The Neutrophil: The Underdog That Packs a Punch in the Fight against Cancer



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
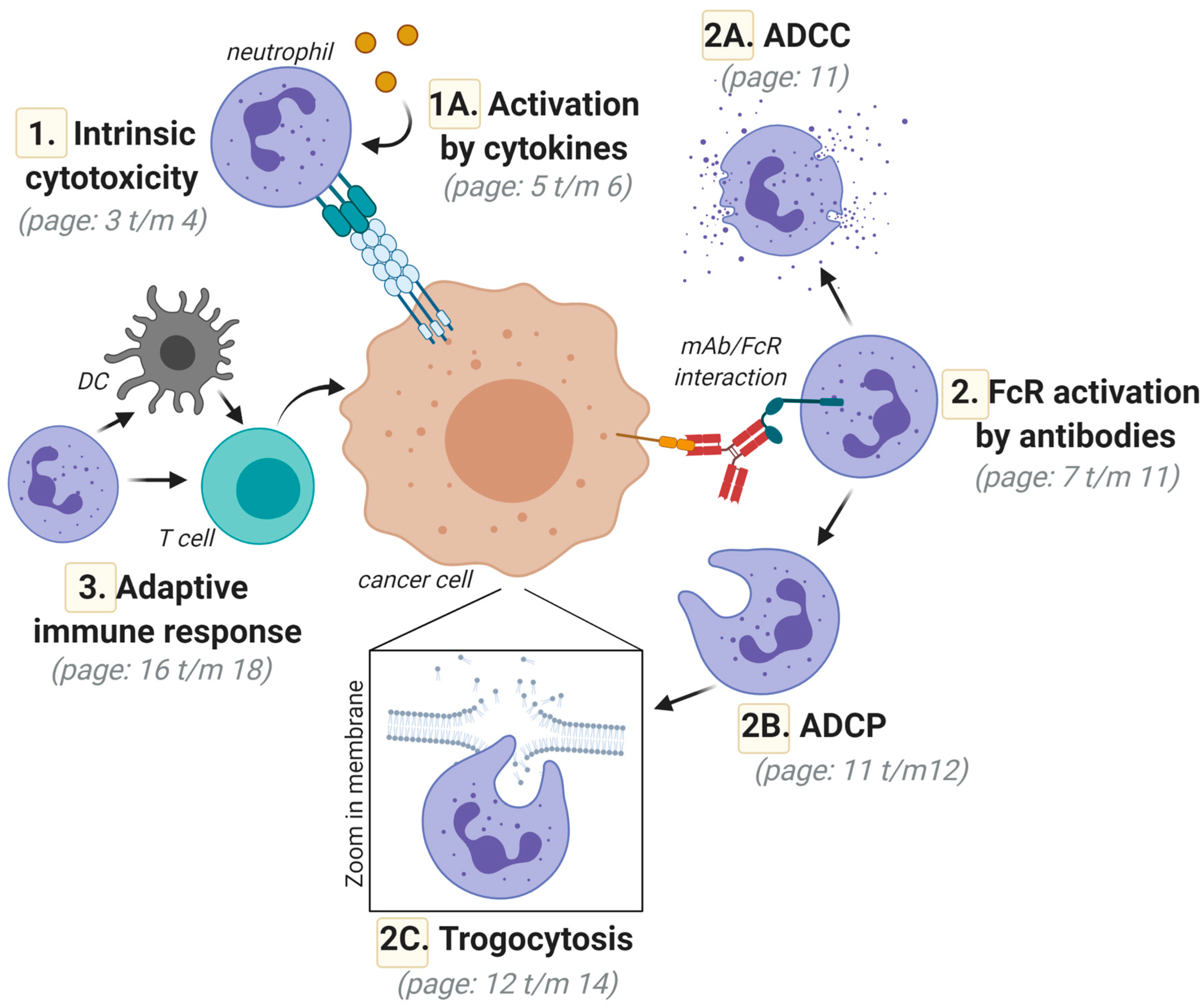
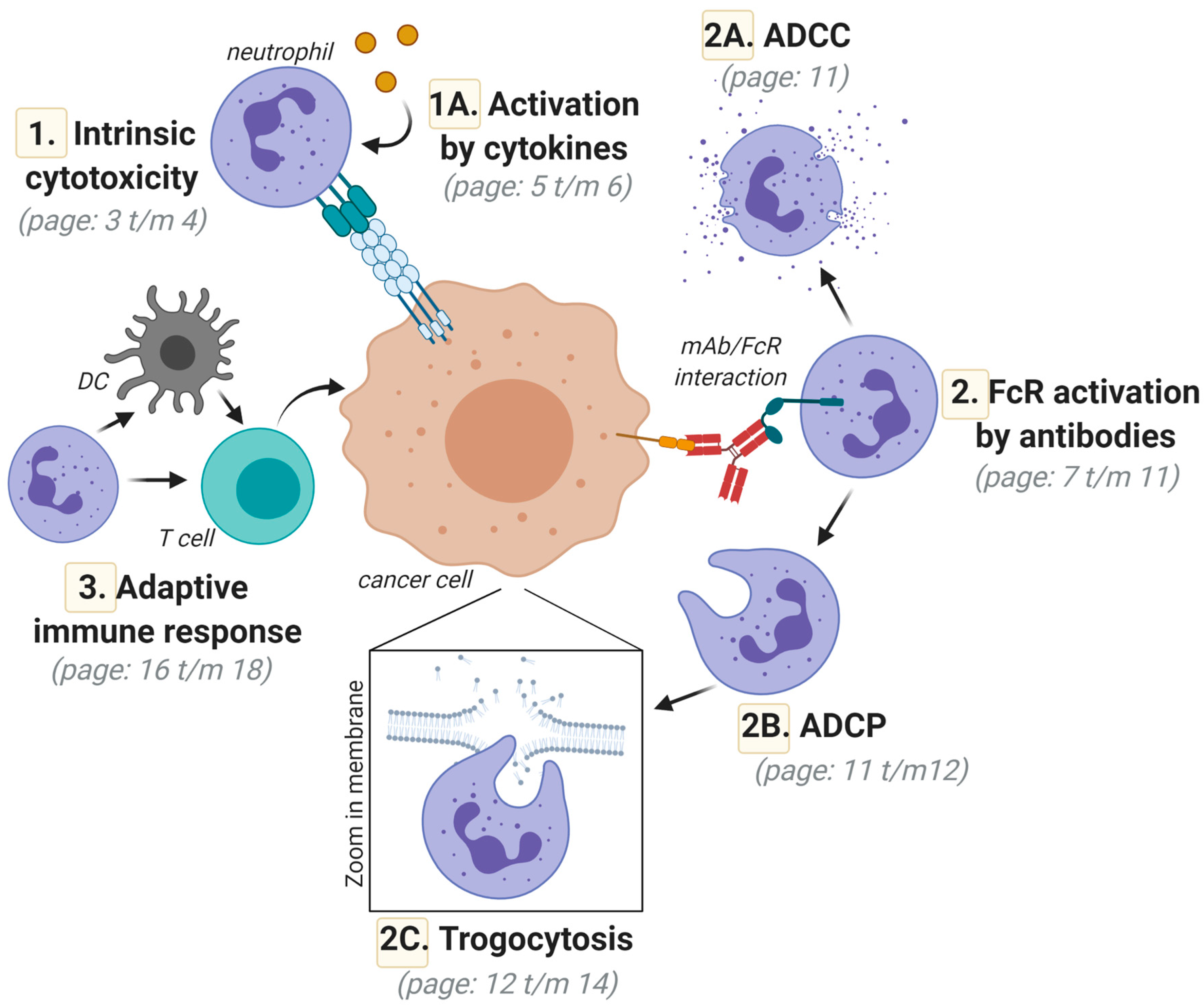
2. Direct Neutrophil-Mediated Cytotoxic Activity toward Cancer Cells; Intrinsic Anticancer Activity via Death-Inducing Ligands of the Tumor Necrosis Factor (TNF) Superfamily
Activation of Neutrophil-Mediated Anticancer Responses by Cytokines
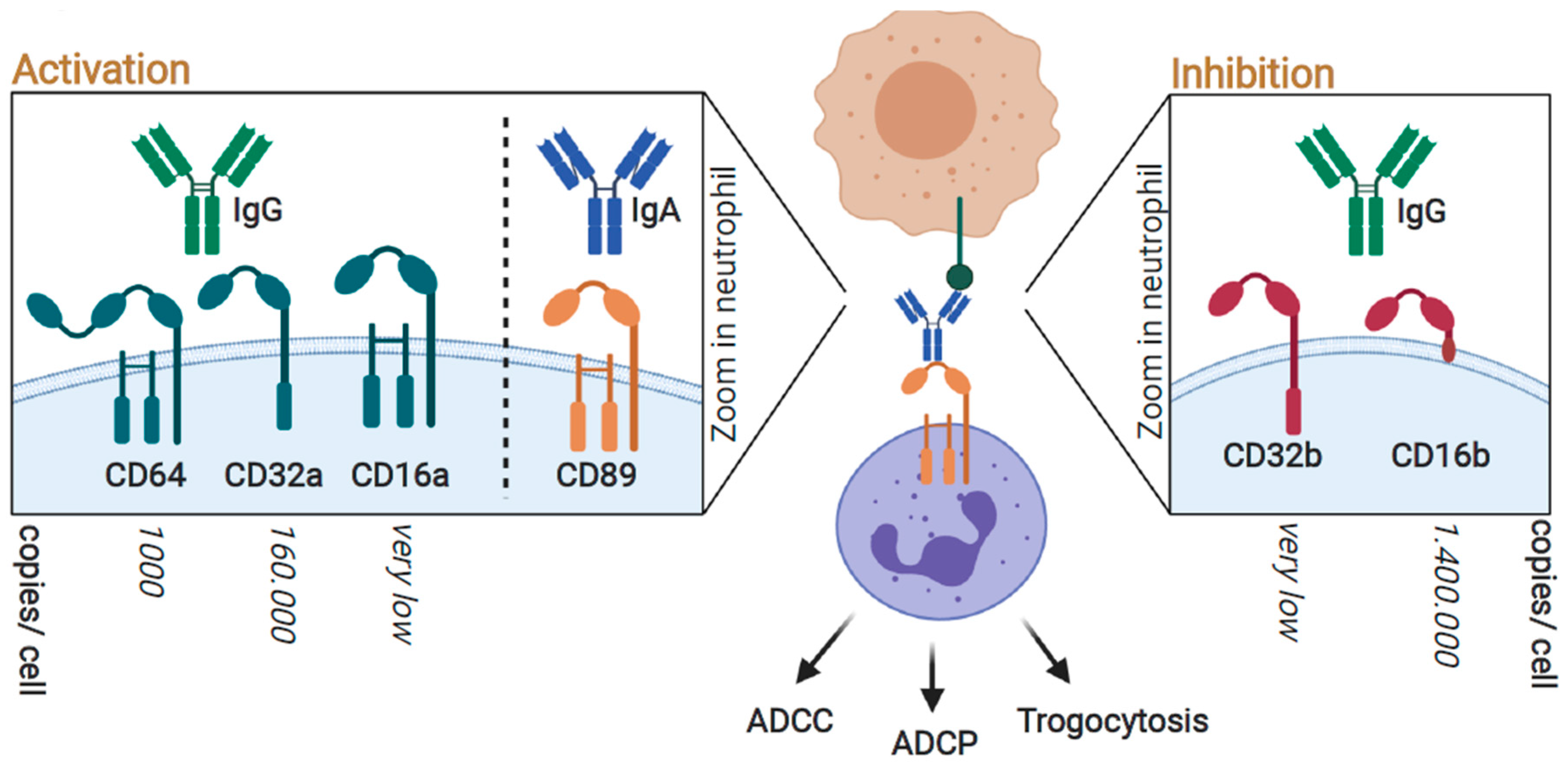
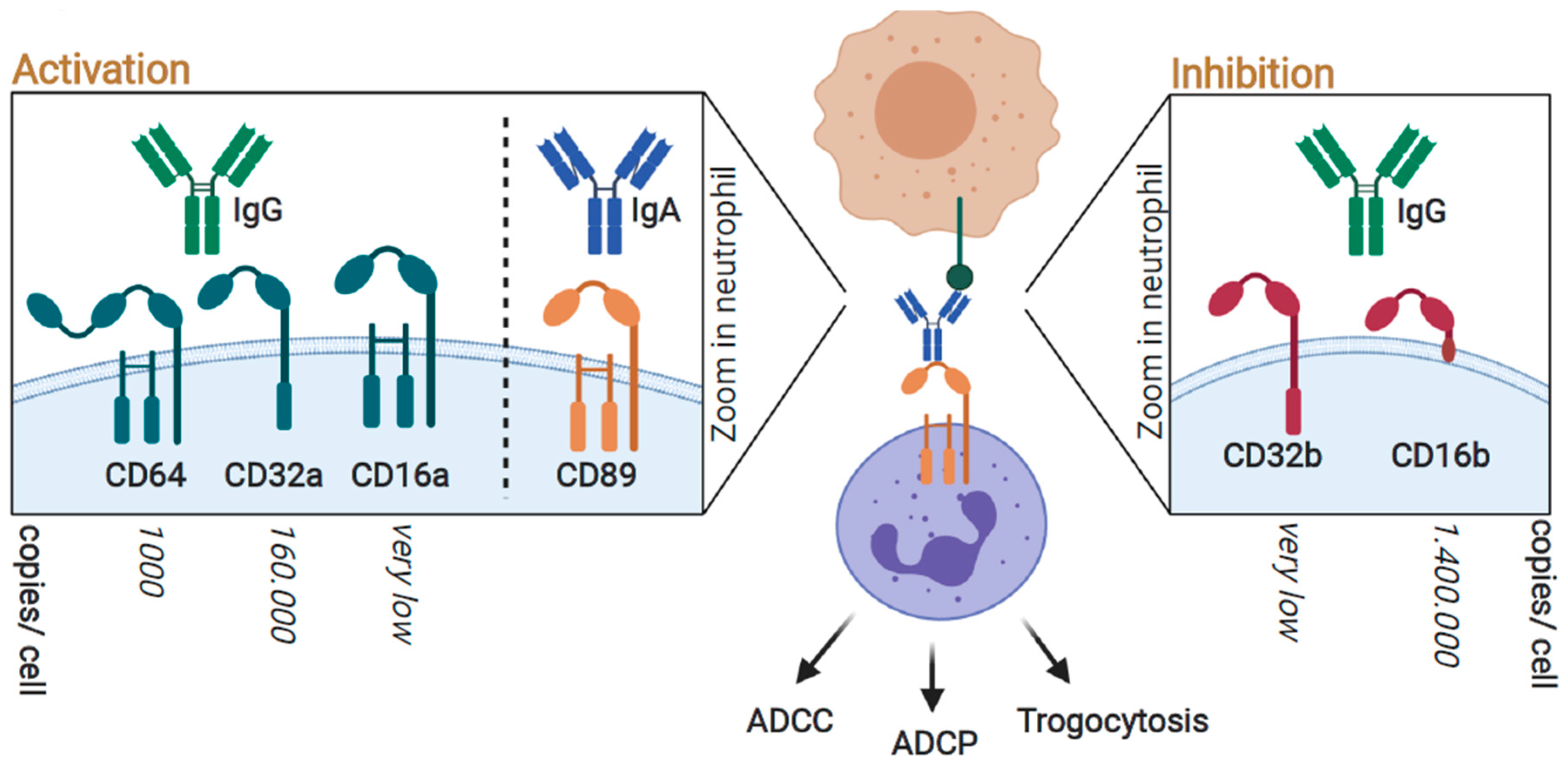
3. FcR-Mediated Neutrophil Activation; Targeting FcγRIIa (CD32a) and FcαRI (CD89) for Optimal Responses
3.1. Neutrophil-Mediated Cytotoxic Activity toward Cancer Cells; Targeted Anticancer Activity by Antibody-Dependent Cellular Cytotoxicity (ADCC)
3.2. Neutrophil-Mediated Engulfment of Cancer Cells; Antibody-Dependent Cellular Phagocytosis (ADCP)
3.3. Trogocytosis; Lysing Cancer Cells by Biting off Parts of the Plasma Membrane
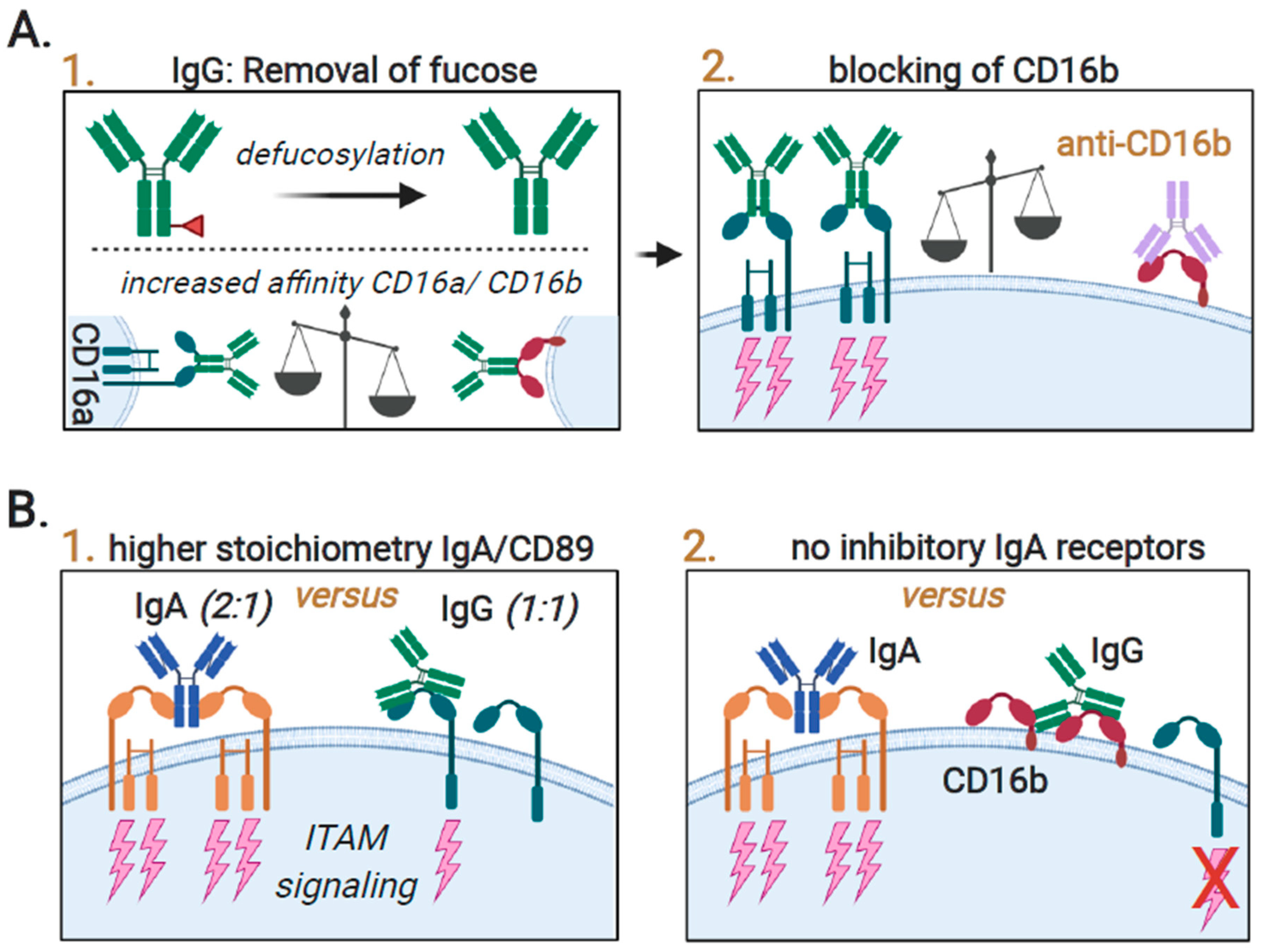
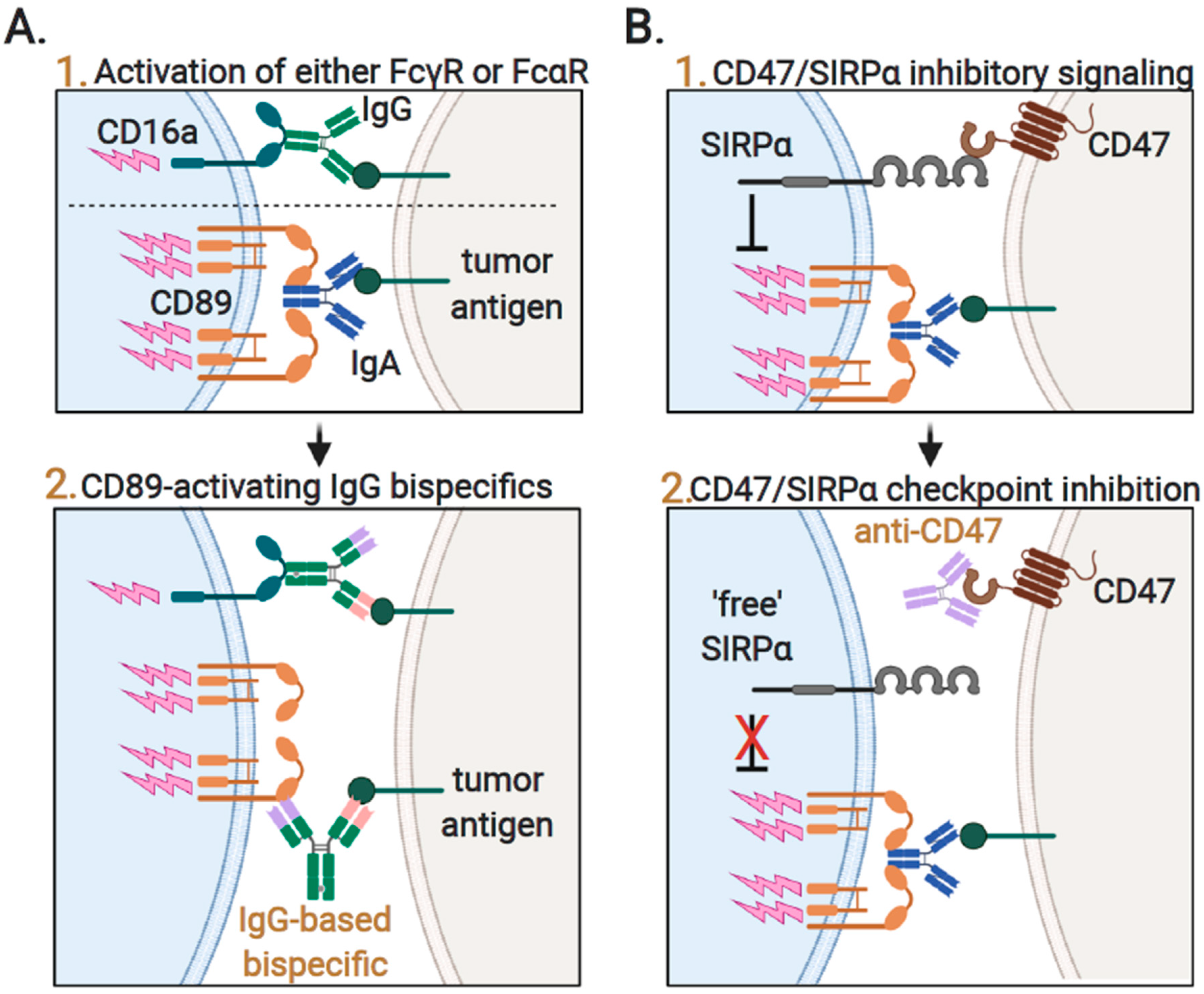
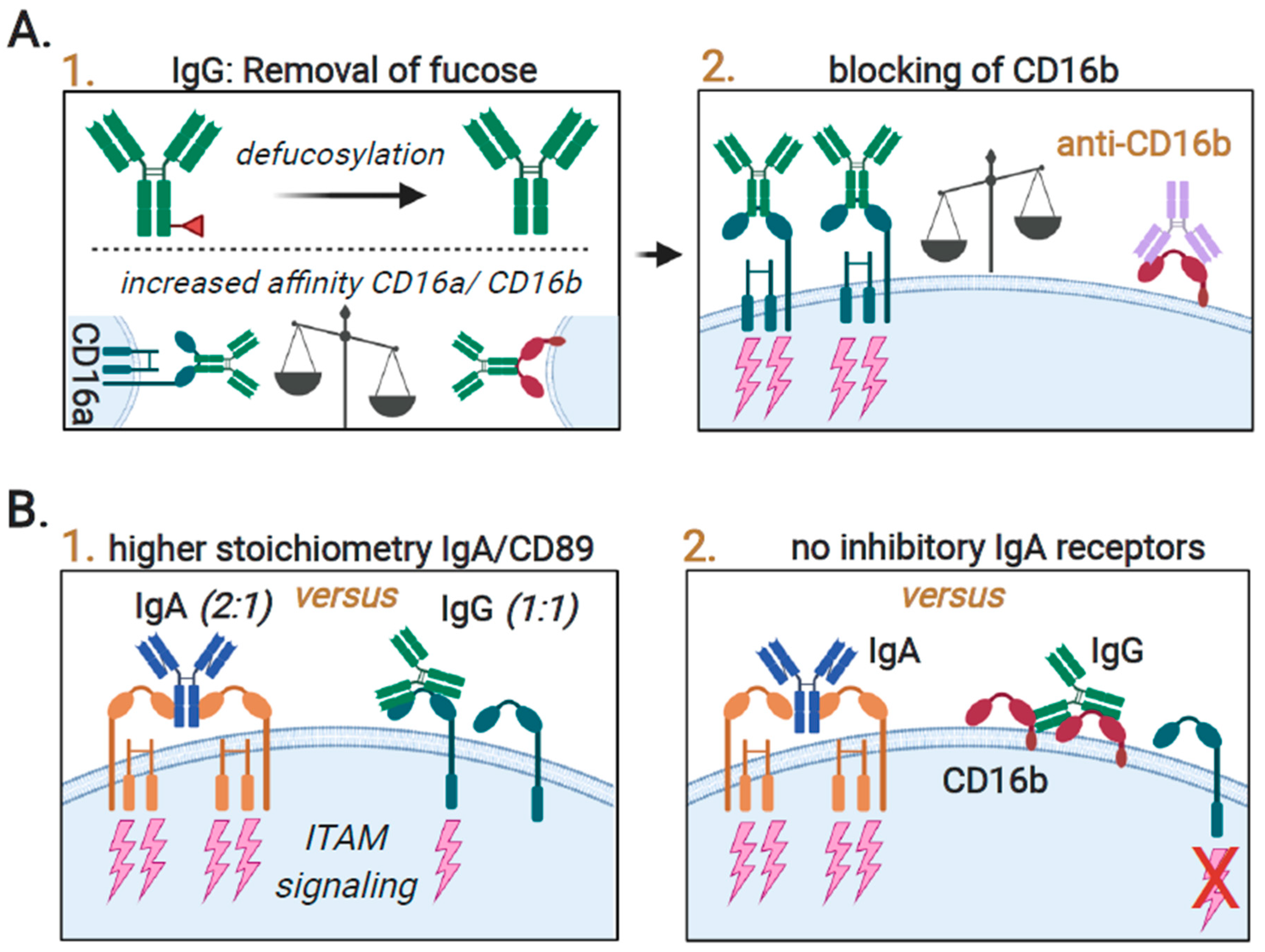
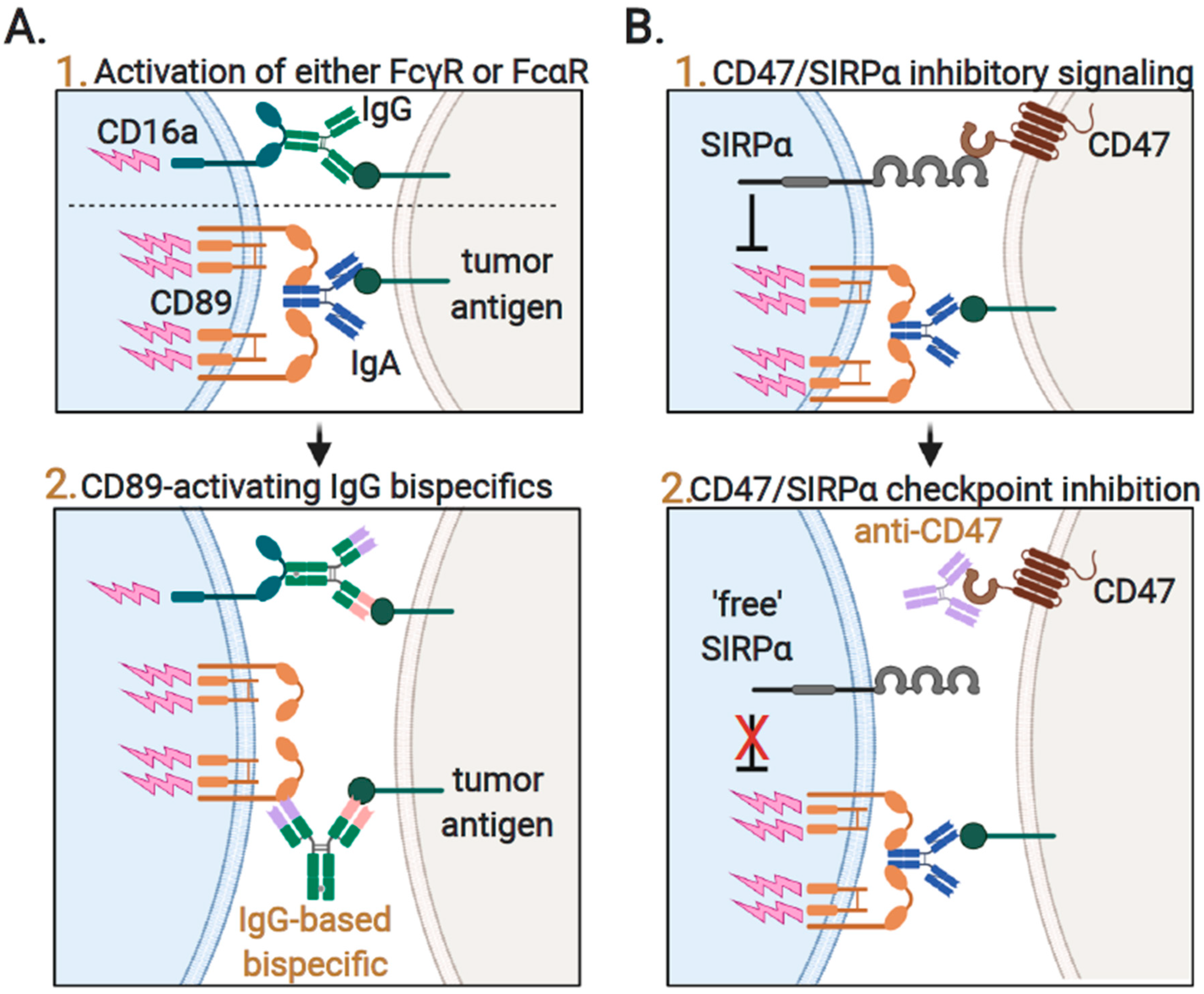
3.4. Augmenting Neutrophil-Mediated ADCP and ADCC by Targeting of Innate Immune Checkpoints
4. Neutrophil-Mediated Induction of Adaptive Anticancer Immune Responses
5. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Brentjens, R.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; Van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.-P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Gerrard, T.L.; Cohen, D.J.; Kaplan, A.M. Human Neutrophil-Mediated Cytotoxicity to Tumor Cells. J. Natl. Cancer Inst. 1981, 66, 483–488. [Google Scholar] [CrossRef]
- Vols, S.; Sionov, R.V.; Granot, Z. Always Look on the Bright Side: Anti-Tumor Functions of Neutrophils. Curr. Pharm. Des. 2017, 23, 4862–4892. [Google Scholar] [CrossRef]
- Eruslanov, E.B.; Bhojnagarwala, P.S.; Quatromoni, J.G.; Stephen, T.L.; Ranganathan, A.; Deshpande, C.; Akimova, T.; Vachani, A.; Litzky, L.; Hancock, W.W.; et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J. Clin. Investig. 2014, 124, 5466–5480. [Google Scholar] [CrossRef] [Green Version]
- Sagiv, J.Y.; Michaeli, J.; Assi, S.; Mishalian, I.; Kisos, H.; Levy, L.; Damti, P.; Lumbroso, D.; Polyansky, L.; Sionov, R.V.; et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep. 2015, 10, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.-P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef]
- Manfroi, B.; Moreaux, J.; Righini, C.; Ghiringhelli, F.; Sturm, N.; Huard, B. Tumor-associated neutrophils correlate with poor prognosis in diffuse large B-cell lymphoma patients. Blood Cancer J. 2018, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Margetts, J.; Ogle, L.F.; Chan, S.L.; Chan, A.W.H.; Chan, K.C.A.; Jamieson, D.; Willoughby, C.E.; Mann, D.A.; Wilson, C.L.; Manas, D.M.; et al. Neutrophils: Driving progression and poor prognosis in hepatocellular carcinoma? Br. J. Cancer 2017, 118, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Hu, P.; Donskov, F.; Wang, G.; Liu, Q.; Du, J. Tumor-Associated Neutrophils as a New Prognostic Factor in Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e98259. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Cong, X.; Gao, H.; Lan, X.; Li, Z.; Wang, W.; Song, S.; Wang, Y.; Li, C.; Zhang, H.; et al. Tumor-associated neutrophils induce EMT by IL-17a to promote migration and invasion in gastric cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Ding, Y.; Li, N.; Wu, L.; Gao, Y.; Xiao, C.; Jiang, H.; Zheng, Y.; Mao, C.; Deng, J.; et al. Prognostic Value of Neutrophil–Lymphocyte Ratio, Platelet–Lymphocyte Ratio, and Combined Neutrophil–Lymphocyte Ratio and Platelet–Lymphocyte Ratio in Stage IV Advanced Gastric Cancer. Front. Oncol. 2020, 10, 841. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Liu, N.; Wang, S.; Guo, J.; Song, X.; Qi, Y.; Qiu, W.; Lv, J. Prognostic significance of the neutrophil-to-lymphocyte and platelet-to-lymphocyte ratio in patients with metastatic gastric cancer. Medicine 2020, 99, e19405. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.S.; Xiong, M.-J.; Greenbaum, A.; Mortaji, P.; Nofchissey, R.A.; Schultz, F.; Martinez, C.; Luo, L.; Morris, K.T.; Hanson, J.A. High levels of tumor-associated neutrophils are associated with improved overall survival in patients with stage II colorectal cancer. PLoS ONE 2017, 12, e0188799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galdiero, M.R.; Bianchi, P.; Grizzi, F.; Di Caro, G.; Basso, G.; Ponzetta, A.; Bonavita, E.; Barbagallo, M.; Tartari, S.; Polentarutti, N.; et al. Occurrence and significance of tumor-associated neutrophils in patients with colorectal cancer. Int. J. Cancer 2016, 139, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Garley, M.; Jabłońska, E. Heterogeneity Among Neutrophils. Archivum Immunologiae Therapiae Experimentalis 2017, 66, 21–30. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Shaul, M.E.; Levy, L.; Sun, J.; Mishalian, I.; Singhal, S.; Kapoor, V.; Horng, W.; Fridlender, G.; Albelda, S.M.; Fridlender, Z.G. Tumor-associated neutrophils display a distinct N1 profile following TGFβ modulation: A transcriptomics analysis of pro- vs. antitumor TANs. OncoImmunology 2016, 5, e1232221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.-F.; Gereke, M.; Von Köckritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type IIFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2015, 138, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Tai, J.A.; Li, S.; Nishikawa, T.; Kaneda, Y. Virus-stimulated neutrophils in the tumor microenvironment enhance T cell-mediated anti-tumor immunity. Oncotarget 2016, 7, 42195–42207. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Assi, S.; Gershkovitz, M.; Sagiv, J.Y.; Polyansky, L.; Mishalian, I.; Fridlender, Z.G.; Granot, Z. Isolation and Characterization of Neutrophils with Anti-Tumor Properties. J. Vis. Exp. 2015, 2015, 52933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, B.E.; Tabariès, S.; Johnson, R.M.; Andrzejewski, S.; Senecal, J.; Lehuédé, C.; Annis, M.G.; Ma, E.H.; Völs, S.; Ramsay, L.; et al. Immature Low-Density Neutrophils Exhibit Metabolic Flexibility that Facilitates Breast Cancer Liver Metastasis. Cell Rep. 2019, 27, 3902–3915.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, M.E.; Eyal, O.; Guglietta, S.; Aloni, P.; Zlotnik, A.; Forkosh, E.; Levy, L.; Weber, L.M.; Levin, Y.; Pomerantz, A.; et al. Circulating neutrophil subsets in advanced lung cancer patients exhibit unique immune signature and relate to prognosis. FASEB J. 2020, 34, 4204–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Poljak-Blazi, M.; Kirac, I.; Borovic, S.; Schaur, R.J.; Zarkovic, N. Granulocytes as effective anticancer agent in experimental solid tumor models. Immunobiology 2010, 215, 1015–1020. [Google Scholar] [CrossRef]
- Jaganjac, M.; Poljak-Blazi, M.; Žarković, K.; Schaur, R.J.; Zarkovic, N. The involvement of granulocytes in spontaneous regression of Walker 256 carcinoma. Cancer Lett. 2008, 260, 180–186. [Google Scholar] [CrossRef]
- Challacombe, J.M.; Suhrbier, A.; Parsons, P.G.; Jones, B.; Hampson, P.; Kavanagh, D.; Rainger, G.E.; Morris, M.; Lord, J.M.; Le, T.T.T.; et al. Neutrophils are a key component of the antitumor efficacy of topical chemotherapy with ingenol-3-angelate. J. Immunol. 2006, 177, 8123–8132. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, M.; Mancardi, D.A.; Jönsson, F.; Iannascoli, B.; Fiette, L.; Di Santo, J.P.; Lowell, C.A.; Bruhns, P. Neutrophils mediate antibody-induced antitumor effects in mice. Blood 2013, 122, 3160–3164. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Kloecker, G.; Fleming, C.; Bousamra, M.; Hansen, R.; Hu, X.; Ding, C.; Cai, Y.; Xiang, D.; Donninger, H.; et al. Human polymorphonuclear neutrophils specifically recognize and kill cancerous cells. OncoImmunology 2014, 3, e950163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissemond, J.; Weimann, T.K.; Schneider, L.A.; Schneeberger, A.; Scharffetter-Kochanek, K.; Goos, M.; Wagner, S.N. Activated Neutrophils Exert Antitumor Activity Against Human Melanoma Cells: Reactive Oxygen Species-Induced Mechanisms and Their Modulation by Granulocyte-Macrophage–Colony-Stimulating Factor. J. Investig. Dermatol. 2003, 121, 936–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Qin, W.; Song, M.; Liu, L.; Yu, Y.; Qi, X.; Sun, H. Neutrophil Suppresses Tumor Cell Proliferation via Fas/Fas Ligand Pathway Mediated Cell Cycle Arrested. Int. J. Biol. Sci. 2018, 14, 2103–2113. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, A.T. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand: A Novel Mechanism for Bacillus Calmette-Guerin-Induced Antitumor Activity. Cancer Res. 2004, 64, 3386–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tecchio, C.; Huber, V.; Scapini, P.; Calzetti, F.; Margotto, D.; Todeschini, G.; Pilla, L.; Martinelli, G.; Pizzolo, G.; Rivoltini, L.; et al. IFNa-stimulated neutrophils and monocytes release a soluble form ofTNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) displaying apoptotic activity on leukemic cells. Blood 2004, 103, 3837–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamohara, H.; Matsuyama, W.; Shimozato, O.; Abe, K.; Galligan, C.; Hashimoto, S.-I.; Matsushima, K.; Yoshimura, T. Regulation of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptor expression in human neutrophils. Immunology 2004, 111, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Matsuzaki, A.; Suminoe, A.; Hattori, H.; Hara, T. Neutrophil-Derived TNF-Related Apoptosis-Inducing Ligand (TRAIL). Cancer Res. 2004, 64, 1037–1043. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, V.R.; De Bruyn, M.; Shi, C.; Gooden, M.J.; Wouters, M.C.; Samplonius, D.F.; Hendriks, D.; Nijman, H.W.; Wei, Y.; Zhou, J.; et al. C-type lectin-like molecule-1 (CLL1)-targeted TRAIL augments the tumoricidal activity of granulocytes and potentiates therapeutic antibody-dependent cell-mediated cytotoxicity. mAbs 2015, 7, 321–330. [Google Scholar] [CrossRef]
- Renshaw, S.A.; Parmar, J.S.; Singleton, V.; Rowe, S.J.; Dockrell, D.H.; Dower, S.K.; Bingle, C.D.; Chilvers, E.R.; Whyte, M.K.B. Acceleration of Human Neutrophil Apoptosis by TRAIL. J. Immunol. 2003, 170, 1027–1033. [Google Scholar] [CrossRef]
- Shigeno, M.; Nakao, K.; Ichikawa, T.; Suzuki, K.; Kawakami, A.; Abiru, S.; Miyazoe, S.; Nakagawa, Y.; Ishikawa, H.; Hamasaki, K.; et al. Interferon-α sensitizes human hepatoma cells to TRAIL-induced apoptosis through DR5 upregulation and NF-κB inactivation. Oncogene 2003, 22, 1653–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toiyama, D.; Takaha, N.; Shinnoh, M.; Ueda, T.; Kimura, Y.; Nakamura, T.; Hongo, F.; Mikami, K.; Kamoi, K.; Kawauchi, A.; et al. Significance of serum tumor necrosis factor-related apoptosis-inducing ligand as a prognostic biomarker for renal cell carcinoma. Mol. Clin. Oncol. 2012, 1, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Kemp, T.J.; Gao, Y.-T.; Corbel, A.; McGee, E.E.; Roa, J.C.; Wang, B.; Araya, J.C.; Shen, M.-C.; Rashid, A.; et al. Circulating Levels of Inflammatory Proteins and Survival in Patients with Gallbladder Cancer. Sci. Rep. 2018, 8, 5671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajant, H.; Moosmayer, D.; Wüest, T.; Bartke, T.; Gerlach, E.; Schönherr, U.; Peters, N.; Scheurich, P.; Pfizenmaier, K. Differential activation of TRAIL-R1 and -2 by soluble and membrane TRAIL allows selective surface antigen-directed activation of TRAIL-R2 by a soluble TRAIL derivative. Oncogene 2001, 20, 4101–4106. [Google Scholar] [CrossRef] [Green Version]
- Kemp, T.J.; Ludwig, A.T.; Earel, J.K.; Moore, J.M.; VanOosten, R.L.; Moses, B.; Leidal, K.; Nauseef, W.M.; Griffith, T.S. Neutrophil stimulation with Mycobacterium bovis bacillus Calmette-Gueérin (BCG) results in the release of functional soluble TRAIL/Apo-2L. Blood 2005, 106, 3474–3482. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.A.; Botella, R.; Galloway, T.H.; Murray, N.; Kramp, J.M.; Song, I.S.; Ansel, J.C. Antitumor effects of granulocyte-macrophage colony-stimulating factor production by melanoma cells. Cancer Res. 1996, 56, 2191–2198. [Google Scholar] [PubMed]
- Zarei, S.; Schwenter, F.; Luy, P.; Aurrand-Lions, M.; Morel, P.; Kopf, M.; Dranoff, G.; Mach, N. Role of GM-CSF signaling in cell-based tumor immunization. Blood 2009, 113, 6658–6668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoppacciaro, A.; Forni, G.; Colombo, M.P. Different tumours, transduced with different cytokine genes as G-CSF and IL-2, show inhibition of tumour take through neutrophil activation but differ in T cell functions. Folia Biol. 1994, 40, 89–99. [Google Scholar]
- Schneider-Merck, T.; Van Bueren, J.J.L.; Berger, S.; Rossen, K.; Van Berkel, P.H.; Derer, S.; Beyer, T.; Lohse, S.; Bleeker, W.K.; Peipp, M.; et al. Human IgG2 Antibodies against Epidermal Growth Factor Receptor Effectively Trigger Antibody-Dependent Cellular Cytotoxicity but, in Contrast to IgG1, Only by Cells of Myeloid Lineage. J. Immunol. 2010, 184, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Wislez, M.; Fleury-Feith, J.; Rabbe, N.; Moreau, J.; Cesari, D.; Milleron, B.; Mayaud, C.; Antoine, M.; Soler, P.; Cadranel, J. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor and Granulocyte Colony-Stimulating Factor Prolong the Survival of Neutrophils Infiltrating Bronchoalveolar Subtype Pulmonary Adenocarcinoma. Am. J. Pathol. 2001, 159, 1423–1433. [Google Scholar] [CrossRef] [Green Version]
- Casbon, A.-J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegué, E.; Werb, Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 2015, 112, E566–E575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-C.; Sun, H.-W.; Chen, H.-T.; Liang, J.; Yu, X.-J.; Wu, C.; Wang, Z.; Zheng, L. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl. Acad. Sci. USA 2014, 111, 4221–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowanetz, M.; Wu, X.; Lee, J.; Tan, M.; Hagenbeek, T.J.; Qu, X.; Yu, L.; Ross, J.; Korsisaari, N.; Cao, T.; et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 21248–21255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallo, F.; Giovarelli, M.; Gulino, A.; Vacca, A.; Stoppacciaro, A.; Modesti, A.; Forni, G. Role of neutrophils and CD4+ T lymphocytes in the primary and memory response to nonimmunogenic murine mammary adenocarcinoma made immunogenic by IL-2 gene. J. Immunol. 1992, 149, 3627–3635. [Google Scholar] [PubMed]
- Musiani, P.; Alione, A.; Modica, A.; Lollini, P.L.; Giovarelli, M.; Cavallo, F.; Belardelli, F.; Forni, G.; Modesti, A. Role of Neutrophils and Lymphocytes in Inhibition of a Mouse Mammary Adenocarcinoma Engineered to Release IL-2, IL-4, IL-7, IL-10, IFN-α, IFN-γ, and TNF-α. Lab. Investig. 1996, 74, 146–157. [Google Scholar]
- Meazza, R.; Marciano, S.; Sforzini, S.; Orengo, A.; Coppolecchia, M.; Musiani, P.; Ardizzoni, A.; Santi, L.; Azzarone, B.; Ferrini, S. Analysis of IL-2 receptor expression and of the biological effects of IL-2 gene transfection in small-cell lung cancer. Br. J. Cancer 1996, 74, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Girard, D.; Gosselin, J.; Heitz, D.; Paquin, R.; Beaulieu, A.D. Effects of interleukin-2 on gene expression in human neutrophils. Blood 1995, 86, 1170–1176. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gyorffy, S.; Lee, S.; Kwok, C.S. Effect of recombinant human interleukin 2 on neutrophil adherence to endothelial cells in vitro. Inflammation 1996, 20, 361–372. [Google Scholar] [CrossRef]
- Comen, E.; Wojnarowicz, P.; Seshan, V.E.; Shah, R.; Coker, C.; Norton, L.; Benezra, R. TNF is a key cytokine mediating neutrophil cytotoxic activity in breast cancer patients. NPJ Breast Cancer 2016, 2, 16009. [Google Scholar] [CrossRef]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.R.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nat. Cell Biol. 2015, 522, 349–353. [Google Scholar] [CrossRef]
- Mantovani, A. The Yin-Yang of Tumor-Associated Neutrophils. Cancer Cell 2009, 16, 173–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Smith, J.M.; Shen, Z.; Eriksson, M.; Sentman, C.; Wira, C.R. Inhibition of human neutrophil degranulation by transforming growth factor-β. Clin. Exp. Immunol. 2007, 149, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor Entrained Neutrophils Inhibit Seeding in the Premetastatic Lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, F.; Liu, X.; Chen, J.; Huang, S.; Wei, W.; Zou, Y.; Liu, X.; Deng, K.; Mo, S.; Chen, J.; et al. Anti-TGF-β attenuates tumor growth via polarization of tumor associated neutrophils towards an anti-tumor phenotype in colorectal cancer. J. Cancer 2020, 11, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Balazovich, K.J.; Fernandez, R.; Hinkovska-Galcheva, V.; Suchard, S.J.; Boxer, L.A. Transforming growth factor-β1 stimulates degranulation and oxidant release by adherent human neutrophils. J. Leukoc. Biol. 1996, 60, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming Growth Factor-β and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology 2019, 69, 222–236. [Google Scholar] [CrossRef] [Green Version]
- Guerin, M.V.; Regnier, F.; Feuillet, V.; Vimeux, L.; Weiss, J.M.; Bismuth, G.; Altan-Bonnet, G.; Guilbert, T.; Thoreau, M.; Finisguerra, V.; et al. TGFβ blocks IFNα/β release and tumor rejection in spontaneous mammary tumors. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pylaeva, E.; Lang, S.; Jablonska, J. The essential role of type I interferons in differentiation and activation of tumor-associated neutrophils. Front. Immunol. 2016, 7, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jablonska, J.; Leschner, S.; Westphal, K.; Lienenklaus, S.; Weiss, S. Neutrophils responsive to endogenous IFN-β regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 2010, 120, 1151–1164. [Google Scholar] [CrossRef]
- Bekisz, J.; Baron, S.; Balinsky, C.; Morrow, A.; Zoon, K.C. Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals 2010, 3, 994–1015. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.-Q.; Tao, N.; Dergay, A.; Moy, P.; Fawell, S.; Davis, A.; Wilson, J.M.; Barsoum, J. Interferon- gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 14411–14416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prantner, D.; Perkins, D.J.; Lai, W.; Williams, M.S.; Sharma, S.; Fitzgerald, K.A.; Vogel, S.N. 5,6-Dimethylxanthenone-4-acetic Acid (DMXAA) Activates Stimulator of Interferon Gene (STING)-dependent Innate Immune Pathways and Is Regulated by Mitochondrial Membrane Potential. J. Biol. Chem. 2012, 287, 39776–39788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkels, K.M.; Frondorf, K.; Gonzalez-Mejia, M.E.; Doseff, A.L.; Gomez-Cambronero, J. IL-8-induced neutrophil chemotaxis is mediated by Janus kinase 3 (JAK3). FEBS Lett. 2011, 585, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altstaedt, J.; Kirchner, H.; Rink, L. Cytokine production of neutrophils is limited to interleukin-8. Immunology 1996, 89, 563–568. [Google Scholar] [CrossRef] [PubMed]
- De Larco, J.E.; Wuertz, B.R.K.; Furcht, L.T. The Potential Role of Neutrophils in Promoting the Metastatic Phenotype of Tumors Releasing Interleukin-8. Clin. Cancer Res. 2004, 10, 4895–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, K.C.; Liu, L.-F.; Gupta, V.; Madireddi, S.; Keerthivasan, S.; Li, C.; Rishipathak, D.; Williams, P.; Kadel, E.E.; Koeppen, H.; et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 2020, 26, 693–698. [Google Scholar] [CrossRef] [PubMed]
- López-Lago, M.A.; Posner, S.; Thodima, V.J.; Molina, A.M.; Motzer, R.J.; Chaganti, R.S.K. Neutrophil chemokines secreted by tumor cells mount a lung antimetastatic response during renal cell carcinoma progression. Oncogene 2013, 32, 1752–1760. [Google Scholar] [CrossRef] [Green Version]
- Jablonska, J.; Wu, C.-F.; Andzinski, L.; Leschner, S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.-S.; Weber, S.; Gan, J.; Rakhmilevich, A.L.; Mahvi, D.M. Granulocyte-macrophage colony-stimulating factor (GM-CSF) secreted by cDNA-transfected tumor cells induces a more potent antitumor response than exogenous GM-CSF. Cancer Gene Ther. 1999. [Google Scholar] [CrossRef] [Green Version]
- Gale, R.P.; Zighelboim, J. Polymorphonuclear leukocytes in antibody-dependent cellular cytotoxicity. J. Immunol. 1975, 114, 1047–1051. [Google Scholar]
- Petroni, K.C.; Shen, L.; Guyre, P.M. Modulation of human polymorphonuclear leukocyte IgG Fc receptors and Fc receptor-mediated functions by IFN-gamma and glucocorticoids. J. Immunol. 1988, 140, 3467–3472. [Google Scholar] [PubMed]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; Van Houdt, M.; Treffers, L.W.; Van Rees, D.J.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep. 2018, 23, 3946–3959. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Shen, L.; Eicher, D.M.; Wewers, M.D.; Gill, J.K. Phagocytosis mediated by three distinct Fcγ receptor classes on human leukocytes. J. Exp. Med. 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golay, J.; Valgardsdottir, R.; Musaraj, G.; Giupponi, D.; Spinelli, O.; Introna, M. Human neutrophils express low levels of FcγRIIIA, which plays a role in PMN activation. Blood 2019, 133, 1395–1405. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcγRIIIB but induces neutrophil extracellular traps via FcγRIIA in vivo. Blood 2012, 120, 4421–4431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alevy, Y.G.; Tucker, J.; Naziruddin, B.; Mohanakumar, T. CD32C (Fcγ RIIC) mRNA expression and regulation. Mol. Immunol. 1993, 30, 775–782. [Google Scholar] [CrossRef]
- Su, K.; Yang, H.; Li, X.; Li, X.; Gibson, A.W.; Cafardi, J.M.; Zhou, T.; Edberg, J.C.; Kimberly, R.P. Expression Profile of FcγRIIb on Leukocytes and Its Dysregulation in Systemic Lupus Erythematosus. J. Immunol. 2007, 178, 3272–3280. [Google Scholar] [CrossRef] [Green Version]
- Treffers, L.W.; Van Houdt, M.; Bruggeman, C.W.; Heineke, M.H.; Zhao, X.W.; Van Der Heijden, J.; Nagelkerke, S.Q.; Verkuijlen, P.J.J.H.; Geissler, J.; Lissenberg-Thunnissen, S.; et al. FcγRIIIb Restricts Antibody-Dependent Destruction of Cancer Cells by Human Neutrophils. Front. Immunol. 2019. [Google Scholar] [CrossRef]
- Treffers, L.W.; Zhao, X.W.; Van Der Heijden, J.; Nagelkerke, S.Q.; Van Rees, D.J.; Gonzalez, P.; Geissler, J.; Verkuijlen, P.; Van Houdt, M.; De Boer, M.; et al. Genetic variation of human neutrophil Fcγ receptors and SIRPα in antibody-dependent cellular cytotoxicity towards cancer cells. Eur. J. Immunol. 2018, 48, 344–354. [Google Scholar] [CrossRef] [Green Version]
- Kerntke, C.; Nimmerjahn, F.; Biburger, M. There Is (Scientific) Strength in Numbers: A Comprehensive Quantitation of Fc Gamma Receptor Numbers on Human and Murine Peripheral Blood Leukocytes. Front. Immunol. 2020. [Google Scholar] [CrossRef]
- Kerst, J.J.; van de Winkel, J.G.; Evans, A.H.; de Haas, M.; Slaper-Cortenbach, I.C.; de Wit, T.P.; Borne, A.E.V.; van der Schoot, C.E.; van Oers, R.H. Granulocyte colony-stimulating factor induces hFc gamma RI (CD64 antigen)-positive neutrophils via an effect on myeloid precursor cells. Blood 1993, 81, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Gericke, G.H.; Ericson, S.G.; Pan, L.; Mills, L.E.; Guyre, P.M.; Ely, P. Mature polymorphonuclear leukocytes express high-affinity receptors for IgG (FcγRI) after stimulation with granulocyte colony-stimulating factor (G-CSF). J. Leukoc. Biol. 1995, 57, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Perussia, B.; Dayton, E.T.; Lazarus, R.; Fanning, V.; Trinchieri, G. Immune interferon induces the receptor for monomeric IgG1 on human monocytic and myeloid cells. J. Exp. Med. 1983, 158, 1092–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibeau, F.; Lopez-Crapez, E.; Di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frébourg, T.; Michel, P.; et al. Impact of FcγRIIa-FcγRIIIa Polymorphisms and KRAS Mutations on the Clinical Outcome of Patients with Metastatic Colorectal Cancer Treated with Cetuximab Plus Irinotecan. J. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.-K.; Levy, R. Two Immunoglobulin G Fragment C Receptor Polymorphisms Independently Predict Response to Rituximab in Patients with Follicular Lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G Fragment C Receptor Polymorphisms and Clinical Efficacy of Trastuzumab-Based Therapy in Patients With HER-2/neu–Positive Metastatic Breast Cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gordon, M.; Schultheis, A.M.; Yang, D.Y.; Nagashima, F.; Azuma, M.; Chang, H.-M.; Borucka, E.; Lurje, G.; Sherrod, A.E.; et al. FCGR2A and FCGR3A Polymorphisms Associated with Clinical Outcome of Epidermal Growth Factor Receptor–Expressing Metastatic Colorectal Cancer Patients Treated with Single-Agent Cetuximab. J. Clin. Oncol. 2007, 25, 3712–3718. [Google Scholar] [CrossRef] [PubMed]
- Shashidharamurthy, R.; Zhang, F.; Amano, A.; Kamat, A.; Panchanathan, R.; Ezekwudo, D.; Zhu, C.; Selvaraj, P. Dynamics of the Interaction of Human IgG Subtype Immune Complexes with Cells Expressing R and H Allelic Forms of a Low-Affinity Fcγ Receptor CD32A. J. Immunol. 2009, 183, 8216–8224. [Google Scholar] [CrossRef] [Green Version]
- Hurvitz, S.A.; Betting, D.J.; Stern, H.M.; Quinaux, E.; Stinson, J.; Seshagiri, S.; Zhao, Y.; Buyse, M.; Mackey, J.; Driga, A.; et al. Analysis of Fc Receptor IIIa and IIa Polymorphisms: Lack of Correlation with Outcome in Trastuzumab-Treated Breast Cancer Patients. Clin. Cancer Res. 2012, 18, 3478–3486. [Google Scholar] [CrossRef] [Green Version]
- Amigorena, S.; Bonnerot, C.; Choquet, D.; Hunziker, W.; Guillet, J.; Webster, P.; Sautes, C.; Mellman, I.; Fridman, W.H. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science 1992, 256, 1808–1812. [Google Scholar] [CrossRef]
- Ravetch, J.V. Fc receptors. Curr. Opin. Immunol. 1997, 9, 121–125. [Google Scholar] [CrossRef]
- Vidarsson, G.; Van De Winkel, J.G. Fc receptor and complement receptor-mediated phagocytosis in host defence. Curr. Opin. Infect. Dis. 1998, 11, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Golay, F.J.; Da Roit, L.; Bologna, C.; Ferrara, C.K.; Leusen, J.H.; Rambaldi, A.; Martino, I. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 2013, 122, 3482–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derer, S.; Glorius, P.; Schlaeth, M.; Lohse, S.; Klausz, K.; Muchhal, U.; DesJarlais, J.R.; Humpe, A.; Valerius, T.; Peipp, M. Increasing FcγRIIa affinity of an FcγRIII-optimized anti-EGFR antibody restores neutrophil-mediated cytotoxicity. mAbs 2014, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, G.; Treffers, L.; Plomp, R.; Bentlage, A.E.H.; De Boer, M.; Koeleman, C.A.M.; Lissenberg-Thunnissen, S.N.; Visser, R.; Brouwer, M.; Mok, J.Y.; et al. Decoding the Human Immunoglobulin G-Glycan Repertoire Reveals a Spectrum of Fc-Receptor- and Complement-Mediated-Effector Activities. Front. Immunol. 2017, 8, 877. [Google Scholar] [CrossRef] [PubMed]
- Subedi, G.P.; Hanson, Q.M.; Barb, A.W. Restricted Motion of the Conserved Immunoglobulin G1 N-Glycan Is Essential for Efficient FcγRIIIa Binding. Structure 2014, 22, 1478–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, C.; Grau, S.; Jäger, C.; Sondermann, P.; Brünker, P.; Waldhauer, I.; Hennig, M.; Ruf, A.; Rufer, A.C.; Stihle, M.; et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between Fc RIII and antibodies lacking core fucose. Proc. Natl. Acad. Sci. USA 2011, 108, 12669–12674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peipp, M.; Van Bueren, J.J.L.; Schneider-Merck, T.; Bleeker, W.W.K.; DeChant, M.; Beyer, T.; Repp, R.; Van Berkel, P.H.C.; Vink, T.; Van De Winkel, J.G.J.; et al. Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood 2008, 112, 2390–2399. [Google Scholar] [CrossRef]
- Shibata-Koyama, M.; Iida, S.; Misaka, H.; Mori, K.; Yano, K.; Shitara, K.; Satoh, M. Nonfucosylated rituximab potentiates human neutrophil phagocytosis through its high binding for FcγRIIIb and MHC class II expression on the phagocytotic neutrophils. Exp. Hematol. 2009, 37, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef]
- Vitolo, U.; Trněný, M.; Belada, D.; Burke, J.M.; Carella, A.M.; Chua, N.; Abrisqueta, P.; Demeter, J.; Flinn, I.; Hong, X.; et al. Obinutuzumab or Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Previously Untreated Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2017, 35, 3529–3537. [Google Scholar] [CrossRef] [PubMed]
- Marois, L.; Paré, G.; Vaillancourt, M.; Rollet-Labelle, E.; Naccache, P.H. FcγRIIIb Triggers Raft-dependent Calcium Influx in IgG-mediated Responses in Human Neutrophils. J. Biol. Chem. 2011, 286, 3509–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wirthmueller, U.; Ravetch, J.V. Reconstitution of human FcγRIII cell type specificity in transgenic mice. J. Exp. Med. 1996, 183, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Meknache, N.; Jönsson, F.; Laurent, J.; Guinnepain, M.-T.; Daëron, M. Human Basophils Express the Glycosylphosphatidylinositol-Anchored Low-Affinity IgG Receptor FcγRIIIB (CD16B). J. Immunol. 2009, 182, 2542–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Parrinello, N.L.; Simeon, V.; Puglisi, F.; La Cava, P.; Bellofiore, C.; Giallongo, C.; Camiolo, G.; D’Auria, F.; Grieco, V.; et al. High-density neutrophils in MGUS and multiple myeloma are dysfunctional and immune-suppressive due to increased STAT3 downstream signaling. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Tridandapani, S.; Wardrop, R.; Baran, C.P.; Wang, Y.; Opalek, J.M.; Caligiuri, M.A.; Marsh, C.B. TGF-β1 Suppresses Myeloid Fcγ Receptor Function by Regulating the Expression and Function of the Common γ-Subunit. J. Immunol. 2003, 170, 4572–4577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wu, J.; Newton, R.; Bahaie, N.S.; Long, C.; Walcheck, B. ADAM17 cleaves CD16b (FcγRIIIb) in human neutrophils. Biochimica Biophysica Acta (BBA) Bioenerg. 2013, 1833, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, S.; Srivastava, R.M.; Concha-Benavente, F.; Ferrone, S.; Garcia-Bates, T.M.; Li, J.; Ferris, R.L. Anti-EGFR Targeted Monoclonal Antibody Isotype Influences Antitumor Cellular Immunity in Head and Neck Cancer Patients. Clin. Cancer Res. 2016, 22, 5229–5237. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.E.; Selvaraj, P.; Zhu, C. Concurrent Binding to Multiple Ligands: Kinetic Rates of CD16b for Membrane-Bound IgG1 and IgG. Biophys. J. 2000, 79, 1858–1866. [Google Scholar] [CrossRef] [Green Version]
- Brandsma, A.M.; Bondza, S.; Evers, M.; Koutstaal, R.; Nederend, M.; Jansen, J.H.M.; Rösner, T.; Valerius, T.; Leusen, J.H.W.; Broeke, T.T. Potent Fc Receptor Signaling by IgA Leads to Superior Killing of Cancer Cells by Neutrophils Compared to IgG. Front. Immunol. 2019, 10, 704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, R.C.; Kubagawa, H.; Cooper, M.D. Cellular distribution, regulation, and biochemical nature of an Fc alpha receptor in humans. J. Exp. Med. 1990, 171, 597–613. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.A. The structure and function of human IgA. Biochem. J. 1990, 271, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangione, B.; Wolfenstein-Todel, C. Partial Duplication in the “Hinge” Region of IgA1 Myeloma Proteins. Proc. Natl. Acad. Sci. USA 1972, 69, 3673–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattu, T.S.; Pleass, R.J.; Willis, A.C.; Kilian, M.; Wormald, M.R.; Lellouch, A.C.; Rudd, P.M.; Woof, J.M.; Dwek, R.A. The Glycosylation and Structure of Human Serum IgA1, Fab, and Fc Regions and the Role ofN-Glycosylation on Fcα Receptor Interactions. J. Biol. Chem. 1998, 273, 2260–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göritzer, K.; Turupcu, A.; Maresch, D.; Novak, J.; Altmann, F.; Oostenbrink, C.; Obinger, C.; Strasser, R. Distinct Fcα receptor N-glycans modulate the binding affinity to immunoglobulin A (IgA) antibodies. J. Biol. Chem. 2019, 294, 13995–14008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, U.; Koeleman, C.A.; Sokolova, M.V.; Bang, H.; Kleyer, A.; Rech, J.; Unterweger, H.; Schicht, M.; Garreis, F.; Hahn, J.; et al. IgA subclasses have different effector functions associated with distinct glycosylation profiles. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- DeChant, M.; Beyer, T.; Schneider-Merck, T.; Weisner, W.; Peipp, M.; Van De Winkel, J.G.J.; Valerius, T. Effector mechanisms of recombinant IgA antibodies against epidermal growth factor receptor. J. Immunol. 2007, 179, 2936–2943. [Google Scholar] [CrossRef] [Green Version]
- Bogart, J.A.; Ungureanu, C.; Shihadeh, E.; Chung, C.T.; King, G.A.; Ryu, S.; Kent, C.; Winfield, J.A. Resection and Permanent I-125 Brachytherapy Without Whole Brain Irradiation for Solitary Brain Metastasis from Non-small Cell Lung Carcinoma. J. Neuro-Oncology 1999, 44, 53–57. [Google Scholar] [CrossRef]
- Stockmeyer, B.; Elsässer, D.; DeChant, M.; Repp, R.; Gramatzki, M.; Glennie, M.J.; Van De Winkel, J.G.; Valerius, T. Mechanisms of G-CSF- or GM-CSF-stimulated tumor cell killing by Fc receptor-directed bispecific antibodies. J. Immunol. Methods 2001, 248, 103–111. [Google Scholar] [CrossRef]
- Boross, P.; Lohse, S.; Nederend, M.; Jansen, J.H.M.; Van Tetering, G.; DeChant, M.; Peipp, M.; Royle, L.; Liew, L.P.; Boon, L.; et al. Ig A EGFR antibodies mediate tumour killing in vivo. EMBO Mol. Med. 2013, 5, 1213–1226. [Google Scholar] [CrossRef]
- Sundarapandiyan, K.; Keler, T.; Behnke, D.; Engert, A.; Barth, S.; Matthey, B.; Deo, Y.M.; Graziano, R.F. Bispecific antibody-mediated destruction of Hodgkin’s lymphoma cells. J. Immunol. Methods 2001, 248, 113–123. [Google Scholar] [CrossRef]
- DeChant, M.; Vidarsson, G.; Stockmeyer, B.; Repp, R.; Glennie, M.J.; Gramatzki, M.; Van De Winkel, J.G.; Valerius, T. Chimeric IgA antibodies against HLA class II effectively trigger lymphoma cell killing. Blood 2002, 100, 4574–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascal, V.; Laffleur, B.; Debin, A.; Cuvillier, A.; Van Egmond, M.; Drocourt, D.; Imbertie, L.; Pangault, C.; Tarte, K.; Tiraby, G.; et al. Anti-CD20 IgA can protect mice against lymphoma development: Evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica 2012, 97, 1686–1694. [Google Scholar] [CrossRef] [PubMed]
- Lohse, S.; Loew, S.; Kretschmer, A.; Jansen, J.H.M.; Meyer, S.; Broeke, T.T.; Rösner, T.; DeChant, M.; Derer, S.; Klausz, K.; et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Br. J. Haematol. 2018, 181, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Otten, M.A.; Rudolph, E.; DeChant, M.; Tuk, C.W.; Reijmers, R.M.; Beelen, R.H.J.; Van De Winkel, J.G.J.; Van Egmond, M. Immature Neutrophils Mediate Tumor Cell Killing via IgA but Not IgG Fc Receptors. J. Immunol. 2005, 174, 5472–5480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazengera, R.L.; Kerr, M.A. The specificity of the IgA receptor purified from human neutrophils. Biochem. J. 1990, 272, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Herr, A.B.; White, C.L.; Milburn, C.; Wu, C.; Bjorkman, P.J. Bivalent Binding of IgA1 to FcαRI Suggests a Mechanism for Cytokine Activation of IgA Phagocytosis. J. Mol. Biol. 2003, 327, 645–657. [Google Scholar] [CrossRef]
- Morell, A.; Skvaril, F.; Noseda, G.; Brandun, S. Metabolic properties of human IgA subclasses. Clin. Exp. Immunol. 1973, 13, 521–528. [Google Scholar]
- Lohse, S.; Meyer, S.; Meulenbroek, L.A.; Jansen, J.M.; Nederend, M.; Kretschmer, A.; Klausz, K.; Möginger, U.; Derer, S.; Rösner, T.; et al. An Anti-EGFR IgA That Displays Improved Pharmacokinetics and Myeloid Effector Cell Engagement In Vivo. Cancer Res. 2016, 76, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Meyer, S.; Nederend, M.; Jansen, J.M.; Reiding, K.R.; Jacobino, S.R.; Meeldijk, J.; Bovenschen, N.; Wuhrer, M.; Valerius, T.; Ubink, R.; et al. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. mAbs 2015, 8, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Egmond, M.; Van Vuuren, A.H.; Van De Winkel, J.G. The human Fc receptor for IgA (FcαRI, CD89) on transgenic peritoneal macrophages triggers phagocytosis and tumor cell lysis. Immunol. Lett. 1999, 68, 83–87. [Google Scholar] [CrossRef]
- Brandsma, A.M.; Broeke, T.T.; Nederend, M.; Meulenbroek, L.A.; Van Tetering, G.; Meyer, S.; Jansen, J.H.M.; Buitrago, M.A.B.; Nagelkerke, S.Q.; Németh, I.; et al. Simultaneous Targeting of Fc Rs and Fc RI Enhances Tumor Cell Killing. Cancer Immunol. Res. 2015, 3, 1316–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrok, M.J.; Luheshi, N.M.; Beyaz, N.; Davies, G.C.; Legg, J.W.; Wu, H.; Dall’Acqua, W.F.; Tsui, P. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcαRI (CD89) binding. mAbs 2015, 7, 743–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Xu, L.; Tao, F.; Xie, K.; Wu, Z.; Li, Y.; Li, J.; Chen, K.; Pi, C.; Mendelsohn, A.; et al. Simultaneous exposure to FcγR and FcαR on monocytes and macrophages enhances antitumor activity in vivo. Oncotarget 2017, 8, 39356–39366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huls, G.; Heijnen, I.A.; Cuomo, E.; Van Der Linden, J.; Boel, E.; Van De Winkel, J.G.; Logtenberg, T. Antitumor immune effector mechanisms recruited by phage display-derived fully human IgG1 and IgA1 monoclonal antibodies. Cancer Res. 1999, 59, 5778–5784. [Google Scholar] [PubMed]
- Pasquier, B.; Launay, P.; Kanamaru, Y.; Moura, I.C.; Pfirsch, S.; Ruffié, C.; Hénin, D.; Benhamou, M.; Pretolani, M.; Blank, U.; et al. Identification of FcαRI as an Inhibitory Receptor that Controls Inflammation. Immunity 2005, 22, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Xu, L.; Pi, C.; Yin, Y.; Xie, K.; Tao, F.; Li, R.; Gu, H.; Fang, J. CD89-mediated recruitment of macrophages via a bispecific antibody enhances anti-tumor efficacy. OncoImmunology 2017, 7, e1380142. [Google Scholar] [CrossRef] [Green Version]
- Stockmeyer, B.; DeChant, M.; Van Egmond, M.; Tutt, A.L.; Sundarapandiyan, K.; Graziano, R.F.; Repp, R.; Kalden, J.R.; Gramatzki, M.; Glennie, M.J.; et al. Triggering FCα-Receptor I (CD89) Recruits Neutrophils as Effector Cells for CD20-Directed Antibody Therapy. J. Immunol. 2000, 165, 5954–5961. [Google Scholar] [CrossRef] [Green Version]
- Guettinger, Y.; Barbin, K.; Peipp, M.; Bruenke, J.; Dechant, M.; Horner, H.; Thierschmidt, D.; Valerius, T.; Repp, R.; Fey, G.H.; et al. A Recombinant Bispecific Single-Chain Fragment Variable Specific for HLA Class II and FcαRI (CD89) Recruits Polymorphonuclear Neutrophils for Efficient Lysis of Malignant B Lymphoid Cells. J. Immunol. 2010, 184, 1210–1217. [Google Scholar] [CrossRef] [Green Version]
- Van Der Kolk, L.E.; De Haas, M.; Grillo-López, A.J.; Baars, J.W.; Van Oers, M.H.J. Analysis of CD20-dependent cellular cytotoxicity by G-CSF-stimulated neutrophils. Leukemia 2002, 16, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, E.M.; Sycz, G.; Arriaga, J.M.; Barrio, M.M.; Von Euw, E.M.; Morales, S.B.; González, M.; Mordoh, J.; Bianchini, M. Cetuximab-mediated cellular cytotoxicity is inhibited by HLA-E membrane expression in colon cancer cells. Innate Immun. 2009, 15, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Derer, S.; Bauer, P.; Lohse, S.; Scheel, A.H.; Berger, S.; Kellner, C.; Peipp, M.; Valerius, T. Impact of Epidermal Growth Factor Receptor (EGFR) Cell Surface Expression Levels on Effector Mechanisms of EGFR Antibodies. J. Immunol. 2012, 189, 5230–5239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.-K.; Sui, J.; Geng, S.; Muvaffak, A.; Bai, M.; Fuhlbrigge, R.C.; Lo, A.; Yammanuru, A.; Hubbard, L.; Sheehan, J.; et al. Humanization of an Anti-CCR4 Antibody That Kills Cutaneous T-Cell Lymphoma Cells and Abrogates Suppression by T-Regulatory Cells. Mol. Cancer Ther. 2012, 11, 2451–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Turner, M.J.; Shields, J.; Gale, M.S.; Hutto, E.; Roberts, B.L.; Siders, W.M.; Kaplan, J.M. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology 2009, 128, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Watanabe, R.; Teague, J.E.; Schlapbach, C.; Tawa, M.C.; Adams, N.; Dorosario, A.A.; Chaney, K.S.; Cutler, C.S.; Leboeuf, N.R.; et al. Skin Effector Memory T Cells Do Not Recirculate and Provide Immune Protection in Alemtuzumab-Treated CTCL Patients. Sci. Transl. Med. 2012, 4, 117ra7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siders, W.M.; Shields, J.; Garron, C.; Hu, Y.; Boutin, P.; Shankara, S.; Weber, W.; Roberts, B.; Kaplan, J.M. Involvement of neutrophils and natural killer cells in the anti-tumor activity of alemtuzumab in xenograft tumor models. Leuk. Lymphoma 2010, 51, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Ilizaliturri, F.J.; Jupudy, V.; Ostberg, J.; Oflazoglu, E.; Huberman, A.; Repasky, E.; Czuczman, M.S. Neutrophils contribute to the biological antitumor activity of rituximab in a non-Hodgkin’s lymphoma severe combined immunodeficiency mouse model. Clin. Cancer Res. 2003, 9, 5866–5873. [Google Scholar]
- Hong, F.; Yan, J.; Baran, J.; Allendorf, D.J.; Hansen, R.D.; Ostroff, G.R.; Xing, P.X.; Cheung, N.-K.V.; Ross, G.D. Mechanism by Which Orally Administered β-1,3-Glucans Enhance the Tumoricidal Activity of Antitumor Monoclonal Antibodies in Murine Tumor Models. J. Immunol. 2004, 173, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Allendorf, D.J.; Hansen, R.; Marroquin, J.; Cramer, D.E.; Harris, C.L.; Yan, J. Combined yeast β-glucan and antitumor monoclonal antibody therapy requires C5a-mediated neutrophil chemotaxis via regulation of decay-accelerating factor CD. Cancer Res. 2007, 67, 7421–7430. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Hansen, R.D.; Yan, J.; Allendorf, D.J.; Baran, J.T.; Ostroff, G.R.; Ross, G.D. β-Glucan Functions as an Adjuvant for Monoclonal Antibody Immunotherapy by Recruiting Tumoricidal Granulocytes as Killer Cells. Cancer Res. 2003, 63, 9023–9031. [Google Scholar] [PubMed]
- Allendorf, D.J.; Yan, J.; Ross, G.D.; Hansen, R.D.; Baran, J.; Subbarao, K.; Wang, L.; Haribabu, B. C5a-Mediated Leukotriene B4-Amplified Neutrophil Chemotaxis Is Essential in Tumor Immunotherapy Facilitated by Anti-Tumor Monoclonal Antibody and β-Glucan. J. Immunol. 2005, 174, 7050–7056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishelson, Z.; Donin, N.; Jurianz, K.; Ziporen, L.; Schultz, S.; Kirschfink, M. Complement resistance of carcinoma cells Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin. Exp. Imunnol. 2003, 131, 254–263. [Google Scholar]
- Moore, G.L.; Chen, H.; Karki, S.; Lazar, G.A. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. mAbs 2010, 2, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microbes Infect. 2003, 5, 1299–1306. [Google Scholar] [CrossRef]
- Ring, N.G.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.-P.; George, B.M.; Lietzenmayer, M.; McKenna, K.M.; Naik, T.J.; Mccarty, A.; et al. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [Green Version]
- Bologna, L.; Gotti, E.; Da Roit, F.; Intermesoli, T.; Rambaldi, A.; Introna, M.; Golay, J. Ofatumumab Is More Efficient than Rituximab in Lysing B Chronic Lymphocytic Leukemia Cells in Whole Blood and in Combination with Chemotherapy. J. Immunol. 2013, 190, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Cornet, S.; Mathé, D.; Chettab, K.; Evesque, A.; Matera, E.-L.; Trédan, O.; Dumontet, C. Pegfilgrastim Enhances the Antitumor Effect of Therapeutic Monoclonal Antibodies. Mol. Cancer Ther. 2016, 15, 1238–1247. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, B.; Singha, A.K.; Maiti, D. Phagocytic activity of neutrophil is induced by granulocyte colony stimulating factor and interleukin-15 in leukemic animal model. J. Cell. Immunother. 2016, 2, 52–57. [Google Scholar] [CrossRef]
- Rybka, J.; Butrym, A.; Wróbel, T.; Jaźwiec, B.; Bogucka-Fedorczuk, A.; Poreba, R.; Kuliczkowski, K. The Expression of Toll-Like Receptors in Patients with B-Cell Chronic Lymphocytic Leukemia. Archivum Immunologiae Therapiae Experimentalis 2016, 64 (Suppl. S1), 147–150. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Cuaxospa, M.; Contreras-Ramos, A.; Pérez-Figueroa, E.; Medina-Sansón, A.; Jiménez-Hernández, E.; Torres-Nava, J.R.; Rojas, E.; Maldonado-Bernal, C. Low expression of Toll-like receptors in peripheral blood mononuclear cells of pediatric patients with acute lymphoblastic leukemia. Int. J. Oncol. 2016, 49, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Kuninaka, N.; Kurata, M.; Yamamoto, K.; Suzuki, S.; Umeda, S.; Kirimura, S.; Arai, A.; Nakagawa, Y.; Suzuki, K.; Kitagawa, M. Expression of Toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia. Exp. Mol. Pathol. 2010, 88, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Van Der Kolk, L.E.; Grillo-López, A.J.; Baars, J.W.; Van Oers, M.H.J. Treatment of relapsed B-cell non-Hodgkin’s lymphoma with a combination of chimeric anti-CD20 monoclonal antibodies (rituximab) and G-CSF: Final report on safety and efficacy. Leukemia 2003, 17, 1658–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerchione, C.; De Renzo, A.; Di Perna, M.; Della Pepa, R.; Pugliese, N.; Catalano, L.; Pane, F.; Picardi, M. Pegfilgrastim in primary prophylaxis of febrile neutropenia following frontline bendamustine plus rituximab treatment in patients with indolent non-Hodgkin lymphoma: A single center, real-life experience. Support. Care Cancer 2017, 25, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wu, B.; Hu, X.; Liu, J.; Zhang, T.; Li, F.; Sun, B.; Cai, L.; Li, X.; Chen, Z.; et al. A randomized multicenter phase II trial of mecapegfilgrastim single administration versus granulocyte colony-stimulating growth factor on treating chemotherapy-induced neutropenia in breast cancer patients. Ann. Transl. Med. 2019, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- Clemons, M.; Fergusson, D.; Simos, D.; Mates, M.; Robinson, A.; Califaretti, N.; Zibdawi, L.; Bahl, M.; Raphael, J.; Ibrahim, M.; et al. A multicentre, randomised trial comparing schedules of G-CSF (filgrastim) administration for primary prophylaxis of chemotherapy-induced febrile neutropenia in early stage breast cancer. Ann. Oncol. 2020, 31, 951–957. [Google Scholar] [CrossRef]
- Doerschuk, C.M.; Beyers, N.; Coxson, H.O.; Wiggs, B.; Hogg, J.C. Comparison of neutrophil and capillary diameters and their relation to neutrophil sequestration in the lung. J. Appl. Physiol. 1993, 74, 3040–3045. [Google Scholar] [CrossRef]
- Laget, S.; Broncy, L.; Hormigos, K.; Dhingra, D.M.; BenMohamed, F.; Capiod, T.; Osteras, M.; Farinelli, L.; Jackson, S.; Paterlini-Bréchot, P. Technical insights into highly sensitive isolation and molecular characterization of fixed and live circulating tumor cells for early detection of tumor invasion. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Valgardsdottir, R.; Cattaneo, I.; Klein, C.; Introna, M.; Figliuzzi, M.; Golay, J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti-CD20 antibodies. Blood 2017, 129, 2636–2644. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.A.; Goldenberg, D.M.; Michel, R.; Rossi, D.L.; Wallace, D.J.; Chang, C.-H. Trogocytosis of multiple B-cell surface markers by CD22 targeting with epratuzumab. Blood 2013, 122, 3020–3029. [Google Scholar] [CrossRef] [Green Version]
- Strizova, Z.; Vachtenheim, J.; Bartunkova, J. The potential role of neutrophil trogocytosis and G-CSF in the loss of HER2 expression. Breast Cancer Res. Treat. 2019, 178, 247–248. [Google Scholar] [CrossRef]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; Van Kessel, B.; Van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.; Zweegman, S.; Van Meerloo, J.; Musters, R.J.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef] [Green Version]
- Horner, H.; Frank, C.; DeChant, C.; Repp, R.; Glennie, M.; Herrmann, M.; Stockmeyer, B. Intimate cell conjugate formation and exchange of membrane lipids precede apoptosis induction in target cells during antibody-dependent, granulocyte-mediated cytotoxicity. J. Immunol. 2007, 179, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zhang, L.; Yang, L.; Li, H.; Li, R.; Yu, J.; Yang, L.; Wei, F.; Yan, C.; Sun, Q.; et al. Anti-CD47 Antibody as a Targeted Therapeutic Agent for Human Lung Cancer and Cancer Stem Cells. Front. Immunol. 2017, 8, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Shi, X.; Chen, C.; He, H.; Liu, L.; Wu, J.; Yan, H. High expression of CD47 in triple negative breast cancer is associated with epithelial-mesenchymal transition and poor prognosis. Oncol. Lett. 2019, 18, 3249–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.W.; Van Beek, E.M.; Schornagel, K.; Van Der Maaden, H.; Van Houdt, M.; Otten, M.A.; Finetti, P.; Van Egmond, M.; Matozaki, T.; Kraal, G.; et al. CD47-signal regulatory protein-(SIRP) interactions form a barrier for antibody-mediated tumor cell destruction. Proc. Natl. Acad. Sci. USA 2011, 108, 18342–18347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwstra, R.; He, Y.; De Boer, J.; Kooistra, H.; Cendrowicz, E.; Fehrmann, R.S.; Ammatuna, E.; Zu Eulenburg, C.; Nijland, M.; Huls, G.; et al. CD47 Expression Defines Efficacy of Rituximab with CHOP in Non–Germinal Center B-cell (Non-GCB) Diffuse Large B-cell Lymphoma Patients (DLBCL), but Not in GCB DLBCL. Cancer Immunol. Res. 2019, 7, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Voets, E.; Paradé, M.; Hulsik, D.L.; Spijkers, S.; Janssen, W.; Rens, J.; Reinieren-Beeren, I.; Tillaart, G.V.D.; Van Duijnhoven, S.; Driessen, L.; et al. Functional characterization of the selective pan-allele anti-SIRPα antibody ADU-1805 that blocks the SIRPα–CD47 innate immune checkpoint. J. Immunother. Cancer 2019, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Gozlan, Y.M.; Hilgendorf, S.; Aronin, A.; Sagiv, Y.; Ben-Gigi-Tamir, L.; Amsili, S.; Tamir, A.; Pecker, I.; Greenwald, S.; Chajut, A.; et al. Abstract A076: DSP107—A novel SIRPα-4-1BBL dual signaling protein (DSP) for cancer immunotherapy. In Proceedings of the Fourth CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference: Translating Science into Survival, New York, NY, USA, 30 September–3 October 2018. [Google Scholar] [CrossRef]
- Treffers, L.W.; Broeke, T.T.; Rösner, T.; Jansen, J.H.M.; Van Houdt, M.; Kahle, S.; Schornagel, K.; Verkuijlen, P.J.; Prins, J.M.; Franke, K.; et al. IgA-Mediated Killing of Tumor Cells by Neutrophils Is Enhanced by CD47–SIRPα Checkpoint Inhibition. Cancer Immunol. Res. 2020, 8, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Merlin, D.; Burst, S.L.; Pochet, M.; Madara, J.L.; Parkos, C.A. The Role of CD47 in Neutrophil Transmigration. J. Biol. Chem. 2001, 276, 40156–40166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Bühring, H.-J.; Zen, K.; Burst, S.L.; Schnell, F.J.; Williams, I.R.; Parkos, C.A. Signal Regulatory Protein (SIRPα), a Cellular Ligand for CD47, Regulates Neutrophil Transmigration. J. Biol. Chem. 2002, 277, 10028–10036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, D.; Lindberg, F.P.; Gamble, J.R.; Brown, E.J.; Vadas, M.A. Transendothelial migration of neutrophils involves integrin-associated protein (CD47). Proc. Natl. Acad. Sci. USA 1995, 92, 3978–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bommel, P.E.; He, Y.; Schepel, I.; Hendriks, M.A.J.M.; Wiersma, V.R.; Van Ginkel, R.J.; Van Meerten, T.; Ammatuna, E.; Huls, G.; Samplonius, D.F.; et al. CD20-selective inhibition of CD47-SIRPα “don’t eat me” signaling with a bispecific antibody-derivative enhances the anticancer activity of daratumumab, alemtuzumab and obinutuzumab. OncoImmunology 2017, 7, e1386361. [Google Scholar] [CrossRef] [Green Version]
- Piccione, E.C.; Juarez, S.; Liu, J.; Tseng, S.; Ryan, C.E.; Narayanan, C.; Wang, L.; Weiskopf, K.; Majeti, R. A bispecific antibody targeting CD47 and CD20 selectively binds and eliminates dual antigen expressing lymphoma cells. mAbs 2015, 7, 946–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dheilly, E.; Moine, V.; Broyer, L.; Salgado-Pires, S.; Johnson, Z.; Papaioannou, A.; Cons, L.; Calloud, S.; Majocchi, S.; Nelson, R.; et al. Selective Blockade of the Ubiquitous Checkpoint Receptor CD47 Is Enabled by Dual-Targeting Bispecific Antibodies. Mol. Ther. 2017, 25, 523–533. [Google Scholar] [CrossRef] [Green Version]
- Hatterer, E.; Barba, L.; Noraz, N.; Daubeuf, B.; Aubry-Lachainaye, J.-P.; Von Der Weid, B.; Richard, F.; Kosco-Vilbois, M.; Ferlin, W.; Shang, L.; et al. Co-engaging CD47 and CD19 with a bispecific antibody abrogates B-cell receptor/CD19 association leading to impaired B-cell proliferation. mAbs 2019, 11, 322–334. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Chai, Y.; Duan, X.; Bi, X.; Huang, Q.; Wang, Q.; Tan, S.; Gao, G.F.; Zhu, J.; Yan, J. The identification of a CD47-blocking “hotspot” and design of a CD47/PD-L1 dual-specific antibody with limited hemagglutination. Signal Transduct. Target. Ther. 2020, 5, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Guo, H.; Xu, J.; Qin, T.; Guo, Q.; Gu, N.; Zhang, D.; Qian, W.; Dai, J.; Hou, S.; et al. Elimination of tumor by CD47/PD-L1 dual-targeting fusion protein that engages innate and adaptive immune responses. mAbs 2018, 10, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, L.; Ren, Z.; Yang, K.; Xu, H.; Luan, Y.; Fu, K.; Guo, J.; Peng, H.; Zhu, M.; et al. Dual Targeting of Innate and Adaptive Checkpoints on Tumor Cells Limits Immune Evasion. Cell Rep. 2018, 24, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Bouwstra, R.; Wiersma, V.R.; De Jong, M.; Lourens, H.J.; Fehrmann, R.; De Bruyn, M.; Ammatuna, E.; Huls, G.; Van Meerten, T.; et al. Cancer cell-expressed SLAMF7 is not required for CD47-mediated phagocytosis. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizcano, A.; Secundino, I.; Döhrmann, S.; Corriden, R.; Rohena, C.; Diaz, S.; Ghosh, P.; Deng, L.; Nizet, V.; Varki, A. Erythrocyte sialoglycoproteins engage Siglec-9 on neutrophils to suppress activation. Blood 2017, 129, 3100–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Läubli, H.; Pearce, O.M.T.; Schwarz, F.; Siddiqui, S.S.; Deng, L.; Stanczak, M.A.; Deng, L.; Verhagen, A.; Secrest, P.; Lusk, C.; et al. Engagement of myelomonocytic Siglecs by tumor-associated ligands modulates the innate immune response to cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 14211–14216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, S.; Akita, K.; Ishida, A.; Mori, Y.; Toda, M.; Inoue, M.; Ohta, M.; Yashiro, M.; Sawada, T.; Hirakawa, K.; et al. Binding of the Sialic Acid-binding Lectin, Siglec-9, to the Membrane Mucin, MUC1, Induces Recruitment of β-Catenin and Subsequent Cell Growth. J. Biol. Chem. 2013, 288, 31842–31852. [Google Scholar] [CrossRef] [Green Version]
- Stanczak, M.A.; Siddiqui, S.S.; Trefny, M.P.; Thommen, D.S.; Boligan, K.F.; Von Gunten, S.; Tzankov, A.; Tietze, L.; Lardinois, D.; Heinzelmann-Schwarz, V.; et al. Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J. Clin. Investig. 2018, 128, 4912–4923. [Google Scholar] [CrossRef] [Green Version]
- Haas, Q.; Boligan, K.F.; Jandus, C.; Schneider, C.; Simillion, C.; Stanczak, M.A.; Haubitz, M.; Jafari, S.M.S.; Zippelius, A.; Baerlocher, G.M.; et al. Siglec-9 Regulates an Effector Memory CD8+ T-cell Subset That Congregates in the Melanoma Tumor Microenvironment. Cancer Immunol. Res. 2019, 7, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients with Advanced Cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Johnson, L.D.S.; Banerjee, S.; Kruglov, O.; Viller, N.N.; Horwitz, S.M.; Lesokhin, A.; Zain, J.; Querfeld, C.; Chen, R.; Okada, C.; et al. Targeting CD47 in Sézary syndrome with SIRPαFc. Blood Adv. 2019, 3, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radsak, M.; Iking-Konert, C.; Stegmaier, S.; Andrassy, K.; Hansch, G.M. Polymorphonuclear neutrophils as accessory cells for T-cell activation: Major histocompatibility complex class II restricted antigen-dependent induction of T-cell proliferation. Immunology 2000, 101, 521–530. [Google Scholar] [CrossRef]
- Fanger, N.A.; Liu, C.; Guyre, P.M.; Wardwell, K.; O’Neil, J.; Guo, T.L.; Christian, T.P.; Mudzinski, S.P.; Gosselin, E.J. Activation of Human T Cells by Major Histocompatability Complex Class II Expressing Neutrophils: Proliferation in the Presence of Superantigen, But Not Tetanus Toxoid. Blood 1997, 89, 4128–4135. [Google Scholar] [CrossRef] [PubMed]
- Potter, N.S.; Harding, C.V. Neutrophils Process Exogenous Bacteria Via an Alternate Class I MHC Processing Pathway for Presentation of Peptides to T Lymphocytes. J. Immunol. 2001, 167, 2538–2546. [Google Scholar] [CrossRef] [PubMed]
- Culshaw, S.; Millington, O.R.; Brewer, J.M.; McInnes, I.B. Murine neutrophils present Class II restricted antigen. Immunol. Lett. 2008, 118, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauvillain, C.; Delneste, Y.; Scotet, M.; Peres, A.; Gascan, H.; Guermonprez, P.; Barnaba, V.; Jeannin, P. Neutrophils efficiently cross-prime naive T cells in vivo. Blood 2007, 110, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Burdick, M.D.; Mehrad, B. Neutrophils Mediate Maturation and Efflux of Lung Dendritic Cells in Response to Aspergillus fumigatus Germ Tubes. Infect. Immun. 2012, 80, 1759–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennouna, S.; Bliss, S.K.; Curiel, T.J.; Denkers, E.Y. Cross-Talk in the Innate Immune System: Neutrophils Instruct Recruitment and Activation of Dendritic Cells during Microbial Infection. J. Immunol. 2003, 171, 6052–6058. [Google Scholar] [CrossRef] [Green Version]
- Morel, C.; Badell, E.; Abadie, V.; Robledo, M.; Setterblad, N.; Gluckman, J.C.; Gicquel, B.; Boudaly, S.; Winter, N. Mycobacterium bovis BCG-infected neutrophils and dendritic cells cooperate to induce specific T cell responses in humans and mice. Eur. J. Immunol. 2008, 38, 437–447. [Google Scholar] [CrossRef]
- Megiovanni, A.M.; Sanchez, F.; Robledo-Sarmiento, M.; Morel, C.; Gluckman, J.C.; Boudaly, S. Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: A new link between leukocytes upstream of T lymphocytes. J. Leukoc. Biol. 2006, 79, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Van Gisbergen, K.P.J.M.; Sanchez-Hernandez, M.; Geijtenbeek, T.B.H.; Van Kooyk, Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation-dependent interactions between Mac-1 and DC-SIGN. J. Exp. Med. 2005, 201, 1281–1292. [Google Scholar] [CrossRef]
- Humbert, M.; Guery, L.; Brighouse, D.; Lemeille, S.; Hugues, S. Intratumoral CpG-B promotes anti-tumoral neutrophil, cDC, and T cell cooperation without reprograming tolerogenic pDC. Cancer Res. 2018, 78, 3280–3292. [Google Scholar] [CrossRef] [Green Version]
- Blomgran, R.; Ernst, J.D. Lung Neutrophils Facilitate Activation of Naive Antigen-Specific CD4+T Cells duringMycobacterium tuberculosisInfection. J. Immunol. 2011, 186, 7110–7119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateda, K.; Moore, T.A.; Deng, J.C.; Newstead, M.W.; Zeng, X.; Matsukawa, A.; Swanson, M.S.; Yamaguchi, K.; Standiford, T.J. Early Recruitment of Neutrophils Determines Subsequent T1/T2 Host Responses in a Murine Model ofLegionella pneumophilaPneumonia. J. Immunol. 2001, 166, 3355–3361. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Hyun, Y.-M.; Lambert-Emo, K.; Capece, T.; Bae, S.; Miller, R.; Topham, D.J.; Kim, M. Neutrophil trails guide influenza-specific CD8+ T cells in the airways. Science 2015, 349, aaa4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, M.D.; Brooks, A.G.; Reading, P.C.; Mintern, J.D. Neutrophils sustain effective CD8 + T-cell responses in the respiratory tract following influenza infection. Immunol. Cell Biol. 2012, 90, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Stoppacciaro, A.; Melani, C.; Parenza, M.; Mastracchio, A.; Bassi, C.; Baroni, C.; Parmiani, G.; Colombo, M.P. Regression of an established tumor genetically modified to release granulocyte colony-stimulating factor requires granulocyte-T cell cooperation and T cell-produced interferon γ. J. Exp. Med. 1993, 178, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Governa, V.; Trella, E.; Mele, V.; Tornillo, L.; Amicarella, F.; Cremonesi, E.; Muraro, M.G.; Xu, H.; Droeser, R.; Däster, S.R.; et al. The Interplay Between Neutrophils and CD8+ T Cells Improves Survival in Human Colorectal Cancer. Clin. Cancer Res. 2017, 23, 3847–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, C.; Botta, C.; Zabaleta, A.; Puig, N.; Cedena, M.-T.; Goicoechea, I.; Alameda, D.; José-Eneriz, E.S.; Merino, J.; Rodríguez-Otero, P.; et al. Immunogenomic identification and characterization of granulocytic myeloid-derived suppressor cells in multiple myeloma. Blood 2020, 136, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Mensurado, S.; Rei, M.; Lança, T.; Ioannou, M.; Gonçalves-Sousa, N.; Kubo, H.; Malissen, M.; Papayannopoulos, V.; Serre, K.; Silva-Santos, B. Tumor-associated neutrophils suppress pro-tumoral IL-17+ γδ T cells through induction of oxidative stress. PLoS Biol. 2018, 16, e2004990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minns, D.; Smith, K.J.; Findlay, E.G. Orchestration of Adaptive T Cell Responses by Neutrophil Granule Contents. Mediat. Inflamm. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [Green Version]
- De La Rosa, G.; Yang, D.; Tewary, P.; Varadhachary, A.; Oppenheim, J.J.; De, Y. Lactoferrin acts as an alarmin to promote the recruitment and activation of antigen-presenting cells and antigen-specific immune responses. J. Immunol. 2008, 180, 6868–6876. [Google Scholar] [CrossRef] [PubMed]
- Territo, M.C.; Ganz, T.; Selsted, M.E.; Lehrer, R. Monocyte-chemotactic activity of defensins from human neutrophils. J. Clin. Investig. 1989, 84, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chen, Q.; Chertov, O.; Oppenheim, J.J. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J. Leukoc. Biol. 2000, 68, 9–14. [Google Scholar] [PubMed]
- Ethuin, F.; Gérard, B.; Benna, J.E.; Boutten, A.; Gougereot-Pocidalo, M.-A.; Jacob, L.; Chollet-Martin, S. Human neutrophils produce interferon gamma upon stimulation by interleukin-12. Lab. Investig. 2004, 84, 1363–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, U.; Reinhold, D.; Schneemilch, C.; Kunz, D.; Synowitz, H.-J.; Ansorge, S. Selective Proteolytic Cleavage of IL-2 Receptor and IL-6 Receptor Ligand Binding Chains by Neutrophil-Derived Serine Proteases at Foci of Inflammation. J. Interf. Cytokine Res. 1999, 19, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Makarenkova, V.P.; Bansal, V.; Matta, B.M.; Perez, L.A.; Ochoa, J.B. CD11b+/Gr-1+ Myeloid Suppressor Cells Cause T Cell Dysfunction after Traumatic Stress. J. Immunol. 2006, 176, 2085–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillack, K.; Breiden, P.; Martin, R.; Sospedra, M. T Lymphocyte Priming by Neutrophil Extracellular Traps Links Innate and Adaptive Immune Responses. J. Immunol. 2012, 188, 3150–3159. [Google Scholar] [CrossRef] [PubMed]
- Millrud, C.R.; Kågedal, Å.; Georén, S.K.; Winqvist, O.; Uddman, R.; Razavi, R.; Munck-Wikland, E.; Cardell, L.-O. NET-producing CD16highCD62Ldimneutrophils migrate to tumor sites and predict improved survival in patients with HNSCC. Int. J. Cancer 2017, 140, 2557–2567. [Google Scholar] [CrossRef] [Green Version]
- Hufford, M.M.; Richardson, G.; Zhou, H.; Manicassamy, B.; García-Sastre, A.; Enelow, R.I.; Braciale, T.J. Influenza-Infected Neutrophils within the Infected Lungs Act as Antigen Presenting Cells for Anti-Viral CD8+ T Cells. PLoS ONE 2012, 7, e46581. [Google Scholar] [CrossRef]
- Sun, R.; Xiong, Y.; Liu, H.; Gao, C.; Su, L.; Weng, J.; Yuan, X.; Zhang, D.; Feng, J. Tumor-associated neutrophils suppress antitumor immunity of NK cells through the PD-L1/PD-1 axis. Transl. Oncol. 2020, 13, 100825. [Google Scholar] [CrossRef]
- Henghui, Z.; Zhang, H.; Zhou, J.; Wang, B.; Chen, Y.; Kong, Y.; Xie, X.; Wang, X.; Fei, R.; Wei, L.; et al. Peritumoural neutrophils negatively regulate adaptive immunity via the PD-L1/PD-1 signalling pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gershkovitz, M.; Yajuk, O.; Fainsod-Levi, T.; Granot, Z. The pd-l1/pd-1 axis blocks neutrophil cytotoxicity in cancer. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Miret, J.J.; Kirschmeier, P.; Koyama, S.; Zhu, M.; Li, Y.Y.; Naito, Y.; Wu, M.; Malladi, V.S.; Huang, W.; Walker, W.; et al. Suppression of Myeloid Cell Arginase Activity leads to Therapeutic Response in a NSCLC Mouse Model by Activating Anti-Tumor Immunity. J. Immunother. Cancer 2019, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boniface, J.; Mao, Y.; Schmidt-Mende, J.; Kiessling, R.; Poschke, I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early-stage breast cancer patients. OncoImmunology 2012, 1, 1305–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polat, M.F.; Taysi, S.; Bakan, E. Elevated Serum Arginase Activity Levels in Patients with Breast Cancer. Surg. Today 2003, 33, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.-F.; Miao, Q.; Zeng, X.-Q.; Luo, T.-C.; Ma, L.-L.; Liu, Y.-M.; Lian, J.-J.; Gao, H.; Chen, S.-Y. Transforming Growth Factor-β1 and -β2 in Gastric Precancer and Cancer and Roles in Tumor-Cell Interactions with Peripheral Blood Mononuclear Cells In Vitro. PLoS ONE 2013, 8, e54249. [Google Scholar] [CrossRef]
- Young, M.I.; Wright, M.A.; Matthews, J.P.; Malik, I.; Prechel, M. Suppression of T cell proliferation by tumor-induced granulocyte-macrophage progenitor cells producing transforming growth factor-beta and nitric oxide. J. Immunol. 1996, 156, 1916–1922. [Google Scholar]
- Mittal, S.K.; Mashaghi, A.; Amouzegar, A.; Li, M.; Foulsham, W.; Sahu, S.K.; Chauhan, S.K. Mesenchymal Stromal Cells Inhibit Neutrophil Effector Functions in a Murine Model of Ocular Inflammation. Investig. Opthalmol. Vis. Sci. 2018, 59, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhou, Y.; Dong, K.; Sun, Z.; Zhao, D.; Wang, W.; Yu, G.; Liu, W.; Xu, G.; Han, Z.; et al. Programming of the Development of Tumor-Promoting Neutrophils by Mesenchymal Stromal Cells. Cell. Physiol. Biochem. 2014, 33, 1802–1814. [Google Scholar] [CrossRef]
- Delaney, C.; Heimfeld, S.; Brashem-Stein, C.; Voorhies, H.; Manger, R.L.; Bernstein, I.D. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat. Med. 2010, 16, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Z.; Zhang, Y.; Wang, C.; Shen, B.; Guan, X.; Ren, Z.; Ding, X.; Dai, W.; Jiang, Y. Large-scale ex vivo generation of human neutrophils from cord blood CD34+ cells. PLoS ONE 2017, 12, e0180832. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, M.K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Nasri, M.; Ritter, M.; Mir, P.; Dannenmann, B.; Aghaallaei, N.; Amend, D.; Makaryan, V.; Xu, Y.; Fletcher, B.; Bernhard, R.; et al. CRISPR/Cas9-mediated ELANE knockout enables neutrophilic maturation of primary hematopoietic stem and progenitor cells and induced pluripotent stem cells of severe congenital neutropenia patients. Haematologica 2019, 105, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.-Y.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Chu, D.; Zhao, Q.; Yu, J.; Zhang, F.; Zhang, H.; Wang, Z. Nanoparticle Targeting of Neutrophils for Improved Cancer Immunotherapy. Adv. Healthc. Mater. 2016, 5, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Chu, D.; Dong, X.; Zhao, Q.; Gu, J.; Wang, Z. Photosensitization Priming of Tumor Microenvironments Improves Delivery of Nanotherapeutics via Neutrophil Infiltration. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef]
- Kang, T.; Zhu, Q.; Wei, D.; Feng, J.; Yao, J.; Jiang, T.; Song, Q.; Wei, X.; Chen, H.; Gao, X.; et al. Nanoparticles Coated with Neutrophil Membranes Can Effectively Treat Cancer Metastasis. ACS Nano 2017, 11, 1397–1411. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ustyanovska Avtenyuk, N.; Visser, N.; Bremer, E.; Wiersma, V.R. The Neutrophil: The Underdog That Packs a Punch in the Fight against Cancer. Int. J. Mol. Sci. 2020, 21, 7820. https://doi.org/10.3390/ijms21217820
Ustyanovska Avtenyuk N, Visser N, Bremer E, Wiersma VR. The Neutrophil: The Underdog That Packs a Punch in the Fight against Cancer. International Journal of Molecular Sciences. 2020; 21(21):7820. https://doi.org/10.3390/ijms21217820
Chicago/Turabian StyleUstyanovska Avtenyuk, Natasha, Nienke Visser, Edwin Bremer, and Valerie R. Wiersma. 2020. "The Neutrophil: The Underdog That Packs a Punch in the Fight against Cancer" International Journal of Molecular Sciences 21, no. 21: 7820. https://doi.org/10.3390/ijms21217820
APA StyleUstyanovska Avtenyuk, N., Visser, N., Bremer, E., & Wiersma, V. R. (2020). The Neutrophil: The Underdog That Packs a Punch in the Fight against Cancer. International Journal of Molecular Sciences, 21(21), 7820. https://doi.org/10.3390/ijms21217820