetes mellitus complications. Hypertens. Res. 2018, 41, 147–156. [Google Scholar] [CrossRef]
- Nishiyama, A.; Kobori, H. Independent regulation of renin–angiotensin–aldosterone system in the kidney. Clin. Exp. Nephrol. 2018, 22, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Konishi, Y.; Morikawa, T.; Zhang, Y.; Kitabayashi, C.; Kobara, H.; Masaki, T.; Nakano, D.; Hitomi, H.; Kobori, H.; et al. Effect of a SGLT2 inhibitor on the systemic and intrarenal renin–angiotensin system in subtotally nephrectomized rats. J. Pharmacol. Sci. 2018, 137, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Haneda, M.; Seino, Y.; Inagaki, N.; Kaku, K.; Sasaki, T.; Fukatsu, A.; Kakiuchi, H.; Sato, Y.; Sakai, S.; Samukawa, Y. Influence of Renal Function on the 52-Week Efficacy and Safety of the Sodium Glucose Cotransporter 2 Inhibitor Luseogliflozin in Japanese Patients with Type 2 Diabetes Mellitus. Clin. Ther. 2016, 38, 66–88. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Kittikulsuth, W.; Fujisawa, Y.; Sufiun, A.; Rafiq, K.; Hitomi, H.; Nakano, D.; Sohara, E.; Uchida, S.; Nishiyama, A. Effects of diuretics on sodium-dependent glucose cotransporter 2 inhibitor-induced changes in blood pressure in obese rats suffering from the metabolic syndrome. J. Hypertens. 2016, 34, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Yale, J.-F.; Bakris, G.; Cariou, B.; Yue, D.; David-Neto, E.; Xi, L.; Figueroa, K.; Wajs, E.; Usiskin, K.; Meininger, G. Efficacy and safety of canagliflozin in subjects with type 2 diabetes and chronic kidney disease. Diabetes Obes. Metab. 2013, 15, 463–473. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Xiao, F.; Zimpelmann, J.; Woerle, H.-J.; Johansen, O.E.; Broedl, U.C.; Von Eynatten, M.; Burns, K.D. Sodium glucose cotransport-2 inhibition and intrarenal RAS activity in people with type 1 diabetes. Kidney Int. 2014, 86, 1057–1058. [Google Scholar] [CrossRef]
- Shin, S.J.; Chung, S.; Kim, S.J.; Lee, E.-M.; Yoo, Y.-H.; Kim, J.-W.; Ahn, Y.-B.; Kim, E.-S.; Moon, S.-D.; Kim, M.-J.; et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0165703. [Google Scholar] [CrossRef]
- Gallo, L.A.; Ward, M.S.; Fotheringham, A.K.; Zhuang, A.; Borg, D.J.; Flemming, N.B.; Harvie, B.M.; Kinneally, T.L.; Yeh, S.-M.; McCarthy, D.A.; et al. Once daily administration of the SGLT2 inhibitor, empagliflozin, attenuates markers of renal fibrosis without improving albuminuria in diabetic db/db mice. Sci. Rep. 2016, 6, 26428. [Google Scholar] [CrossRef]
- Mori, I.; Ishizuka, T. Effects of SGLT2 Inhibitors on Renin-Aldosterone System for One Month and Six Months in Type 2 Diabetes. Diabetes 2018, 67, 1196. [Google Scholar] [CrossRef]
- Ansary, T.M.; Nakano, D.; Nishiyama, A. Diuretic Effects of Sodium Glucose Cotransporter 2 Inhibitors and Their Influence on the Renin-Angiotensin System. Int. J. Mol. Sci. 2019, 20, 629. [Google Scholar] [CrossRef]
- Matsusaka, T.; Niimura, F.; Shimizu, A.; Pastan, I.; Saito, A.; Kobori, H.; Nishiyama, A.; Ichikawa, I. Liver Angiotensinogen Is the Primary Source of Renal Angiotensin II. J. Am. Soc. Nephrol. 2012, 23, 1181–1189. [Google Scholar] [CrossRef]
- Kobori, H.; Harrison-Bernard, L.M.; Navar, L.G. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int. 2002, 61, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Nishiyama, A.; Harrison-Bernard, L.M.; Navar, L.G. Urinary angiotensinogen as an indicator of intrarenal Angiotensin status in hypertension. Hypertensension 2003, 41, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shibayama, Y.; Kobori, H.; Liu, Y.; Kobara, H.; Masaki, T.; Wang, Z.; Nishiyama, A. High glucose augments angiotensinogen in human renal proximal tubular cells through hepatocyte nuclear factor-5. PLoS ONE 2017, 12, e0185600. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-Y.; Kobori, H.; Nakano, D.; Hitomi, H.; Mori, H.; Masaki, T.; Sun, Y.-X.; Zhi, N.; Zhang, L.; Huang, W.; et al. Aberrant Activation of the Intrarenal Renin-Angiotensin System in the Developing Kidneys of Type 2 Diabetic Rats. Horm. Metab. Res. 2013, 45, 338–343. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Furuki, T.; Kobori, H.; Miyakawa, M.; Imachi, H.; Murao, K.; Nishiyama, A. Effects of sodium-glucose cotransporter 2 inhibitors on urinary excretion of intact and total angiotensinogen in patients with type 2 diabetes. J. Investig. Med. 2017, 65, 1057–1061. [Google Scholar] [CrossRef]
- Lee, M.J.; Kim, S.S.; Kim, I.J.; Song, S.H.; Kim, E.H.; Seo, J.Y.; Kim, J.H.; Kim, S.; Jeon, Y.K.; Kim, B.H.; et al. Changes in Urinary Angiotensinogen Associated with Deterioration of Kidney Function in Patients with Type 2 Diabetes Mellitus. J. Korean Med. Sci. 2017, 32, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Wan, N.; Rahman, A.; Hitomi, H.; Nishiyama, A. The Effects of Sodium-Glucose Cotransporter 2 Inhibitors on Sympathetic Nervous Activity. Front. Endocrinol. 2018, 9, 421. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Straznicky, N.; Lambert, E.; Lambert, G. Metabolic syndrome: A sympathetic disease? Lancet Diabetes Endocrinol. 2015, 3, 148–157. [Google Scholar] [CrossRef]
- Masuo, K.; Rakugi, H.; Ogihara, T.; Esler, M.D.; Lambert, G.W. Cardiovascular and renal complications of type 2 diabetes in obesity: Role of sympathetic nerve activity and insulin resistance. Curr. Diabetes Rev. 2010, 6, 58–67. [Google Scholar] [CrossRef]
- Baker, W.L.; Smyth, L.R.; Riche, D.M.; Bourret, E.M.; Chamberlin, K.W.; White, W.B. Effects of sodium-glucose co-transporter 2 inhibitors on blood pressure: A systematic review and meta-analysis. J. Am. Soc. Hypertens. 2014, 8, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.W. Impact of sodium–glucose cotransporter 2 inhibitors on blood pressure. Vasc. Health Risk Manag. 2016, 12, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Jackson, K.L.; Sata, Y.; Head, G.A. Factors Responsible for Obesity-Related Hypertension. Curr. Hypertens. Rep. 2017, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, A.; Martínez-González, M.Á.; Alonso-Gómez, A.; Rekondo, J.; Salas-Salvadó, J.; Corella, D.; Ros, E.; Fitó, M.; Estruch, R.; Lapetra, J.; et al. Mediterranean diet and risk of heart failure: Results from the PREDIMED randomized controlled trial. Eur. J. Hear. Fail. 2017, 19, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, Y.; Fujisawa, Y.; Rahman, A.; Kittikulsuth, W.; Nakano, D.; Mori, H.; Masaki, T.; Ohmori, K.; Kohno, M.; Ogata, H.; et al. A sodium-glucose co-transporter 2 inhibitor empagliflozin prevents abnormality of circadian rhythm of blood pressure in salt-treated obese rats. Hypertens. Res. 2016, 39, 415–422. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Chen, G.; Zhang, C.; Wu, X. SGLT2i: Beyond the glucose-lowering effect. Cardiovasc. Diabetol. 2020, 19, 98. [Google Scholar] [CrossRef]
- Chilton, R.; Tikkanen, I.; Crowe, S.; Johansen, O.E.; Broedl, U.C.; Woerle, H.J.; Hach, T. 4B.03: Empagliflozin reduces systolic blood pressure in dipper and non-dipper patients with type 2 diabetes and hypertension. J. Hypertens. 2015, 33, e53. [Google Scholar] [CrossRef]
- Rahman, A.; Hitomi, H.; Nishiyama, A. Cardioprotective effects of SGLT2 inhibitors are possibly associated with normalization of the circadian rhythm of blood pressure. Hypertens. Res. 2017, 40, 535–540. [Google Scholar] [CrossRef]
- Chiba, Y.; Yamada, T.; Tsukita, S.; Takahashi, K.; Munakata, Y.; Shirai, Y.; Kodama, S.; Asai, Y.; Sugisawa, T.; Uno, K.; et al. Dapagliflozin, a Sodium-Glucose Co-Transporter 2 Inhibitor, Acutely Reduces Energy Expenditure in BAT via Neural Signals in Mice. PLoS ONE 2016, 11, e0150756. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Kishi, T.; Shinohara, K.; Takesue, K.; Shibata, R.; Sonoda, N.; Inoguchi, T.; Sunagawa, K.; Tsutsui, H.; Hirooka, Y. Arterial pressure lability is improved by sodium-glucose cotransporter 2 inhibitor in streptozotocin-induced diabetic rats. Hypertens. Res. 2017, 40, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Matthews, V.B.; Elliot, R.H.; Rudnicka, C.; Hricova, J.; Herat, L.; Schlaich, M.P. Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J. Hypertens. 2017, 35, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.; Jordan, J.; Heusser, K.; Heise, T.; Wanner, C.; Heer, M.; Macha, S.; Mattheus, M.; Lund, S.S.; Woerle, H.J.; et al. The effect of empagliflozin on muscle sympathetic nerve activity in patients with type II diabetes mellitus. J. Am. Soc. Hypertens. 2017, 11, 604–612. [Google Scholar] [CrossRef]
- Kusaka, H.; Koibuchi, N.; Hasegawa, Y.; Ogawa, H.; Kim-Mitsuyama, S. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome. Cardiovasc. Diabetol. 2016, 15, 157. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; De Zeeuw, D.; Wie, L.; Leslie, B.; List, J. Dapagliflozin a glucose-regulating drug with diuretic properties in subjects with type 2 diabetes. Diabetes Obes. Metab. 2013, 15, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Kimmerly, D.S.; Shoemaker, J.K. Hypovolemia and MSNA discharge patterns: Assessing and interpreting sympathetic responses. Am. J. Physiol. Circ. Physiol. 2003, 284, H1198–H1204. [Google Scholar] [CrossRef][Green Version]
- Esler, M. The 2009 Carl Ludwig Lecture: Pathophysiology of the human sympathetic nervous system in cardiovascular diseases: The transition from mechanisms to medical management. J. Appl. Physiol. 2010, 108, 227–237. [Google Scholar] [CrossRef]
- Trombetta, I.C.; Batalha, L.T.; Rondon, M.U.P.B.; Laterza, M.C.; Kuniyoshi, F.H.S.; Gowdak, M.M.G.; Barretto, A.C.P.; Halpern, A.; Villares, S.M.F.; Negrão, C.E. Weight loss improves neurovascular and muscle metaboreflex control in obesity. Am. J. Physiol. Circ. Physiol. 2003, 285, H974–H982. [Google Scholar] [CrossRef]
- Scheen, A. Effect of SGLT2 Inhibitors on the Sympathetic Nervous System and Blood Pressure. Curr. Cardiol. Rep. 2019, 21, 70. [Google Scholar] [CrossRef]
- Kario, K.; Ferdinand, K.C.; O’Keefe, J.H. Control of 24-hour blood pressure with SGLT2 inhibitors to prevent cardiovascular disease. Prog. Cardiovasc. Dis. 2020, 63, 249–262. [Google Scholar] [CrossRef]
- Elliott, R.H.; Matthews, V.B.; Rudnicka, C.; Schlaich, M.P. Is it time to think about the sodium glucose co-transporter 2 sympathetically? Nephrology 2016, 21, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Chiba, Y.; Yamada, T.; Katagiri, H. Dapagliflozin, a Sodium-Glucose Co-transporter-2 Inhibitor, Acutely Reduces Energy Expenditure in Brown Adipose Tissue via Neural Signals in Mice. YAKUGAKU ZASSHI 2018, 138, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Wen, S.; Gong, M.; Yuan, X.; Xu, D.; Wang, C.; Jin, J.; Zhou, L. Dapagliflozin Activates Neurons in the Central Nervous System and Regulates Cardiovascular Activity by Inhibiting SGLT-2 in Mice. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 2781–2799. [Google Scholar] [CrossRef] [PubMed]
- Merovci, A.; Solis-Herrera, C.; Daniele, G.; Eldor, R.; Fiorentino, T.V.; Tripathy, D.; Xiong, J.; Perez, Z.; Norton, L.; Abdul-Ghani, M.A.; et al. Dapagliflozin improves muscle insulin sensitivity but enhances endogenous glucose production. J. Clin. Investig. 2014, 124, 509–514. [Google Scholar] [CrossRef]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.-J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508. [Google Scholar] [CrossRef]
- Bonner, C.C.; Kerr-Conte, J.J.; Gmyr, V.V.; Queniat, G.G.; Moerman, E.E.; Thévenet, J.J.; Beaucamps, C.C.; Delalleau, N.N.; Popescu, I.I.; Malaisse, W.J.; et al. Inhibition of the glucose transporter SGLT2 with dapagliflozin in pancreatic alpha cells triggers glucagon secretion. Nat. Med. 2015, 21, 512–517. [Google Scholar] [CrossRef]
- Suga, T.; Kikuchi, O.; Kobayashi, M.; Matsui, S.; Yokota-Hashimoto, H.; Wada, E.; Kohno, D.; Sasaki, T.; Takeuchi, K.; Kakizaki, S.; et al. SGLT1 in pancreatic α cells regulates glucagon secretion in mice, possibly explaining the distinct effects of SGLT2 inhibitors on plasma glucagon levels. Mol. Metab. 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Solini, A.; Sebastiani, G.; Nigi, L.; Santini, E.; Rossi, C.; Dotta, F. Dapagliflozin modulates glucagon secretion in an SGLT2-independent manner in murine alpha cells. Diabetes Metab. 2017, 43, 512–520. [Google Scholar] [CrossRef]
- Borghi, C.; Agabiti-Rosei, E.; Johnson, R.J.; Kielstein, J.T.; Lurbe, E.; Mancia, G.; Redon, J.; Stack, A.G.; Tsioufis, K.P. Hyperuricaemia and gout in cardiovascular, metabolic and kidney disease. Eur. J. Intern. Med. 2020, 80, 1–11. [Google Scholar] [CrossRef]
- Lima, W.G.; Martins-Santos, M.E.S.; Chaves, V.E. Uric acid as a modulator of glucose and lipid metabolism. Biochim. 2015, 116, 17–23. [Google Scholar] [CrossRef] [PubMed]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.-Y.; Lee, Y.-T.; Kuo, W.-H.; Huang, P.-C.; Lee, W.-C.; Lee, C.-T. Alterations of Renal Epithelial Glucose and Uric Acid Transporters in Fructose Induced Metabolic Syndrome. Kidney Blood Press. Res. 2018, 43, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Preitner, F.; Bonny, O.; Laverrière, A.; Rotman, S.; Firsov, D.; Da Costa, A.; Metref, S.; Thorens, B. Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proc. Natl. Acad. Sci. USA 2009, 106, 15501–15506. [Google Scholar] [CrossRef]
- Anzai, N.; Jutabha, P.; Amonpatumrat-Takahashi, S.; Sakurai, H. Recent advances in renal urate transport: Characterization of candidate transporters indicated by genome-wide association studies. Clin. Exp. Nephrol. 2011, 16, 89–95. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Shen, W.; Boulton, D.W.; Leslie, B.R.; Griffen, S.C. Interaction Between the Sodium-Glucose–Linked Transporter 2 Inhibitor Dapagliflozin and the Loop Diuretic Bumetanide in Normal Human Subjects. J. Am. Hear. Assoc. 2018, 7, e007046. [Google Scholar] [CrossRef]
- Kutoh, E.; Wada, A.; Kuto, A.N.; Hayashi, J.; Kurihara, R. Link between body weight changes and metabolic parameters in drugs naïve subjects with type 2 diabetes treated with canagliflozin monotherapy. Hosp. Pr. 2020, 48, 68–74. [Google Scholar] [CrossRef]
- Candler, T.P.; McGregor, D.; Narayan, K.; Moudiotis, C.; Burren, C.P. Improvement in glycaemic parameters using SGLT-2 inhibitor and GLP-1 agonist in combination in an adolescent with diabetes mellitus and Prader-Willi syndrome: A case report. J. Pediatr. Endocrinol. Metab. 2020, 33, 951–955. [Google Scholar] [CrossRef]
- Tahara, A.; Takasu, T. Therapeutic Effects of SGLT2 Inhibitor Ipragliflozin and Metformin on NASH in Type 2 Diabetic Mice. Endocr. Res. 2020, 45, 147–161. [Google Scholar] [CrossRef]
- Gharaibeh, N.E.; Rahhal, M.-N.; Rahimi, L.; Ismail-Beigi, F. SGLT-2 inhibitors as promising therapeutics for non-alcoholic fatty liver disease: Pathophysiology, clinical outcomes, and future directions. Diabetes Metab. Syndr. Obes. 2019, 12, 1001–1012. [Google Scholar] [CrossRef]
- Xu, L.; Fu, Z. Emerging roles of SGLT2 inhibitors in obesity and insulin resistance: Focus on fat browning and macrophage polarization. Adipocyte 2017, 7, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Nagata, N.; Chen, G.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Sakai, Y.; Kaneko, S.; Ota, T. Empagliflozin reverses obesity and insulin resistance through fat browning and alternative macrophage activation in mice fed a high-fat diet. BMJ Open Diabetes Res. Care 2019, 7, e000783. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Matsui, T.; Ishibashi, Y.; Yamagishi, S.-I. Insulin stimulates SGLT2-mediated tubular glucose absorption via oxidative stress generation. Diabetol. Metab. Syndr. 2015, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Larocque, L.M.; Efe, O.; Wang, J.; Sands, J.M.; Klein, J.D. Effect of Dapagliflozin Treatment on Fluid and Electrolyte Balance in Diabetic Rats. Am. J. Med. Sci. 2016, 352, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Muto, S.; Fukuda, K.; Watanabe, M.; Ohara, K.; Koepsell, H.; Vallon, V.; Nagata, D. Osmotic diuresis by SGLT2 inhibition stimulates vasopressin-induced water reabsorption to maintain body fluid volume. Physiol. Rep. 2020, 8, e14360. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Zhang, L.; Bondar, A.; Svensson, D.; Wernerson, A.; Brismar, H.; Scott, L.; Aperia, A. Prompt apoptotic response to high glucose in SGLT-expressing renal cells. Am. J. Physiol. Physiol. 2019, 316, F1078–F1089. [Google Scholar] [CrossRef]
- Gembardt, F.; Bartaun, C.; Jarzebska, N.; Mayoux, E.; Todorov, V.T.; Hohenstein, B.; Hugo, C. The SGLT2 inhibitor empagliflozin ameliorates early features of diabetic nephropathy in BTBR ob/ob type 2 diabetic mice with and without hypertension. Am. J. Physiol. Physiol. 2014, 307, F317–F325. [Google Scholar] [CrossRef]
- Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. Int. J. Mol. Sci. 2016, 17, 1826. [Google Scholar] [CrossRef]
- Pickering, T.G. Stress, Inflammation, and Hypertension. J. Clin. Hypertens. 2007, 9, 567–571. [Google Scholar] [CrossRef]
- Wei, L.; Xiao, Y.; Li, L.; Xiong, X.; Han, Y.; Zhu, X.; Sun, L. The Susceptibility Genes in Diabetic Nephropathy. Kidney Dis. 2018, 4, 226–237. [Google Scholar] [CrossRef]
- Oraby, M.A.; El-Yamany, M.F.; Safar, M.M.; Assaf, N.; Ghoneim, H.A. Dapagliflozin attenuates early markers of diabetic nephropathy in fructose-streptozotocin-induced diabetes in rats. Biomed. Pharmacother. 2019, 109, 910–920. [Google Scholar] [CrossRef]
- Das, N.A.; Carpenter, A.J.; Belenchia, A.; Aroor, A.R.; Noda, M.; Siebenlist, U.; Chandrasekar, B.; Demarco, V.G. Empagliflozin reduces high glucose-induced oxidative stress and miR-21-dependent TRAF3IP2 induction and RECK suppression, and inhibits human renal proximal tubular epithelial cell migration and epithelial-to-mesenchymal transition. Cell. Signal. 2020, 68, 109506. [Google Scholar] [CrossRef]
- Guebre-Egziabher, F.; Alix, P.M.; Koppe, L.; Pelletier, C.; Kalbacher, E.; Fouque, D.; Soulage, C.O. Ectopic lipid accumulation: A potential cause for metabolic disturbances and a contributor to the alteration of kidney function. Biochimie. 2013, 95, 1971–1979. [Google Scholar] [CrossRef]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [PubMed]
- Korbut, A.I.; Taskaeva, I.S.; Bgatova, N.P.; Muraleva, N.A.; Orlov, N.B.; Dashkin, M.V.; Khotskina, A.S.; Zavyalov, E.L.; Konenkov, V.I.; Klein, T.; et al. SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. Int. J. Mol. Sci. 2020, 21, 2987. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Shin, E.; Kim, G.; Lee, J.-Y.; Lee, Y.-H.; Lee, B.-W.; Kang, E.S.; Cha, B.-S. Combining SGLT2 Inhibition With a Thiazolidinedione Additively Attenuate the Very Early Phase of Diabetic Nephropathy Progression in Type 2 Diabetes Mellitus. Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Krishan, P.; Singh, G.; Bedi, O. Carbohydrate restriction ameliorates nephropathy by reducing oxidative stress and upregulating HIF-1α levels in type-1 diabetic rats. J. Diabetes Metab. Disord. 2017, 16, 47. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nat. Cell Biol. 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Ramakrishnan, S.K.; Shah, Y.M. A central role for hypoxia-inducible factor (HIF)-2α in hepatic glucose homeostasis. J. Nutr. Health Aging 2017, 4, 207–216. [Google Scholar] [CrossRef]
- Gillum, M.P.; Erion, D.M.; Shulman, G.I. Sirtuin-1 regulation of mammalian metabolism. Trends Mol. Med. 2011, 17, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Capra, J.A.; Pollard, K.S.; Verdin, E. SIRT1 and SIRT3 Deacetylate Homologous Substrates: AceCS1,2 and HMGCS1,2. Aging 2011, 3, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Schönenberger, M.J. Hypoxia signaling pathways: Modulators of oxygen-related organelles. Front. Cell Dev. Biol. 2015, 3, 42. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Xu, C.; Wang, W.; Zhong, J.; Lei, F.; Xu, N.; Zhang, Y.; Xie, W. Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem. Pharmacol. 2018, 152, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, F.; Scheen, A.J. Effects of SGLT2 inhibitors on systemic and tissue low-grade inflammation: The potential contribution to diabetes complications and cardiovascular disease. Diabetes Metab. 2018, 44, 457–464. [Google Scholar] [CrossRef]
- Tan, S.A.; Tan, L. Empagliflozin And Canagliflozin Attenuate Inflammatory Cytokines Interferon-Λ, Tumor Necrosis Factor-A, Interleukin-6: Possible Mechanism Of Decreasing Cardiovascular Risk In Diabetes Mellitus. J. Am. Coll. Cardiol. 2018, 71, A1830. [Google Scholar] [CrossRef]
- Lee, T.-M.; Chang, N.-C.; Lin, S.-Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free. Radic. Biol. Med. 2017, 104, 298–310. [Google Scholar] [CrossRef]
- Oelze, M.; Kröller-Schön, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinßius, E.; Agdauletova, S.; et al. The Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin Improves Diabetes-Induced Vascular Dysfunction in the Streptozotocin Diabetes Rat Model by Interfering with Oxidative Stress and Glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Lin, B.; Koibuchi, N.; Hasegawa, Y.; Sueta, D.; Toyama, K.; Uekawa, K.; Ma, M.; Nakagawa, T.; Kusaka, H.; Kim-Mitsuyama, S. Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc. Diabetol. 2014, 13, 148. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Thomson, S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: The pleiotropic effects of SGLT2 inhibition. Diabetology 2016, 60, 215–225. [Google Scholar] [CrossRef]
- Scheen, A.J. Pharmacokinetic drug evaluation of saxagliptin plus dapagliflozin for the treatment of type 2 diabetes. Expert Opin. Drug Metab. Toxicol. 2017, 13, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Kushiyama, A.; Tanaka, K.; Hara, S.; Kawazu, S. Linking uric acid metabolism to diabetic complications. World J. Diabetes 2014, 5, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013, 27, 41–53. [Google Scholar] [CrossRef]
- O’Neill, J.; Fasching, A.; Pihl, L.; Patinha, D.; Franzén, S.; Palm, F. Acute SGLT inhibition normalizes O2 tension in the renal cortex but causes hypoxia in the renal medulla in anaesthetized control and diabetic rats. Am. J. Physiol. Physiol. 2015, 309, F227–F234. [Google Scholar] [CrossRef]
- Sano, M.; Takei, M.; Shiraishi, Y.; Suzuki, Y. Increased Hematocrit During Sodium-Glucose Cotransporter 2 Inhibitor Therapy Indicates Recovery of Tubulointerstitial Function in Diabetic Kidneys. J. Clin. Med. Res. 2016, 8, 844–847. [Google Scholar] [CrossRef]
- Sano, M. Inter-organ Communication Pathway Manifested by Non-physiological Stress to the Kidney in Type II Diabetic Patients -Why Are Diabetic Patients Prone to Develop Heart Failure? Intern. Med. 2019, 59, 2870-19. [Google Scholar] [CrossRef]
- Chang, Y.-K.; Choi, H.; Jeong, J.Y.; Na, K.-R.; Lee, K.W.; Lim, B.J.; Choi, D.E. Dapagliflozin, SGLT2 Inhibitor, Attenuates Renal Ischemia-Reperfusion Injury. PLoS ONE 2016, 11, e0158810. [Google Scholar]
- Zhang, Y.; Nakano, D.; Guan, Y.; Hitomi, H.; Uemura, A.; Masaki, T.; Kobara, H.; Sugaya, T.; Nishiyama, A. A sodium-glucose cotransporter 2 inhibitor attenuates renal capillary injury and fibrosis by a vascular endothelial growth factor–dependent pathway after renal injury in mice. Kidney Int. 2018, 94, 524–535. [Google Scholar] [CrossRef]
- Lin, D.P.L.; Dass, C.R. Weak bones in diabetes mellitus—an update on pharmaceutical treatment options. J. Pharm. Pharmacol. 2017, 70, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-I.; Liu, C.-S.; Lin, W.-Y.; Meng, N.-H.; Chen, C.-C.; Yang, S.-Y.; Chen, H.-J.; Lin, C.-C.; Li, T.-C. Glycated Hemoglobin Level and Risk of Hip Fracture in Older People with Type 2 Diabetes: A Competing Risk Analysis of Taiwan Diabetes Cohort Study. J. Bone Miner. Res. 2015, 30, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-C.; Liu, W.-C.; Zheng, C.-M.; Zheng, J.-Q.; Yen, T.-H.; Lu, K.-C. Role of Vitamin D in Uremic Vascular Calcification. BioMed Res. Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, Ö.; Bolinder, J.; Johansson, L.; Wilding, J.; Langkilde, A.M.; Sjöström, C.D.; Sugg, J.; Parikh, S. Dapagliflozin has no effect on markers of bone formation and resorption or bone mineral density in patients with inadequately controlled type 2 diabetes mellitus on metformin. Diabetes Obes. Metab. 2012, 14, 990–999. [Google Scholar] [CrossRef]
- Bilezikian, J.P.; Watts, N.B.; Usiskin, K.; Polidori, D.; Fung, A.; Sullivan, D.; Rosenthal, N. Evaluation of Bone Mineral Density and Bone Biomarkers in Patients With Type 2 Diabetes Treated With Canagliflozin. J. Clin. Endocrinol. Metab. 2016, 101, 44–51. [Google Scholar] [CrossRef]
- De Jong, M.A.; Petrykiv, S.I.; Laverman, G.D.; Van Herwaarden, A.E.; De Zeeuw, D.; Bakker, S.J.L.; Heerspink, H.J.L.; De Borst, M.H. Effects of Dapagliflozin on Circulating Markers of Phosphate Homeostasis. Clin. J. Am. Soc. Nephrol. 2018, 14, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Meier, C.; Schwartz, A.V.; Egger, A.; Lecka-Czernik, B. Effects of diabetes drugs on the skeleton. Bone 2016, 82, 93–100. [Google Scholar] [CrossRef]
- Qian, Q. Salt, water and nephron: Mechanisms of action and link to hypertension and chronic kidney disease. Nephrolology 2018, 23, 44–49. [Google Scholar] [CrossRef]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.-K.; Beck, F.-X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C–dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Marvar, P.J.; Gordon, F.J.; Harrison, D.G. Blood pressure control: Salt gets under your skin. Nat. Med. 2009, 15, 487–488. [Google Scholar] [CrossRef]
- Lankhorst, S.; Severs, D.; Markó, L.; Rakova, N.; Titze, J.; Müller, D.N.; Danser, A.J.; Meiracker, A.; van den Meiracker, A.H. Salt Sensitivity of Angiogenesis Inhibition–Induced Blood Pressure Rise. Hypertension 2017, 69, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.P.; Raff, U.; Kopp, C.; Scheppach, J.B.; Toncar, S.; Wanner, C.; Schlieper, G.; Saritas, T.; Floege, J.; Schmid, M.; et al. Skin Sodium Concentration Correlates with Left Ventricular Hypertrophy in CKD. J. Am. Soc. Nephrol. 2017, 28, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Kannenkeril, D.; Karg, M.V.; Bosch, A.; Ott, C.; Linz, P.; Nagel, A.M.; Uder, M.; Schmieder, R.E. Tissue sodium content in patients with type 2 diabetes mellitus. J. Diabetes Complicat. 2019, 33, 485–489. [Google Scholar] [CrossRef]
- Karg, M.V.; Bosch, A.; Kannenkeril, D.; Striepe, K.; Ott, C.; Schneider, M.P.; Boemke-Zelch, F.; Linz, P.; Nagel, A.M.; Titze, J.; et al. SGLT-2-inhibition with dapagliflozin reduces tissue sodium content: A randomised controlled trial. Cardiovasc. Diabetol. 2018, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2018, 20, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Sato, T.; Sato, H.; Yamaguchi, Y.; Obara, K.; Kurihara, I.; Sato, K.; Hotta, O.; Seino, J.; Miyata, M.; et al. Different clinical outcomes for cardiovascular events and mortality in chronic kidney disease according to underlying renal disease: The Gonryo study. Clin. Exp. Nephrol. 2010, 14, 333–339. [Google Scholar] [CrossRef]
- Liu, W.-C.; Tomino, Y.; Lu, K.-C. Impacts of Indoxyl Sulfate and p-Cresol Sulfate on Chronic Kidney Disease and Mitigating Effects of AST-120. Toxins 2018, 10, 367. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Kanemitsu, Y.; Saigusa, D.; Mukawa, C.; Asaji, K.; Matsumoto, Y.; Tsukamoto, H.; Tachikawa, T.; Tsukimi, T.; et al. Canagliflozin reduces plasma uremic toxins and alters the intestinal microbiota composition in a chronic kidney disease mouse model. Am. J. Physiol. Physiol. 2018, 315, F824–F833. [Google Scholar] [CrossRef]
- McMurray, J.J.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefansson, B.V.; Chertow, G.M.; Correa-Rotter, R.; Greene, T.; Hou, F.-F.; Lindberg, M.; McMurray, J.; Rossing, P.; Toto, R.; et al. Rationale and protocol of the Dapagliflozin And Prevention of Adverse outcomes in Chronic Kidney Disease (DAPA-CKD) randomized controlled trial. Nephrol. Dial. Transplant. 2020, 35, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.; McMurray, J.J.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, D.L.; Cherney, D.Z.I.; Bjornstad, P.; Castilla-Guerra, L.; González, J.P.M. Antihyperglycemic agents as novel natriuretic therapies in diabetic kidney disease. Am. J. Physiol. Physiol. 2018, 315, F1406–F1415. [Google Scholar] [CrossRef]
- Ansary, T.M.; Fujisawa, Y.; Rahman, A.; Nakano, D.; Hitomi, H.; Kobara, H.; Masaki, T.; Titze, J.M.; Kitada, K.; Nishiyama, A. Responses of renal hemodynamics and tubular functions to acute sodium–glucose cotransporter 2 inhibitor administration in non-diabetic anesthetized rats. Sci. Rep. 2017, 7, 9555. [Google Scholar] [CrossRef]
- Górriz, J.L.; Navarro-González, J.F.; Ortiz, A.; Vergara, A.; Nuñez, J.; Jacobs-Cachá, C.; Martínez-Castelao, A.; Soler, M.J. Sodium-glucose cotransporter 2 inhibition: Towards an indication to treat diabetic kidney disease. Nephrol. Dial. Transplant. 2020, 35, i13–i23. [Google Scholar] [CrossRef]
- Jackson, A.M.; Dewan, P.; Anand, I.S.; Bělohlávek, J.; Bengtsson, O.; De Boer, R.A.; Böhm, M.; Boulton, D.W.; Chopra, V.K.; DeMets, D.L.; et al. Dapagliflozin and Diuretic Use in Patients with Heart Failure and Reduced Ejection Fraction in DAPA-HF. Circulation 2020, 142, 1040–1054. [Google Scholar] [CrossRef]
- Douros, A.; Lix, L.M.; Fralick, M.; Dell’Aniello, S.; Shah, B.R.; Ronksley, P.E.; Tremblay, É.; Hu, N.; Alessi-Severini, S.; Fisher, A.; et al. Sodium–Glucose Cotransporter-2 Inhibitors and the Risk for Diabetic Ketoacidosis. Ann. Intern. Med. 2020, 173, 417–425. [Google Scholar] [CrossRef]
- Patoulias, D.; Manafis, A.; Mitas, C.; Avranas, K.; Lales, G.; Zografou, I.; Sambanis, C.; Karagiannis, A. Sodium-glucose Cotransporter 2 Inhibitors and the Risk of Diabetic Ketoacidosis; from Pathophysiology to Clinical Practice. Cardiovasc. Hematol. Disord. Targets 2018, 18, 139–146. [Google Scholar] [CrossRef]
- Burke, K.R.; Schumacher, C.A.; Harpe, S.E. SGLT2 Inhibitors: A Systematic Review of Diabetic Ketoacidosis and Related Risk Factors in the Primary Literature. Pharmacotherapy J. Hum. Pharmacol. Drug Ther. 2017, 37, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Dull, R.B.; Spangler, M.L.; Knezevich, E.L.; Lau, B.M. Euglycemic Diabetic Ketoacidosis Associated With Sodium–Glucose Cotransporter Type 2 Inhibitors in Patients With Type 2 Diabetes Mellitus Receiving Oral Therapy. J. Pharm. Pr. 2017, 32, 240–243. [Google Scholar] [CrossRef]
- Egan, A.M.; Montori, V.M. Review: In adults with type 1 diabetes, SGLT-2 inhibitors reduce HbA1c but increase diabetic ketoacidosis. Ann. Intern. Med. 2018, 169, Jc3. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Rabin-Court, A.; Song, J.D.; Cardone, R.L.; Wang, Y.; Kibbey, R.G.; Shulman, G.I. Dehydration and insulinopenia are necessary and sufficient for euglycemic ketoacidosis in SGLT2 inhibitor-treated rats. Nat. Commun. 2019, 10, 548. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.-C.; Zheng, C.-M.; Yen, T.-H.; Lu, K.-C. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. Int. J. Mol. Sci. 2020, 21, 7833. https://doi.org/10.3390/ijms21217833
Hou Y-C, Zheng C-M, Yen T-H, Lu K-C. Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. International Journal of Molecular Sciences. 2020; 21(21):7833. https://doi.org/10.3390/ijms21217833
Chicago/Turabian StyleHou, Yi-Chou, Cai-Mei Zheng, Tzung-Hai Yen, and Kuo-Cheng Lu. 2020. "Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection" International Journal of Molecular Sciences 21, no. 21: 7833. https://doi.org/10.3390/ijms21217833
APA StyleHou, Y.-C., Zheng, C.-M., Yen, T.-H., & Lu, K.-C. (2020). Molecular Mechanisms of SGLT2 Inhibitor on Cardiorenal Protection. International Journal of Molecular Sciences, 21(21), 7833. https://doi.org/10.3390/ijms21217833