Phosphoglycerate Mutase 1 Prevents Neuronal Death from Ischemic Damage by Reducing Neuroinflammation in the Rabbit Spinal Cord

 ,
,
Abstract
1. Introduction
2. Results
2.1. Tat-PGAM1, but Not Control-PGAM1, Was Effectively Delivered into NSC34 Cells
2.2. Tat-PGAM1, but Not Control-PGAM1, Protects NSC34 Cells from Oxidative Damage
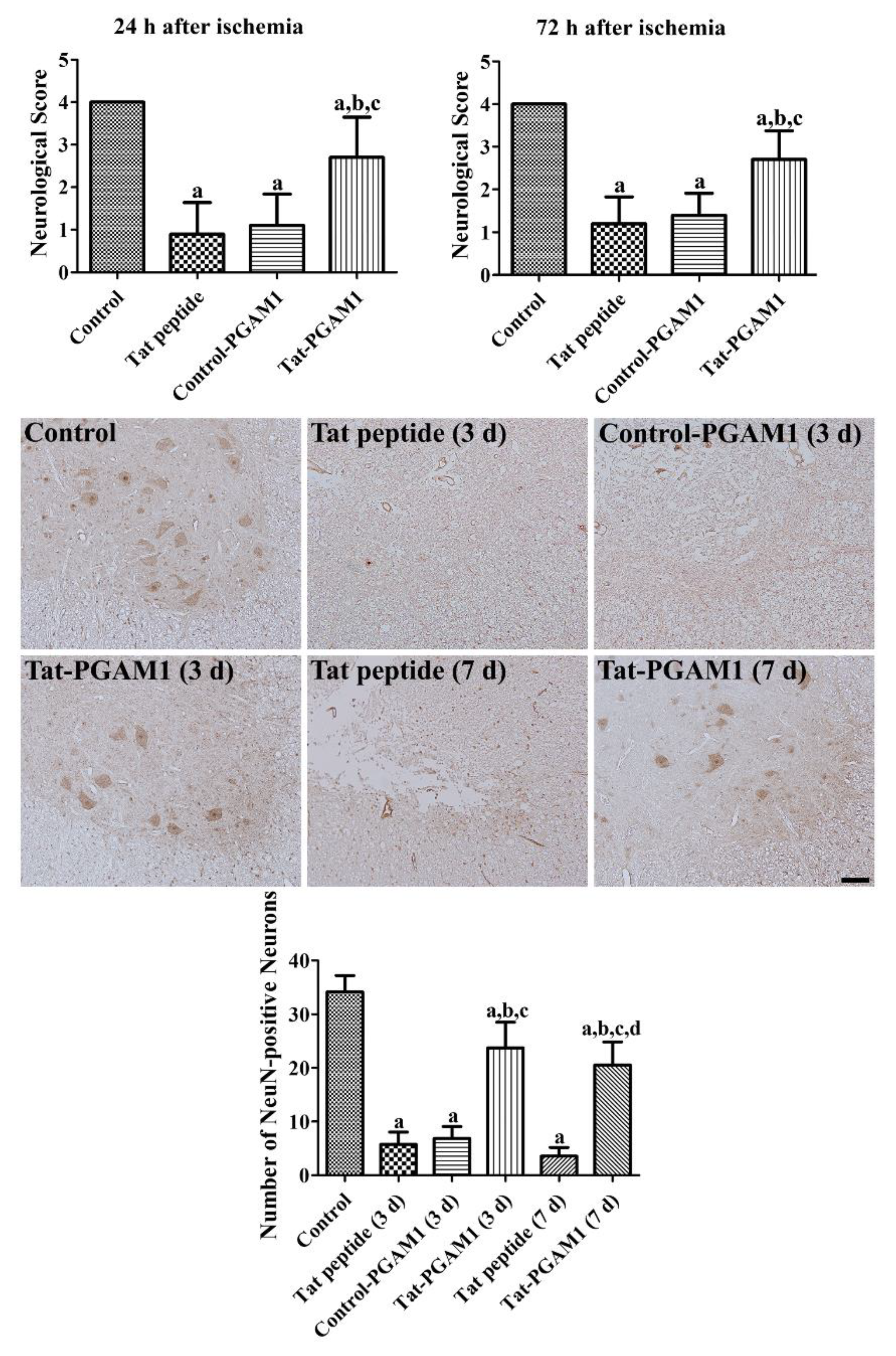
2.3. Tat-PGAM1, but Not Control-PGAM1, Prevents Neuronal Damage in the Ventral Horn of the Rabbit Spinal Cord
2.4. Tat-PGAM1, but Not Control-PGAM1, Reduces Inflammatory Responses and Oxidative Damage after Ischemia
3. Discussion
4. Materials and Methods
4.1. Concentration- and Time-Dependent Delivery of Tat-PGAM1 into NSC34 Cells
4.2. Visualization of Intracellular Delivery of Tat-PGAM1 in NSC34 Cells
4.3. Neuroprotective Effects of Tat-PGAM1 against Oxidative Damage in NSC34 Cells
4.4. Neuroprotective Effects of Tat-PGAM1 against Ischemic Damage in Rabbits
4.5. Anti-Oxidative and Anti-Inflammatory Effects of Tat-PGAM1 Against Ischemic Damage in Rabbits
4.6. Data Quantification and Analysis
Author Contributions
Funding
Conflicts of Interest
References
- De Lavor, M.S.; Binda, N.S.; Fukushima, F.B.; Caldeira, F.M.; da Silva, J.F.; Silva, C.M.; de Oliveira, K.M.; Martins Bde, C.; Torres, B.B.; Rosado, I.R.; et al. Ischemia-reperfusion model in rat spinal cord: Cell viability and apoptosis signaling study. Int. J. Clin. Exp. Pathol. 2015, 8, 9941–9949. [Google Scholar] [PubMed]
- Xiao, W.; Wen, J.; Huang, Y.C.; Yu, B.S. Development of a modified model of spinal cord ischemia injury by selective ligation of lumbar arteries in rabbits. Spinal Cord 2017, 55, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Romi, F.; Naess, H. Spinal cord infarction in clinical neurology: A review of characteristics and long-term prognosis in comparison to cerebral infarction. Eur. Neurol. 2016, 76, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Dawson, T.M.; Raps, E.C.; Galetta, S.L. Risk factors for the neurologic complications associated with aortic aneurysms. Arch. Neurol. 1992, 49, 284–288. [Google Scholar] [CrossRef]
- Basu, S.; Hellberg, A.; Ulus, A.T.; Westman, J.; Karacagil, S. Biomarkers of free radical injury during spinal cord ischemia. FEBS Lett. 2001, 508, 36–38. [Google Scholar] [CrossRef]
- Smith, P.D.; Puskas, F.; Meng, X.; Lee, J.H.; Cleveland, J.C., Jr.; Weyant, M.J.; Fullerton, D.A.; Reece, T.B. The evolution of chemokine release supports a bimodal mechanism of spinal cord ischemia and reperfusion injury. Circulation 2012, 126, S110–S117. [Google Scholar] [CrossRef]
- Garcia, E.; Aguilar-Cevallos, J.; Silva-Garcia, R.; Ibarra, A. Cytokine and growth factor activation in vivo and in vitro after spinal cord injury. Mediat. Inflamm. 2016, 2016, 9476020. [Google Scholar] [CrossRef]
- Kim, W.; Cho, S.B.; Jung, H.Y.; Yoo, D.Y.; Oh, J.K.; Choi, G.M.; Cho, T.G.; Kim, D.W.; Hwang, I.K.; Choi, S.Y.; et al. Phosphatidylethanolamine-binding protein 1 ameliorates ischemia-induced inflammation and neuronal damage in the rabbit spinal cord. Cells 2019, 8, 1370. [Google Scholar] [CrossRef]
- Chen, C.; Shaw, Y.S. Cyclic metabolic pathway of a butylated hydroxytoluene by rat liver microsomal fractions. Biochem. J. 1974, 144, 497–501. [Google Scholar] [CrossRef]
- Fothergill-Gilmore, L.A.; Watson, H.C. The phosphoglycerate mutases. Adv. Enzymol. Relat. Areas Mol. Biol. 1989, 62, 227–313. [Google Scholar]
- Sultana, R.; Perluigi, M.; Newman, S.F.; Pierce, W.M.; Cini, C.; Coccia, R.; Butterfield, D.A. Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer’s disease. Antioxid. Redox Signal. 2010, 12, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Sharif, F.; Rasul, A.; Ashraf, A.; Hussain, G.; Younis, T.; Sarfraz, I.; Chaudhry, M.A.; Bukhari, S.A.; Ji, X.Y.; Selamoglu, Z.; et al. Phosphoglycerate mutase 1 in cancer: A promising target for diagnosis and therapy. IUBMB Life 2019, 71, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Gil, J.; Wang, J.; Degan, P.; Peters, G.; Martinez, D.; Carnero, A.; Beach, D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005, 65, 177–185. [Google Scholar] [PubMed]
- Takahashi, Y.; Takahashi, S.; Yoshimi, T.; Miura, T. Hypoxia-induced expression of phosphoglycerate mutase B in fibroblasts. Eur. J. Biochem. 1998, 254, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, H.; Lleonart, M.E.; Bernard, D.; Gil, J. Protection from oxidative stress by enhanced glycolysis; a possible mechanism of cellular immortalization. Histol. Histopathol. 2007, 22, 85–90. [Google Scholar] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef]
- Niizeki, H.; Kobayashi, M.; Horiuchi, I.; Akakura, N.; Chen, J.; Wang, J.; Hamada, J.I.; Seth, P.; Katoh, H.; Watanabe, H.; et al. Hypoxia enhances the expression of autocrine motility factor and the motility of human pancreatic cancer cells. Br. J. Cancer 2002, 86, 1914–1919. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Wang, F. Intracellular transduction and potential of Tat PTD and its analogs: From basic drug delivery mechanism to application. Expert Opin. Drug Deliv. 2012, 9, 457–472. [Google Scholar] [CrossRef]
- Jung, H.Y.; Kwon, H.J.; Kim, W.; Nam, S.M.; Kim, J.W.; Hahn, K.R.; Yoo, D.Y.; Won, M.H.; Yoon, Y.S.; Kim, D.W.; et al. Phosphoglycerate mutase 1 promotes cell proliferation and neuroblast differentiation in the dentate gyrus by facilitating the phosphorylation of cAMP response element-binding protein. Neurochem. Res. 2019, 44, 323–332. [Google Scholar] [CrossRef]
- Kim, W.; Kwon, H.J.; Jung, H.Y.; Yoo, D.Y.; Kim, D.W.; Hwang, I.K. Phosphoglycerate mutase 1 reduces neuronal damage in the hippocampus following ischemia/reperfusion through the facilitation of energy utilization. Neurochem. Int. 2020, 133, 104631. [Google Scholar] [CrossRef] [PubMed]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Boyd-Kimball, D.; Sultana, R.; Poon, H.F.; Lynn, B.C.; Casamenti, F.; Pepeu, G.; Klein, J.B.; Butterfield, D.A. Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1-42) into rat brain: Implications for Alzheimer’s disease. Neuroscience 2005, 132, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Sethy, N.K.; Bhargava, K. Comparative proteome analysis reveals differential regulation of glycolytic and antioxidant enzymes in cortex and hippocampus exposed to short-term hypobaric hypoxia. J. Proteom. 2013, 79, 277–298. [Google Scholar] [CrossRef]
- Peinado, M.Á.; Hernández, R.; Peragón, J.; Ovelleiro, D.; Pedrosa, J.Á.; Blanco, S. Proteomic characterization of nitrated cell targets after hypobaric hypoxia and reoxygenation in rat brain. J. Proteomics 2014, 109, 309–321. [Google Scholar] [CrossRef]
- Imperlini, E.; Orrù, S.; Corbo, C.; Daniele, A.; Salvatore, F. Altered brain protein expression profiles are associated with molecular neurological dysfunction in the PKU mouse model. J. Neurochem. 2014, 129, 1002–1012. [Google Scholar] [CrossRef]
- Yu, H.; Jiang, X.; Lin, X.; Zhang, Z.; Wu, D.; Zhou, L.; Liu, J.; Yang, X. Hippocampal subcellular organelle proteomic alteration of copper-treated mice. Toxicol. Sci. 2018, 164, 250–263. [Google Scholar] [CrossRef]
- Guyton, K.Z.; Liu, Y.; Gorospe, M.; Xu, Q.; Holbrook, N.J. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996, 271, 4138–4142. [Google Scholar] [CrossRef]
- Lander, H.M. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997, 11, 118–124. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, X.; Zhang, M.; Zhao, C.; Mei, C.; Li, P. MAPK Pathway Inhibitors Attenuated Hydrogen Peroxide Induced Damage in Neural Cells. Biomed Res. Int. 2019, 2019, 5962014. [Google Scholar] [CrossRef]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prévost, M.C.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Zwerschke, W.; Mazurek, S.; Stöckl, P.; Hütter, E.; Eigenbrodt, E.; Jansen-Dürr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, M.; Takaki, E.; Yamashita, K.; Watanabe, K.; Tabuchi, S.; Watanabe, T.; Satoh, K. Nonenzymatic derived lipid peroxide, 8-iso-PGF2 alpha, participates in the pathogenesis of delayed cerebral vasospasm in a canine SAH model. Neurol. Res. 2002, 24, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Kertmen, H.; Celikoglu, E.; Ozturk, O.C.; Gürer, B.; Bozkurt, H.; Kanat, M.A.; Arikok, A.T.; Erguder, B.I.; Sargon, M.F.; Sekerci, Z. Comparative effects of methylprednisolone and tetracosactide (ACTH1-24) on ischemia/reperfusion injury of the rabbit spinal cord. Arch. Med. Sci. 2018, 14, 1459–1470. [Google Scholar] [CrossRef]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J.; et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Gaber, T.; Strehl, C.; Buttgereit, F. Metabolic regulation of inflammation. Nat. Rev. Rheumatol. 2017, 13, 267–279. [Google Scholar] [CrossRef]
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Teixeira, J.M.; de Oliveira-Fusaro, M.C.; Parada, C.A.; Tambeli, C.H. Peripheral P2 × 7 receptor-induced mechanical hyperalgesia is mediated by bradykinin. Neuroscience 2014, 277, 163–173. [Google Scholar] [CrossRef]
- Sun, S.; Chen, D.; Lin, F.; Chen, M.; Yu, H.; Hou, L.; Li, C. Role of interleukin-4, the chemokine CCL3 and its receptor CCR5 in neuropathic pain. Mol. Immunol. 2016, 77, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, M.; Tang, H.; Ma, Z.; Liang, Y.; Li, Z. The over-production of TNF-α via Toll-like receptor 4 in spinal dorsal horn contributes to the chronic postsurgical pain in rat. J. Anesth. 2015, 29, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Capone, M.; Matzelle, D.; Cox, A.; Banik, N.L. Targeting enolase in reducing secondary damage in acute spinal cord injury in rats. Neurochem. Res. 2017, 42, 2777–2787. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Yang, Y.W.; Wang, Y.L.; Lu, J.K.; Tian, L.; Jin, M.; Cheng, W.P. Delayed xenon post-conditioning mitigates spinal cord ischemia/reperfusion injury in rabbits by regulating microglial activation and inflammatory factors. Neural Regen. Res. 2018, 13, 510–517. [Google Scholar]
- Shechter, R.; London, A.; Varol, C.; Raposo, C.; Cusimano, M.; Yovel, G.; Rolls, A.; Mack, M.; Pluchino, S.; Martino, G.; et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009, 6, e1000113. [Google Scholar] [CrossRef]
- Sahin, M.A.; Onan, B.; Guler, A.; Oztas, E.; Uysal, B.; Arslan, S.; Demirkilic, U.; Tatar, H. Cilostazol, a type III phosphodiesterase inhibitor, reduces ischemia/reperfusion-induced spinal cord injury. Heart Surg. Forum 2011, 14, E171–E177. [Google Scholar] [CrossRef]
- Song, J.; Baek, I.J.; Chun, C.H.; Jin, E.J. Dysregulation of the NUDT7-PGAM1 axis is responsible for chondrocyte death during osteoarthritis pathogenesis. Nat. Commun. 2018, 9, 3427. [Google Scholar] [CrossRef]
- Kim, W.; Kwon, H.J.; Jung, H.Y.; Yoo, D.Y.; Moon, S.M.; Kim, D.W.; Hwang, I.K. Tat-HSP70 protects neurons from oxidative damage in the NSC34 cells and ischemic damage in the ventral horn of rabbit spinal cord. Neurochem. Int. 2019, 129, 104477. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]
- Tarlov, I.M. Spinal Cord Compression; Mechanism of Paralysis and Treatment; Charles C. Thomas Publisher: Springfield, IL, USA, 1957. [Google Scholar]
- Moore, W.M., Jr.; Hollier, L.H. The influence of severity of spinal cord ischemia in the etiology of delayed-onset paraplegia. Ann. Surg. 1991, 213, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Wisselink, W.; Patetsios, P.; Panetta, T.F.; Ramirez, J.A.; Rodino, W.; Kirwin, J.D.; Zikria, B.A. Medium molecular weight pentastarch reduces reperfusion injury by decreasing capillary leak in an animal model of spinal cord ischemia. J. Vasc. Surg. 1998, 27, 109–116. [Google Scholar] [CrossRef][Green Version]
- Jung, H.Y.; Kim, D.W.; Kwon, H.J.; Yoo, D.Y.; Hwang, I.K.; Won, M.H.; Cho, T.G.; Choi, S.Y.; Moon, S.M. SUMO-1 delays neuronal damage in the spinal cord following ischemia/reperfusion. Mol. Med. Rep. 2017, 15, 4312–4318. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Morrow, J.D.; Hill, K.E.; Burk, R.F.; Nammour, T.M.; Badr, K.F.; Roberts, L.J., 2nd. A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. USA 1990, 87, 9383–9387. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Distal Mean Arterial Pressure (MAP) (mmHg) | PaCO2 (mmHg) | PaO2 (mmHg) | Glucose (mM) | |
|---|---|---|---|---|---|
| Pre-ischemia | |||||
| Control | 7.39 ± 0.027 | 83.8 ± 9.14 | 37.2 ± 4.13 | 104.6 ± 9.15 | 6.43 ± 0.98 |
| Vehicle | 7.42 ± 0.033 | 84.2 ± 8.81 | 36.9 ± 3.80 | 103.5 ± 8.72 | 6.52 ± 1.29 |
| Control-PGAM1 | 7.41 ± 0.035 | 84.1 ± 9.42 | 37.1 ± 3.75 | 106.4 ± 11.3 | 6.38 ± 0.81 |
| Tat-PGAM1 | 7.39 ± 0.032 | 84.0 ± 8.06 | 36.8 ± 4.37 | 103.9 ± 10.4 | 6.41 ± 1.05 |
| Reperfusion 10 min | |||||
| Control | 7.41 ± 0.032 | 84.1 ± 8.60 | 37.0 ± 4.42 | 103.7 ± 9.71 | 6.37 ± 1.08 |
| Vehicle | 7.35 ± 0.068 | 88.7 ± 11.3 | 39.3 ± 5.95 | 111.4 ± 11.9 | 7.10 ± 1.42 |
| Control-PGAM1 | 7.36 ± 0.092 | 87.8 ± 10.2 | 39.7 ± 5.28 | 106.6 ± 12.7 | 6.83 ± 1.27 |
| Tat-PGAM1 | 7.37 ± 0.088 | 86.3 ± 9.92 | 38.1 ± 5.60 | 114.1 ± 11.1 | 7.25 ± 1.62 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, H.Y.; Kwon, H.J.; Kim, W.; Hahn, K.R.; Moon, S.M.; Yoon, Y.S.; Kim, D.W.; Hwang, I.K. Phosphoglycerate Mutase 1 Prevents Neuronal Death from Ischemic Damage by Reducing Neuroinflammation in the Rabbit Spinal Cord. Int. J. Mol. Sci. 2020, 21, 7425. https://doi.org/10.3390/ijms21197425
Jung HY, Kwon HJ, Kim W, Hahn KR, Moon SM, Yoon YS, Kim DW, Hwang IK. Phosphoglycerate Mutase 1 Prevents Neuronal Death from Ischemic Damage by Reducing Neuroinflammation in the Rabbit Spinal Cord. International Journal of Molecular Sciences. 2020; 21(19):7425. https://doi.org/10.3390/ijms21197425
Chicago/Turabian StyleJung, Hyo Young, Hyun Jung Kwon, Woosuk Kim, Kyu Ri Hahn, Seung Myung Moon, Yeo Sung Yoon, Dae Won Kim, and In Koo Hwang. 2020. "Phosphoglycerate Mutase 1 Prevents Neuronal Death from Ischemic Damage by Reducing Neuroinflammation in the Rabbit Spinal Cord" International Journal of Molecular Sciences 21, no. 19: 7425. https://doi.org/10.3390/ijms21197425
APA StyleJung, H. Y., Kwon, H. J., Kim, W., Hahn, K. R., Moon, S. M., Yoon, Y. S., Kim, D. W., & Hwang, I. K. (2020). Phosphoglycerate Mutase 1 Prevents Neuronal Death from Ischemic Damage by Reducing Neuroinflammation in the Rabbit Spinal Cord. International Journal of Molecular Sciences, 21(19), 7425. https://doi.org/10.3390/ijms21197425