The Best Peptidomimetic Strategies to Undercover Antibacterial Peptides

 ,
,  , ,
, ,
Abstract
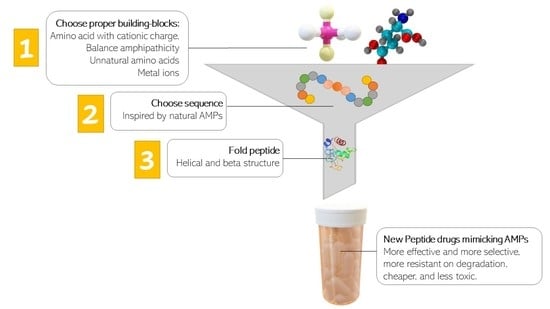
1. Introduction
2. Samsons Hair of AMPs
3. AMPs: Achilles’ Heel
4. Bacterial Membranes vs. Human Cell Membranes
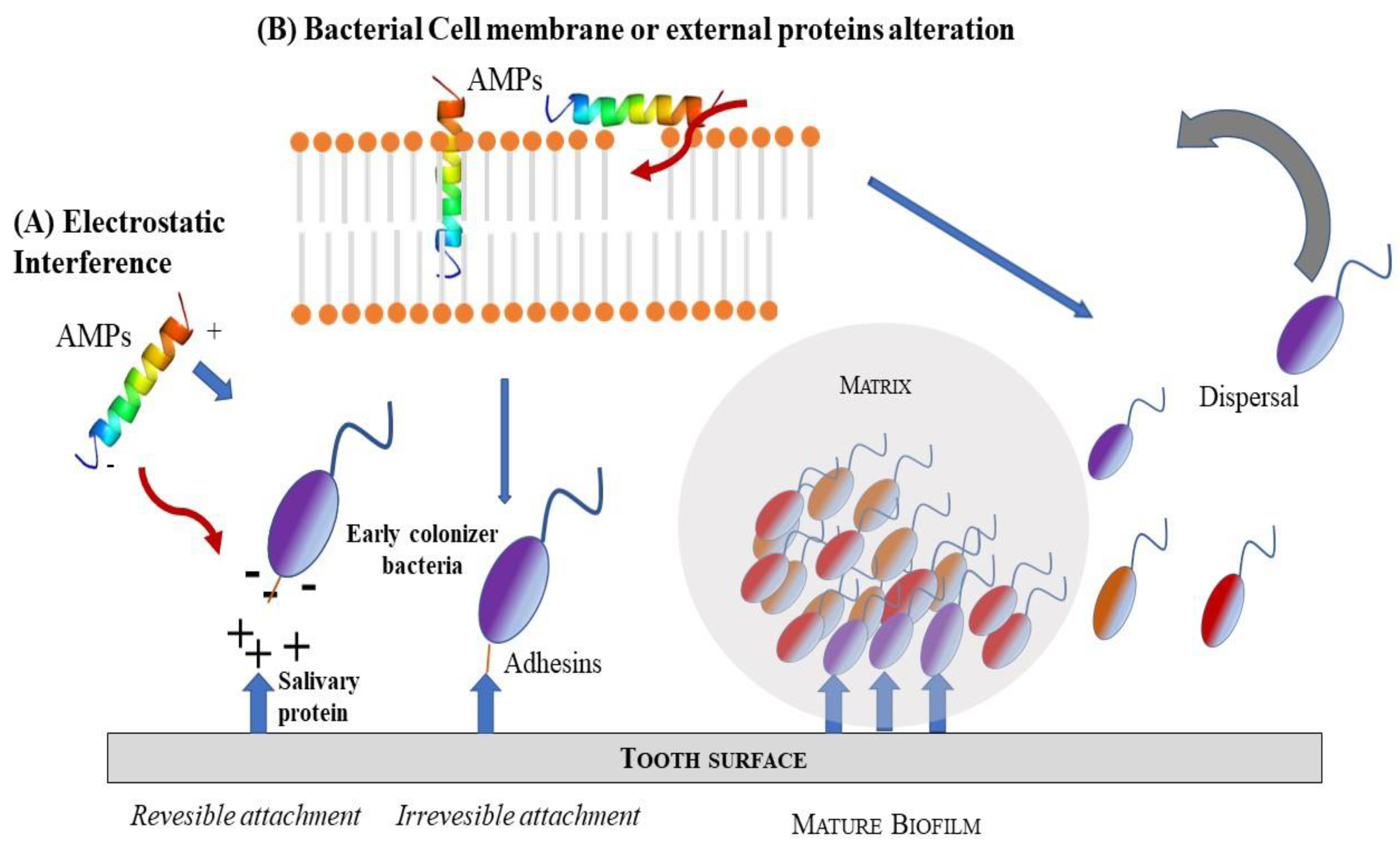
5. Antibacterial Peptides in the Management of Oral Infections with Biofilm Production
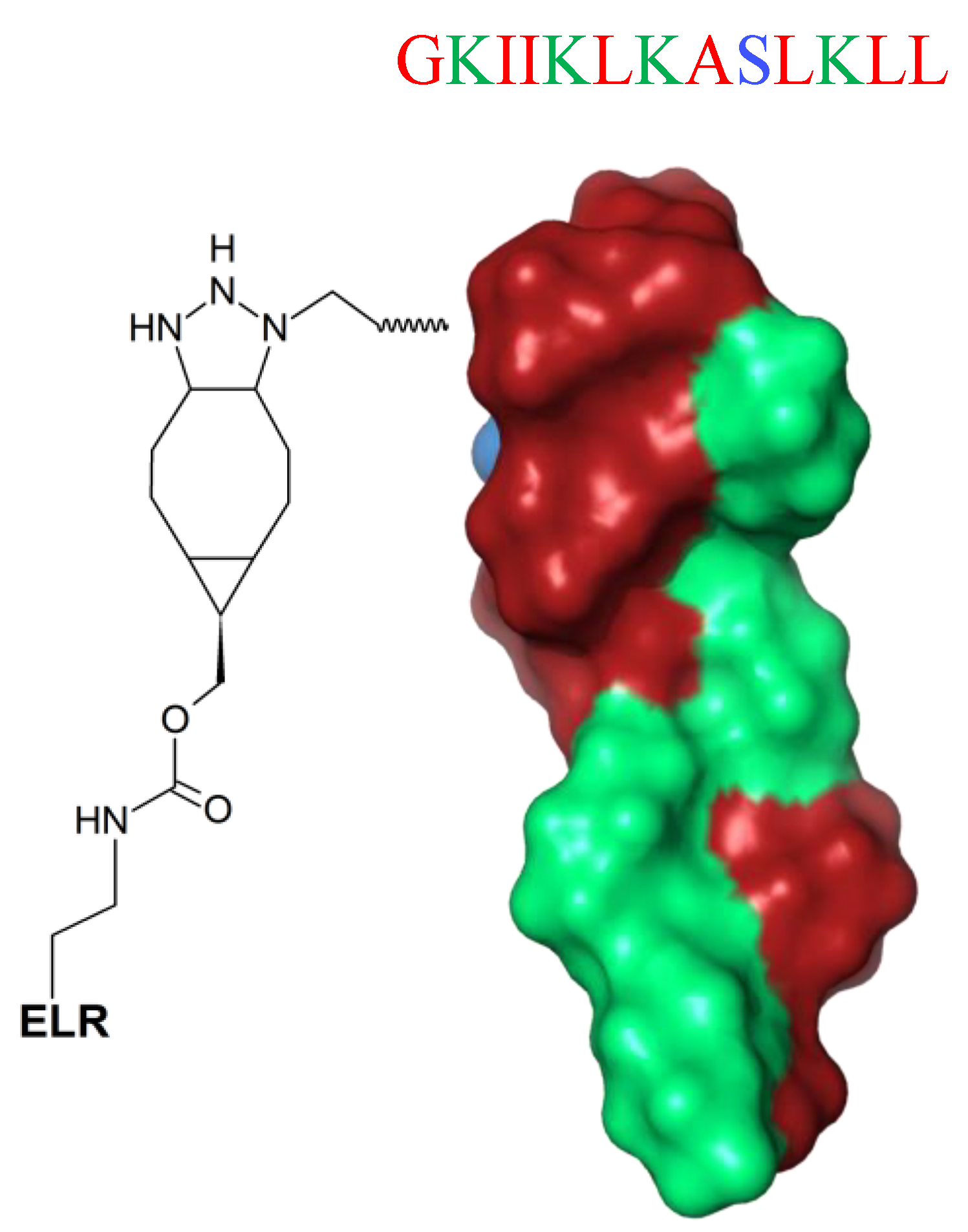
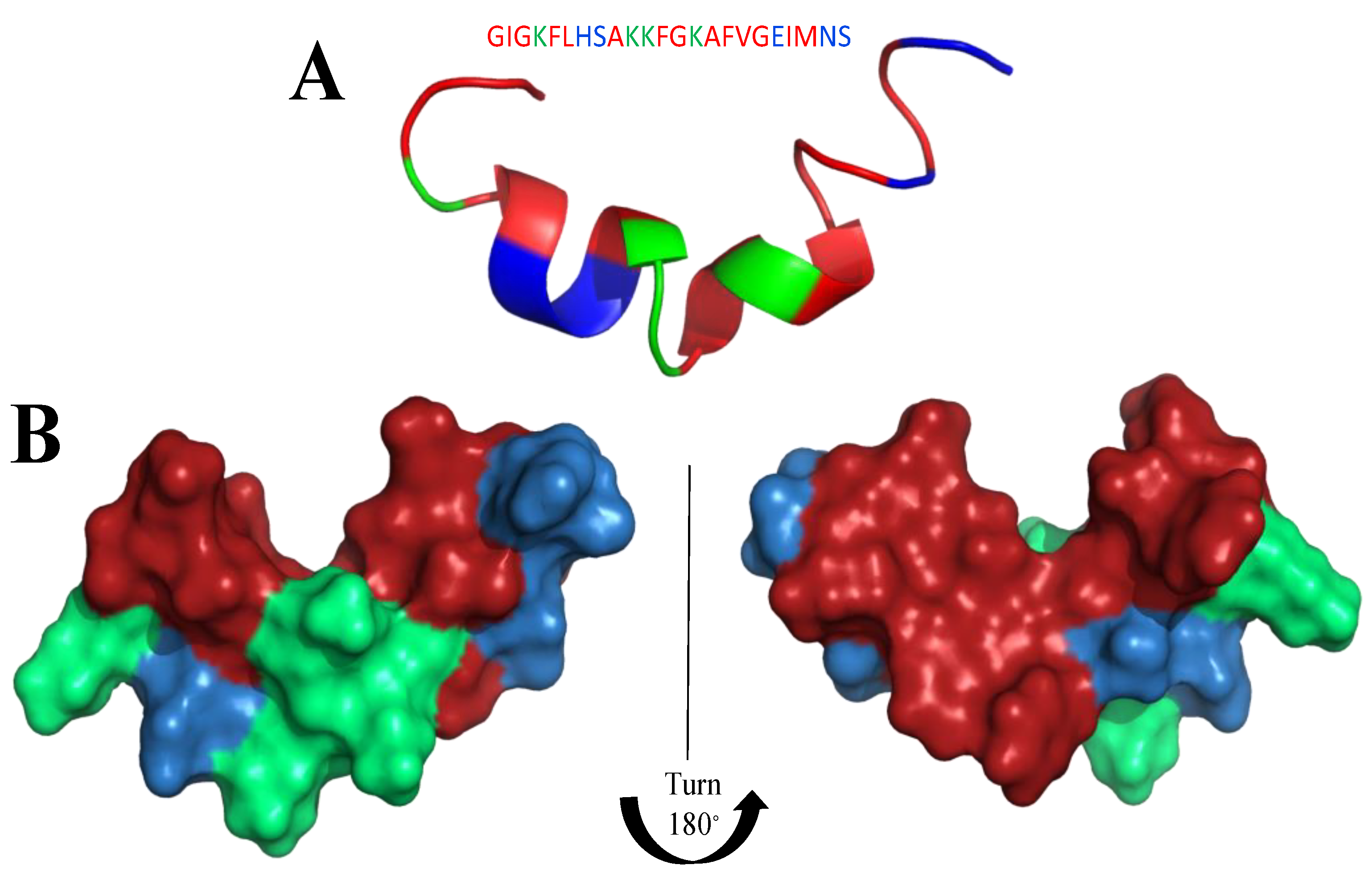
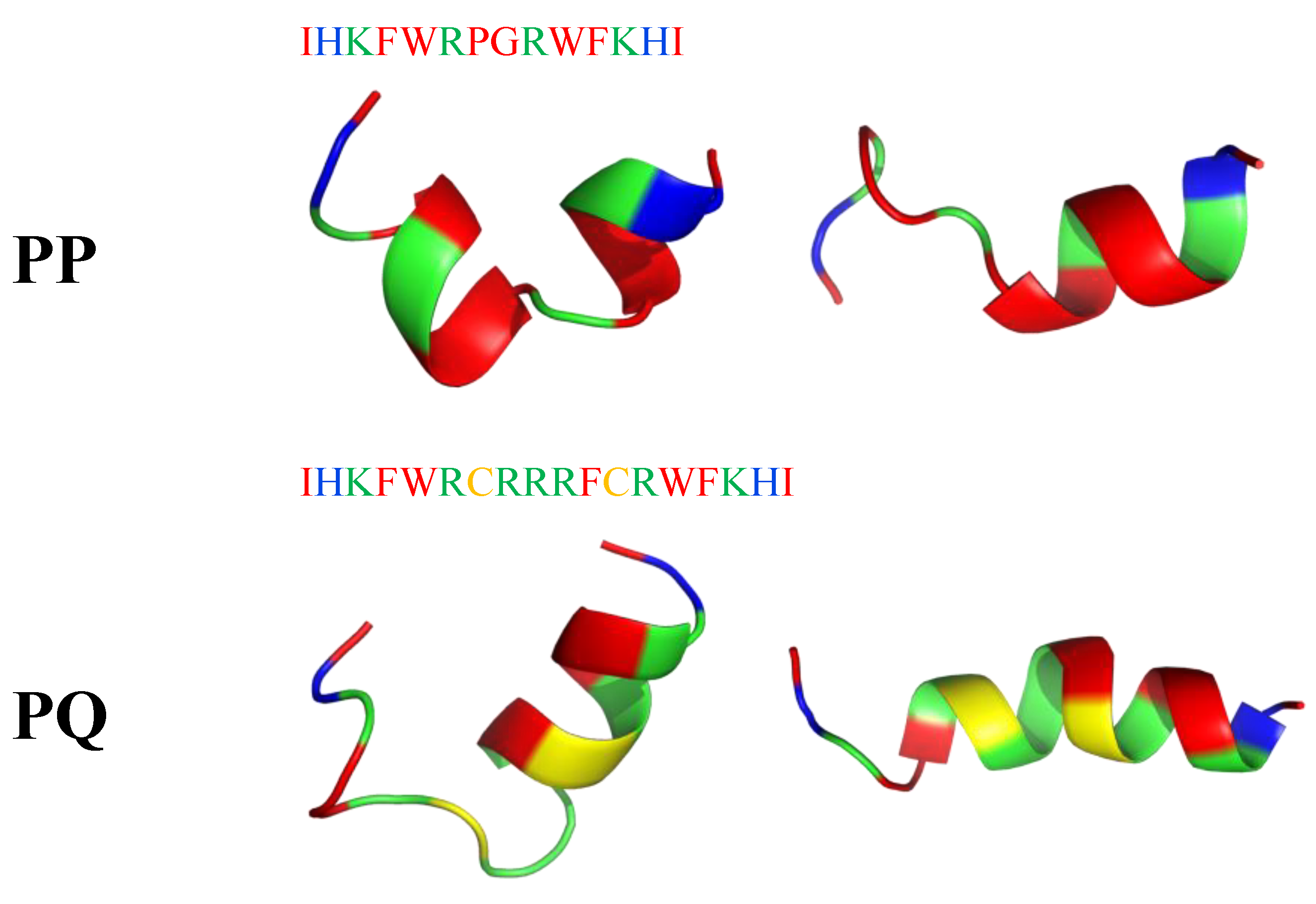
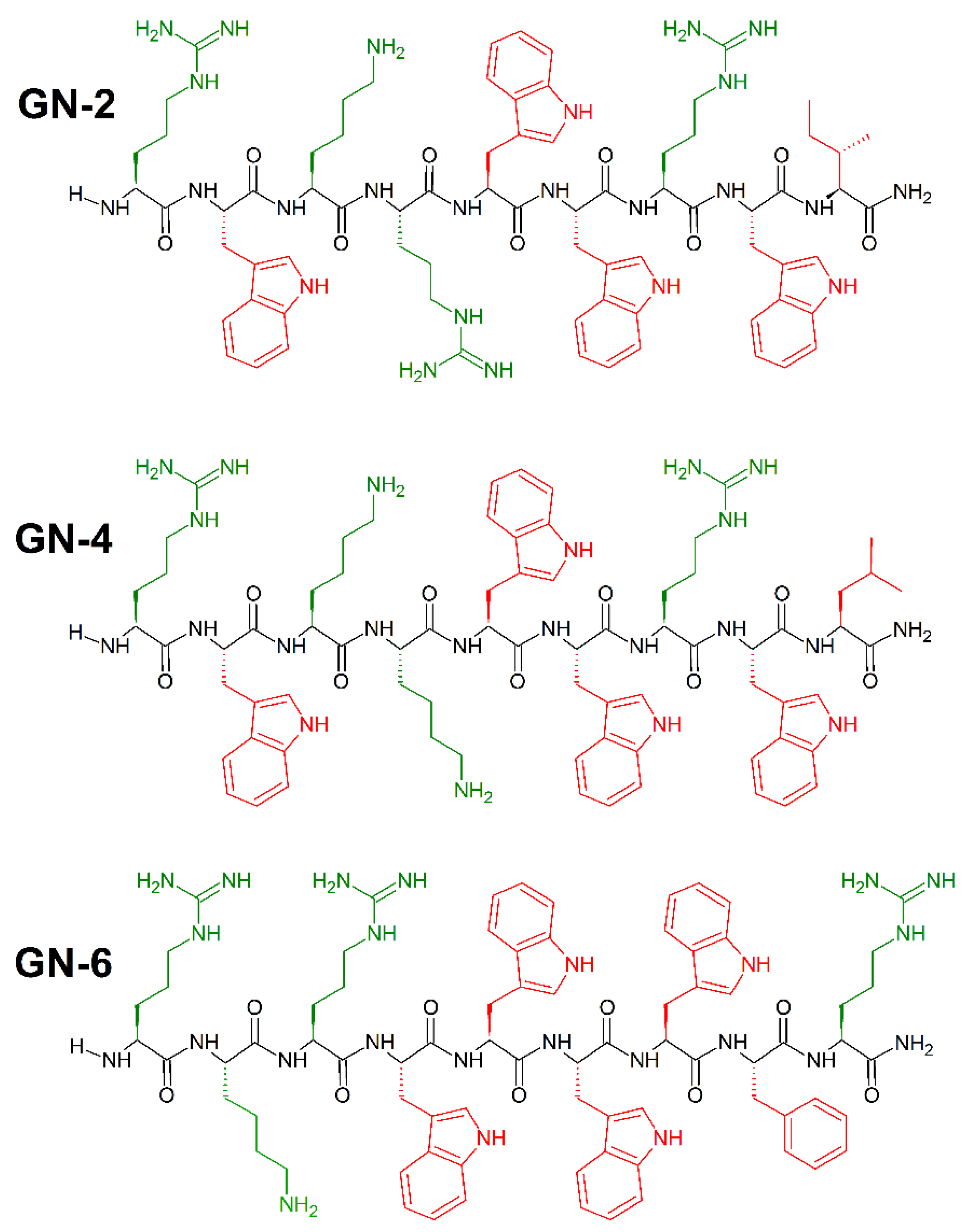
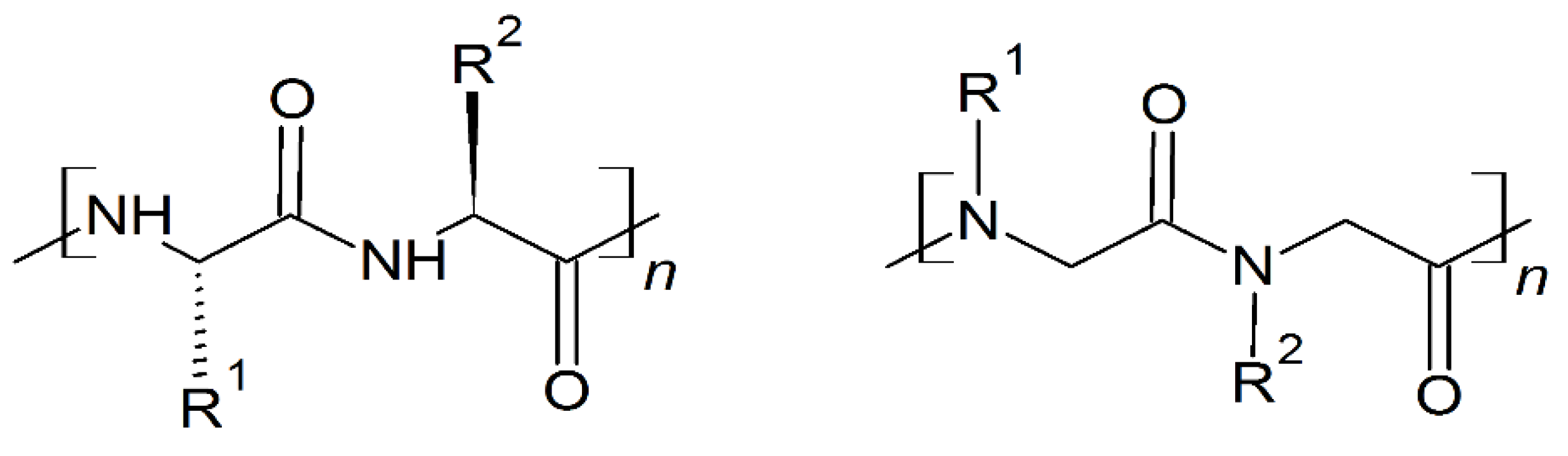
6. Mimicking Amino Acid Sequence
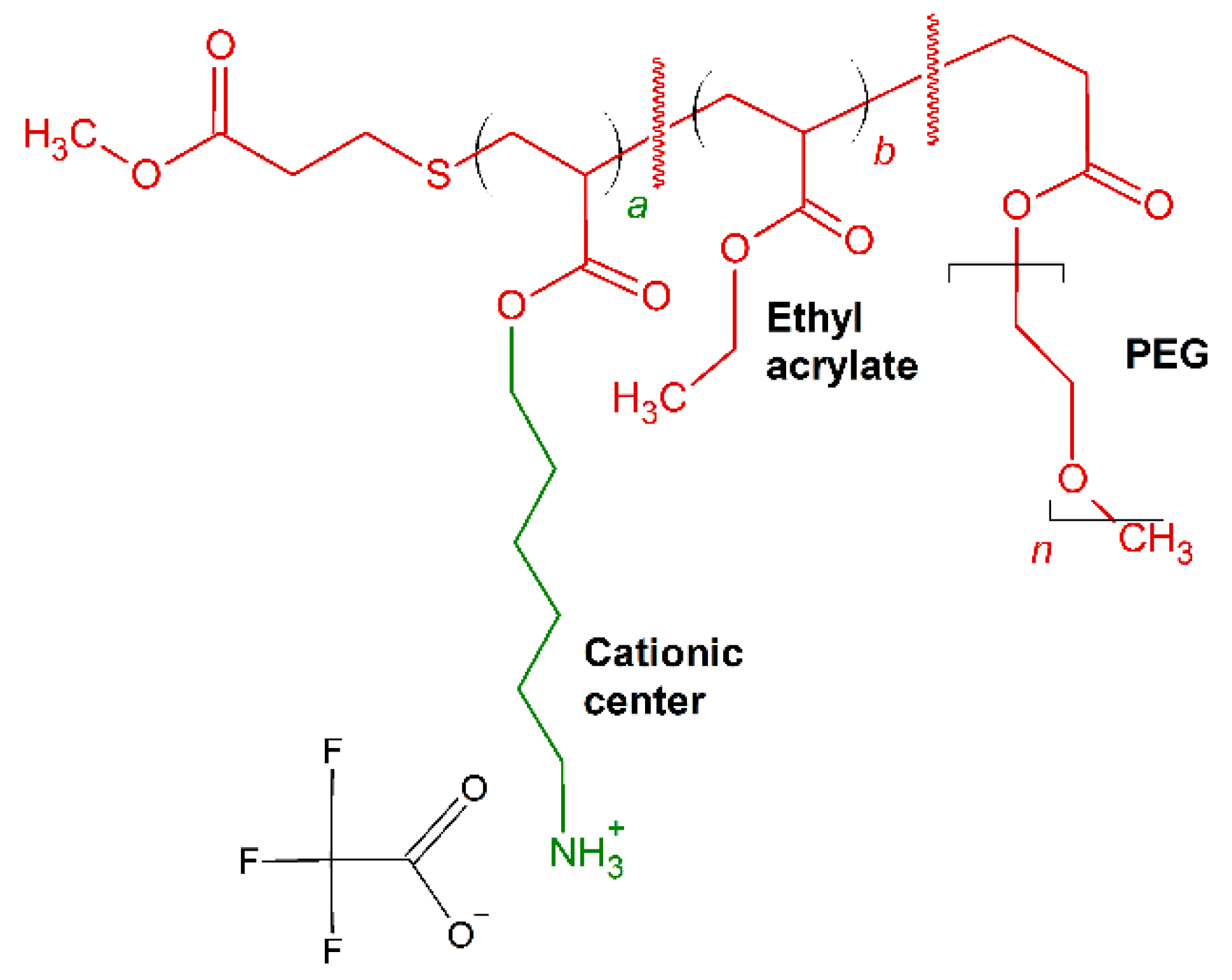
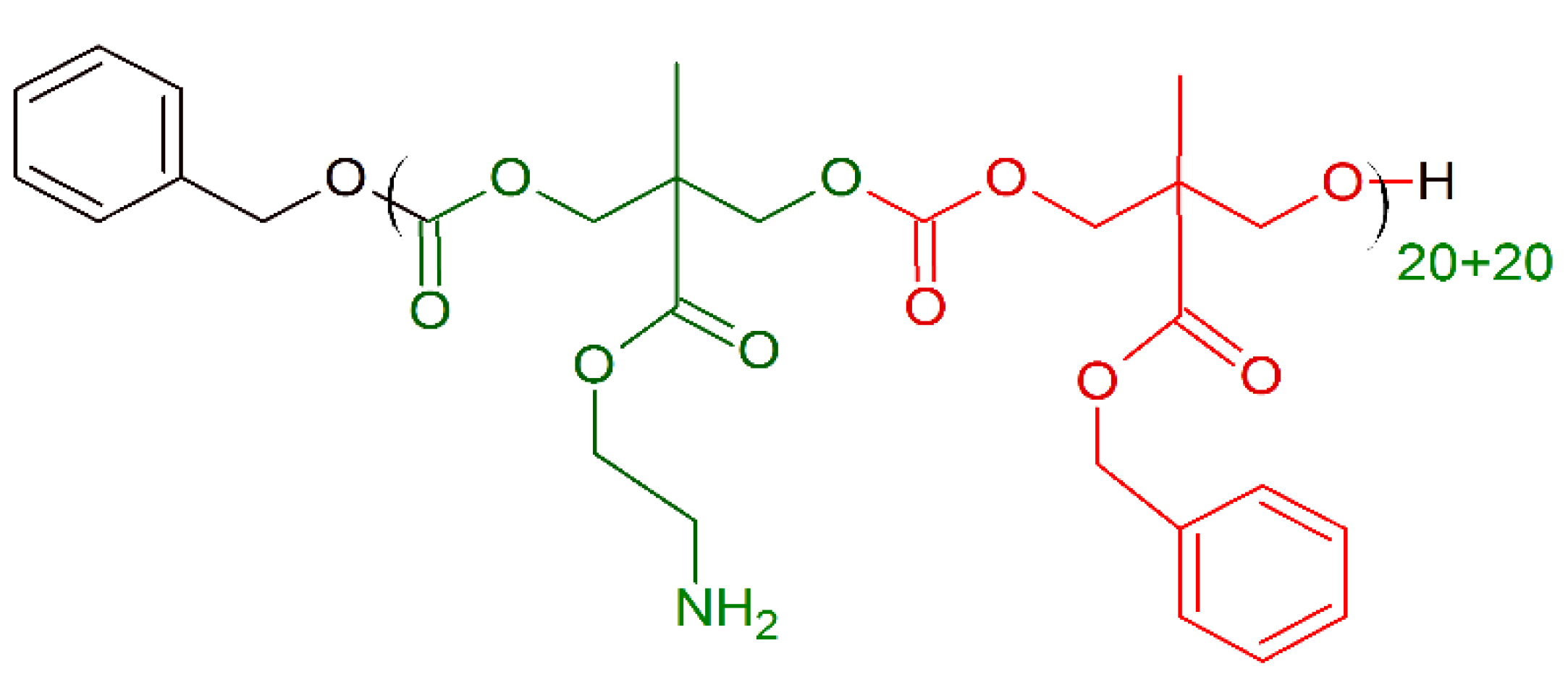
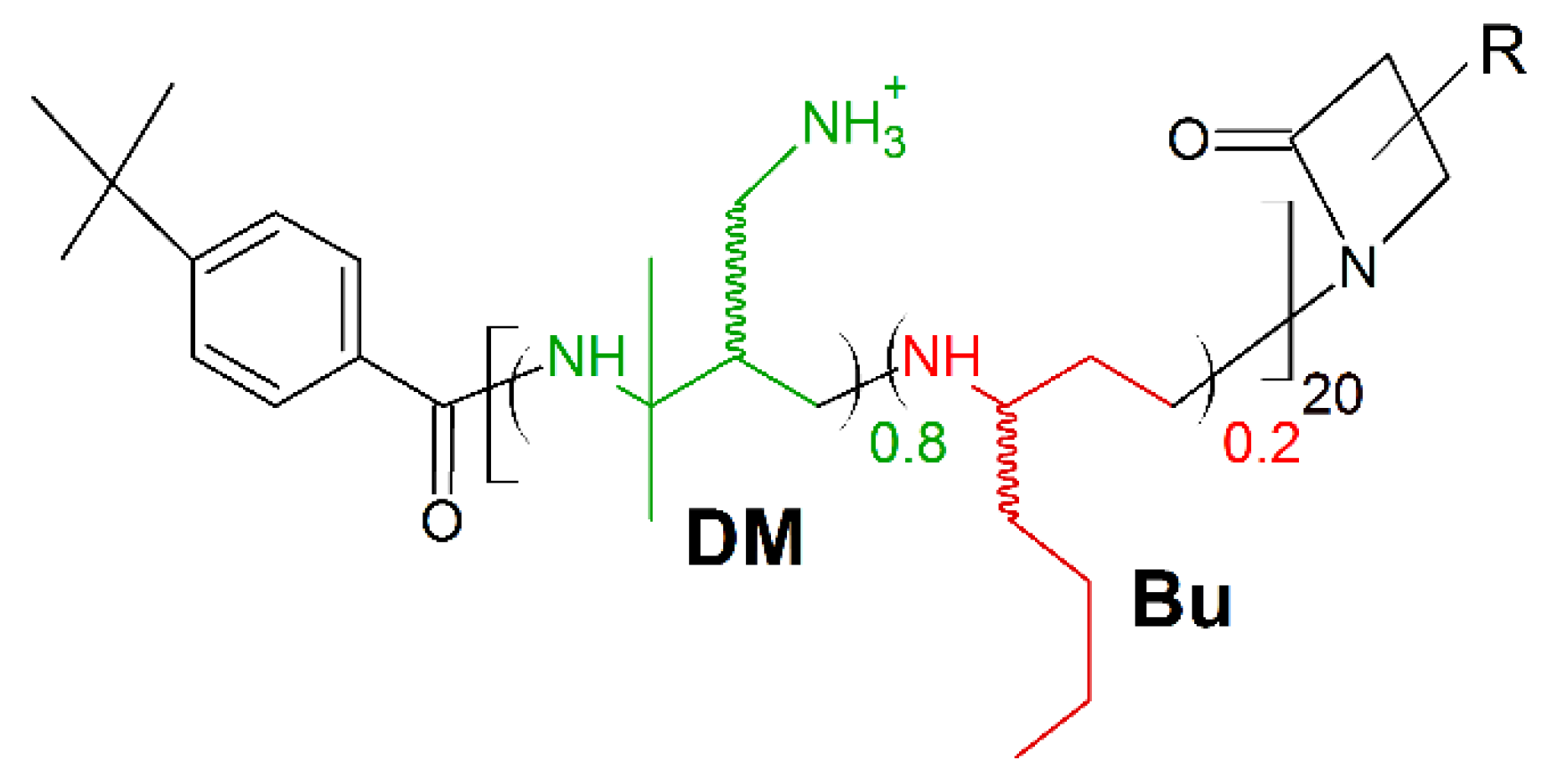
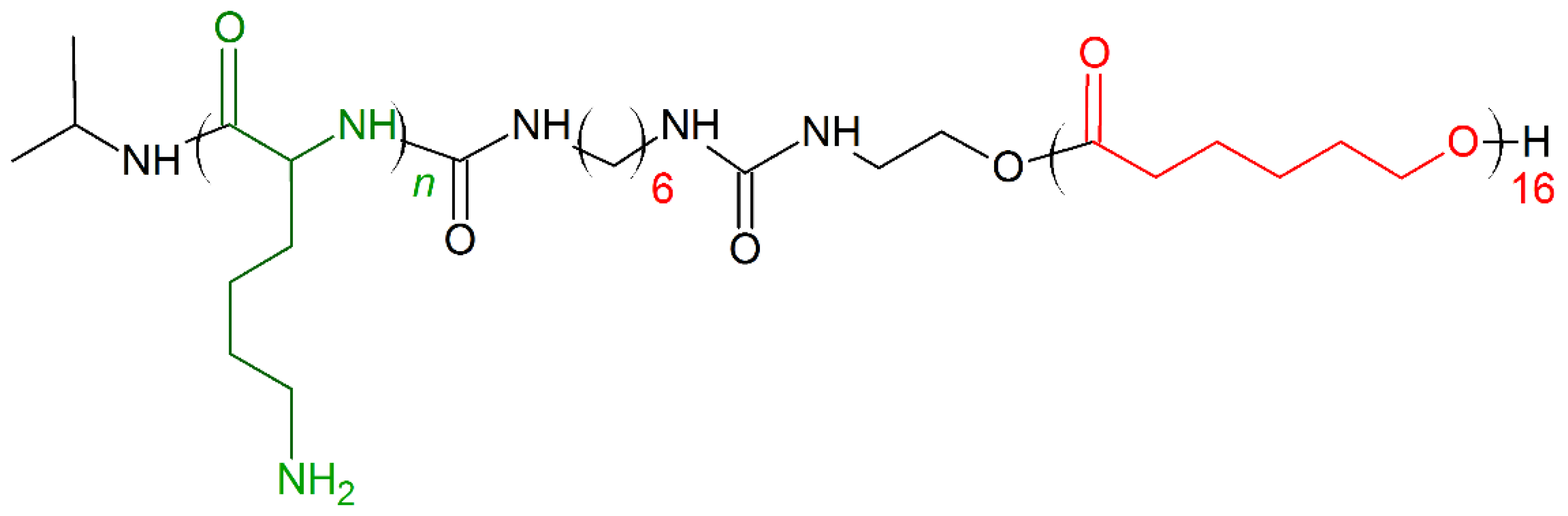
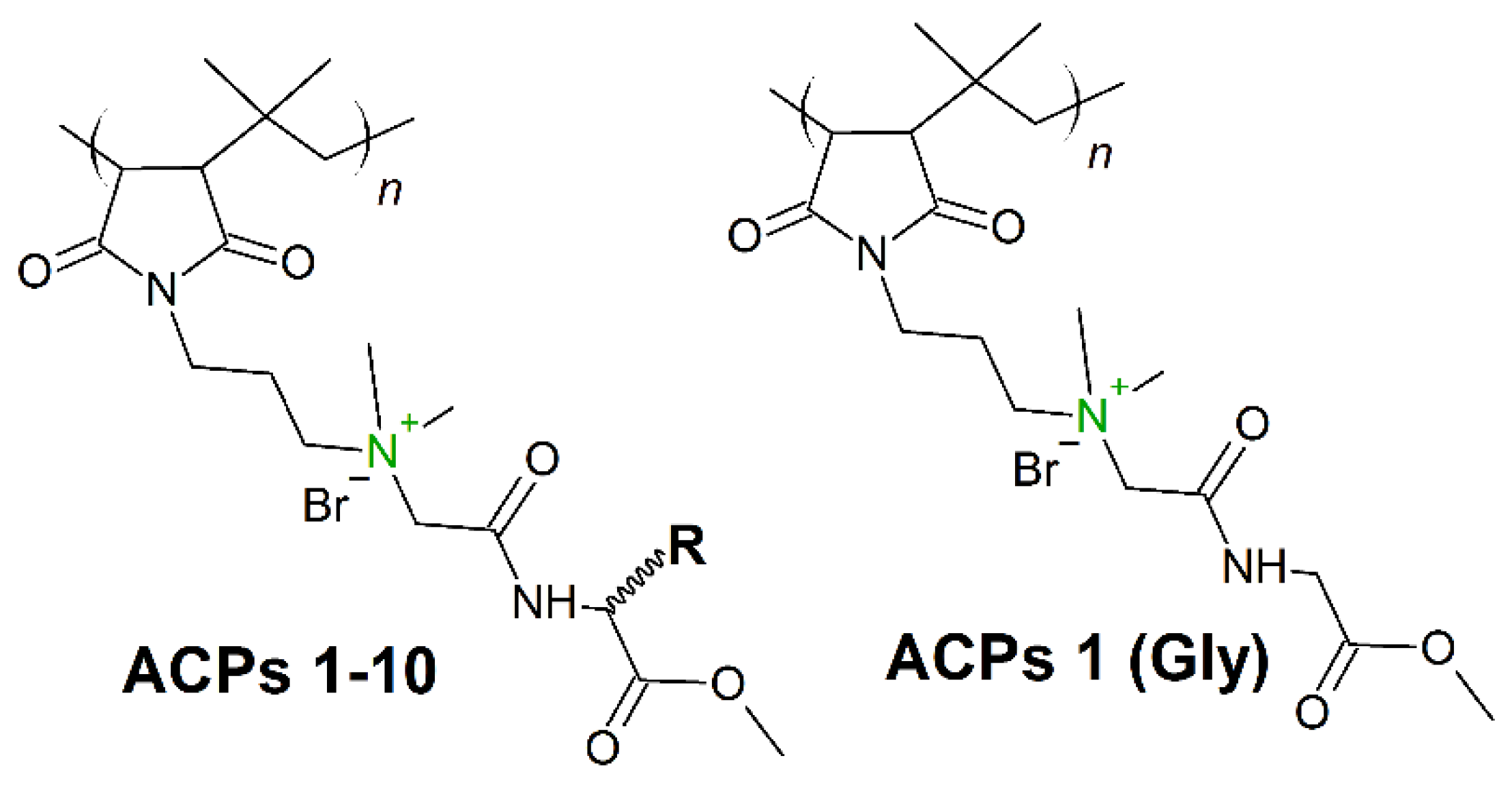
7. Amino Acid Conjugated Polymers
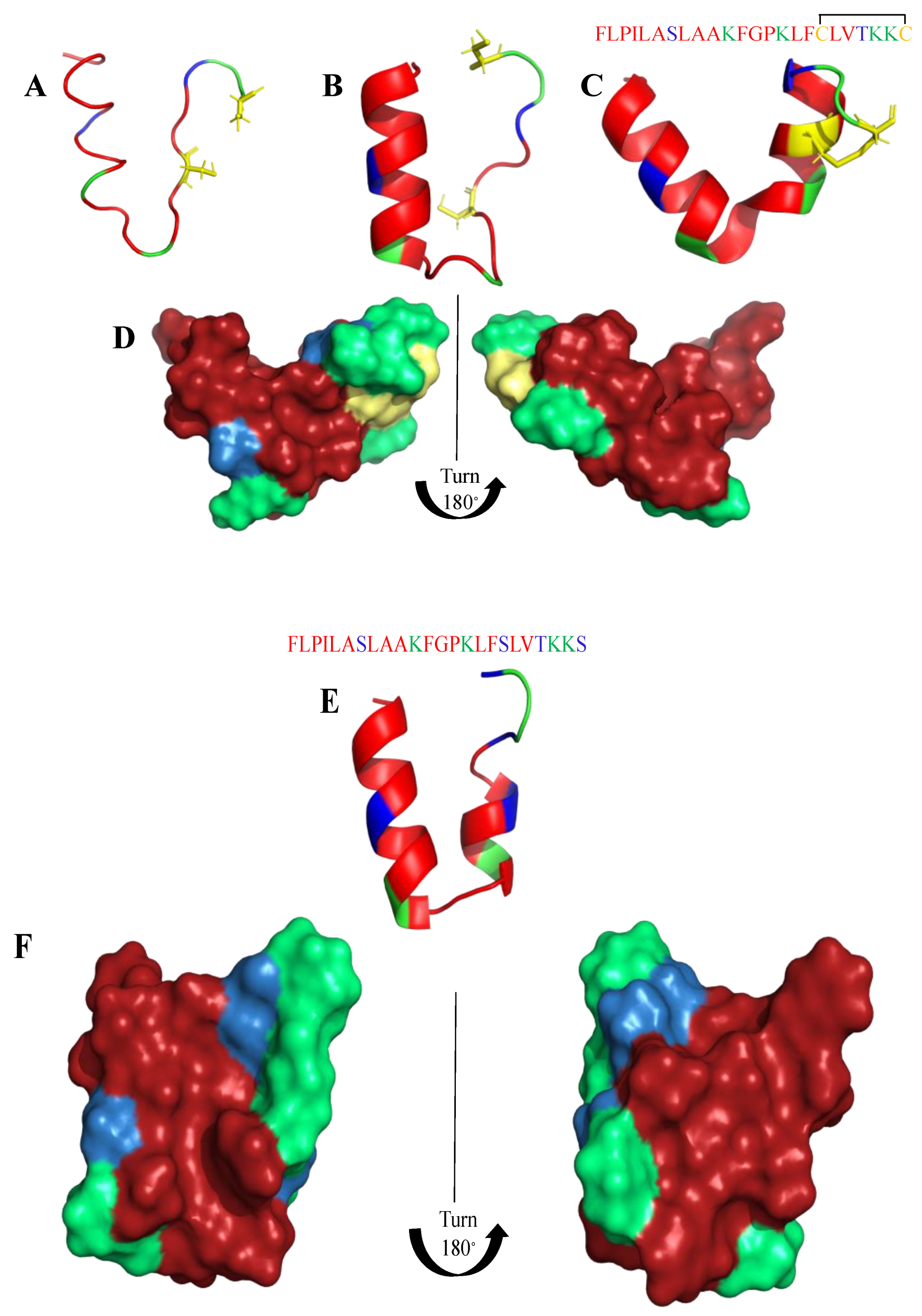
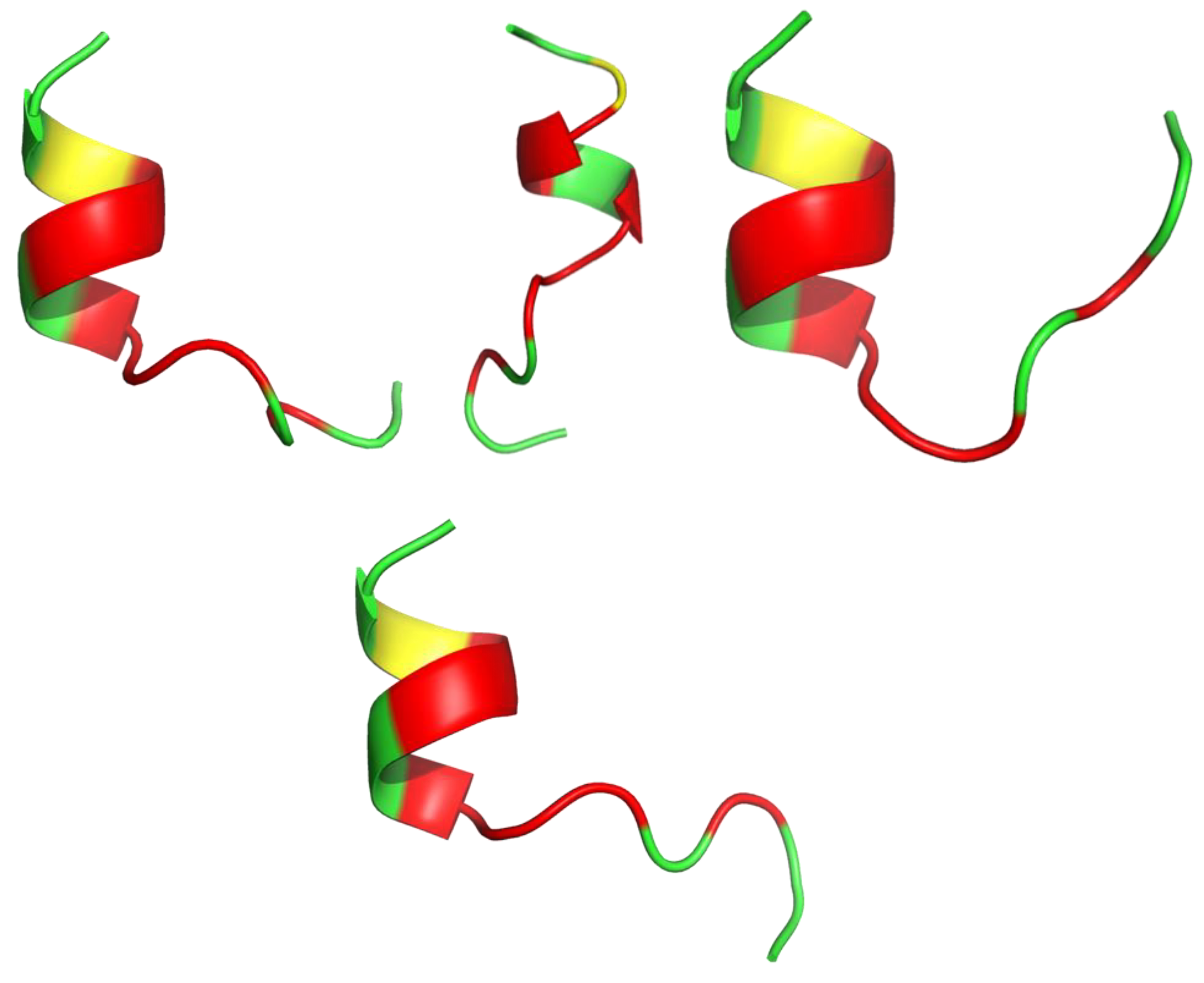
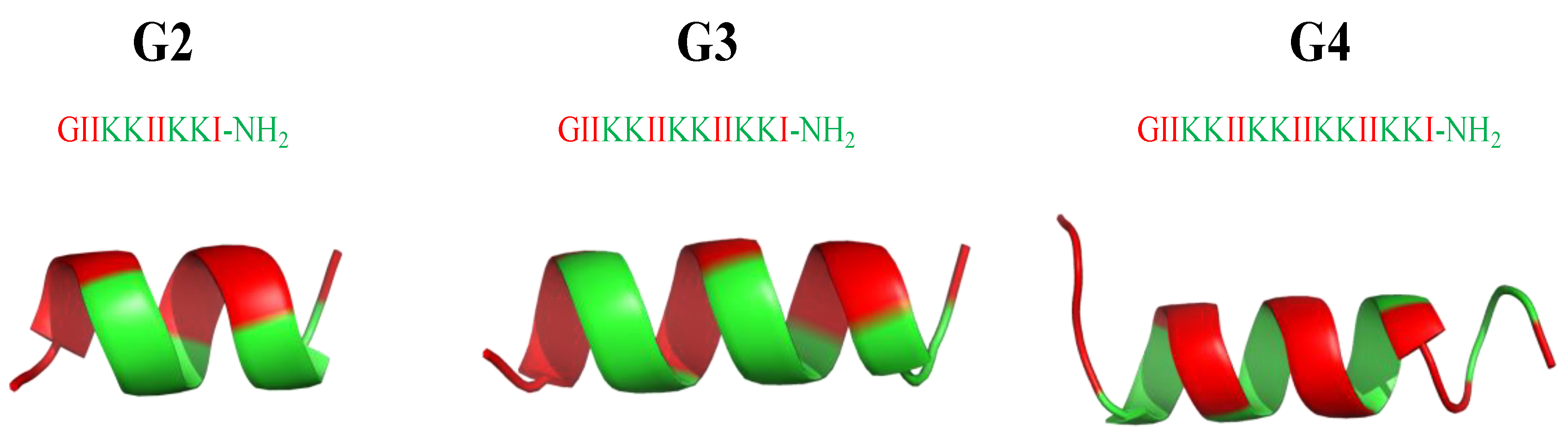
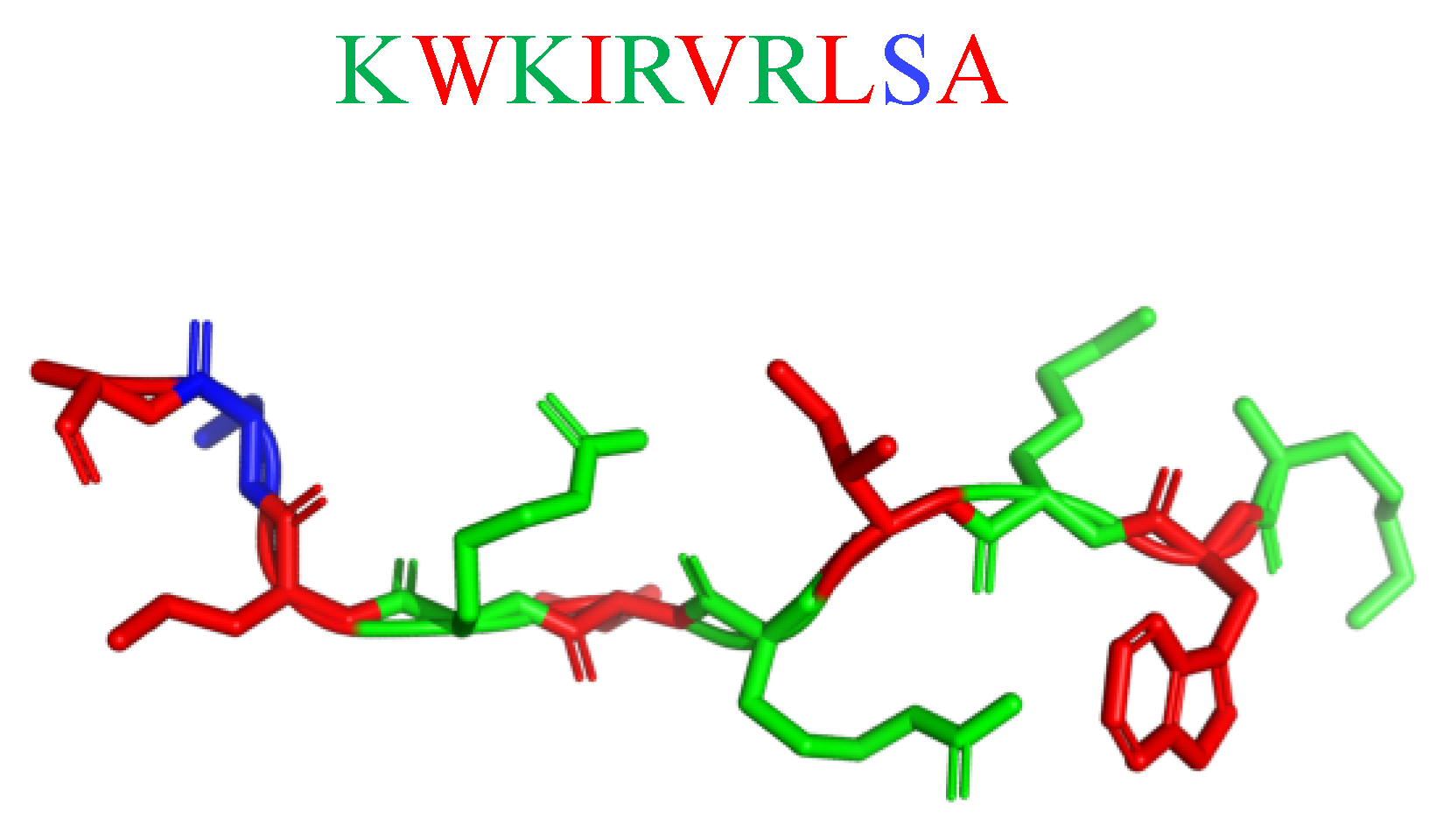
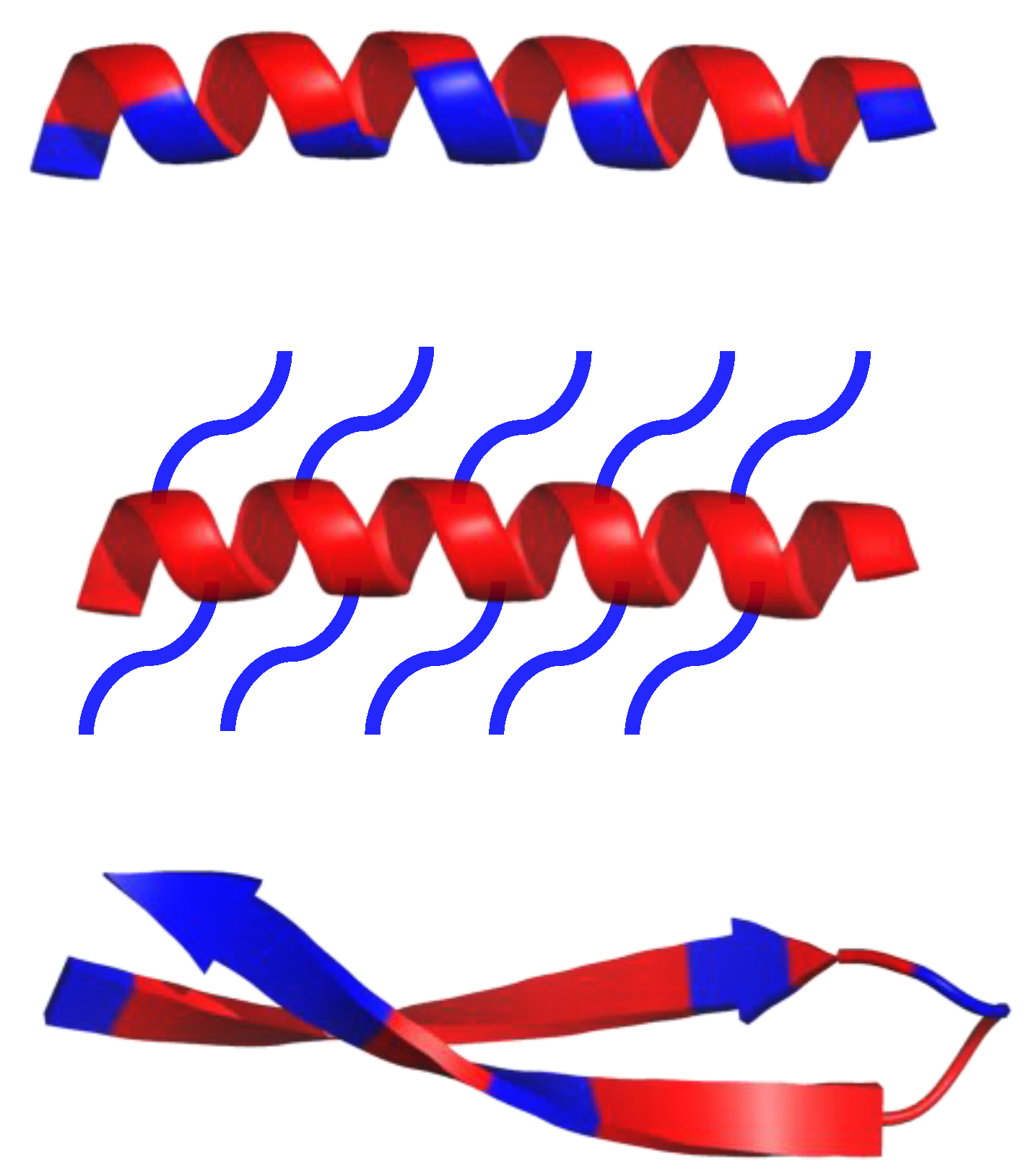
8. Mimicking the Structure of AMPs
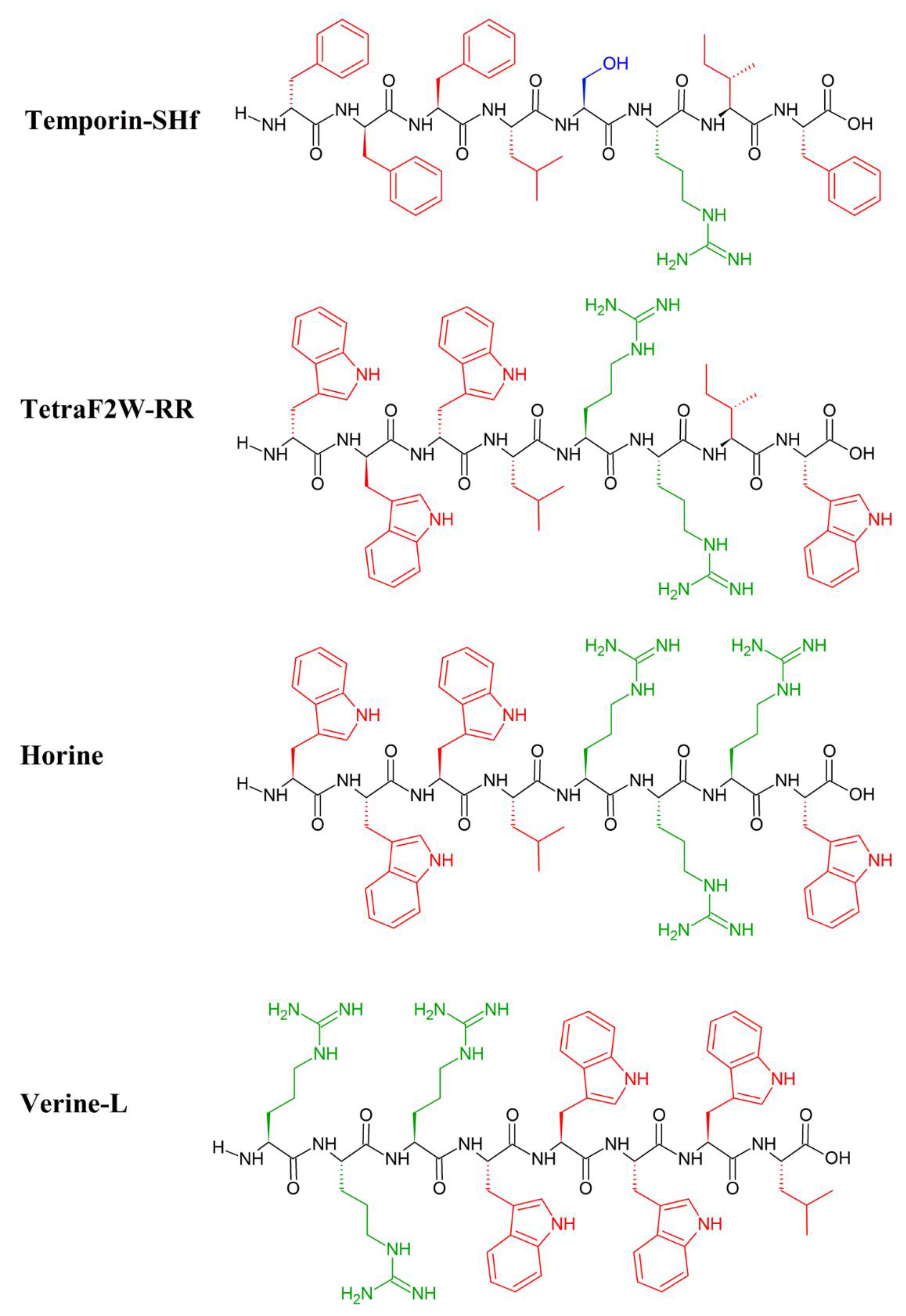
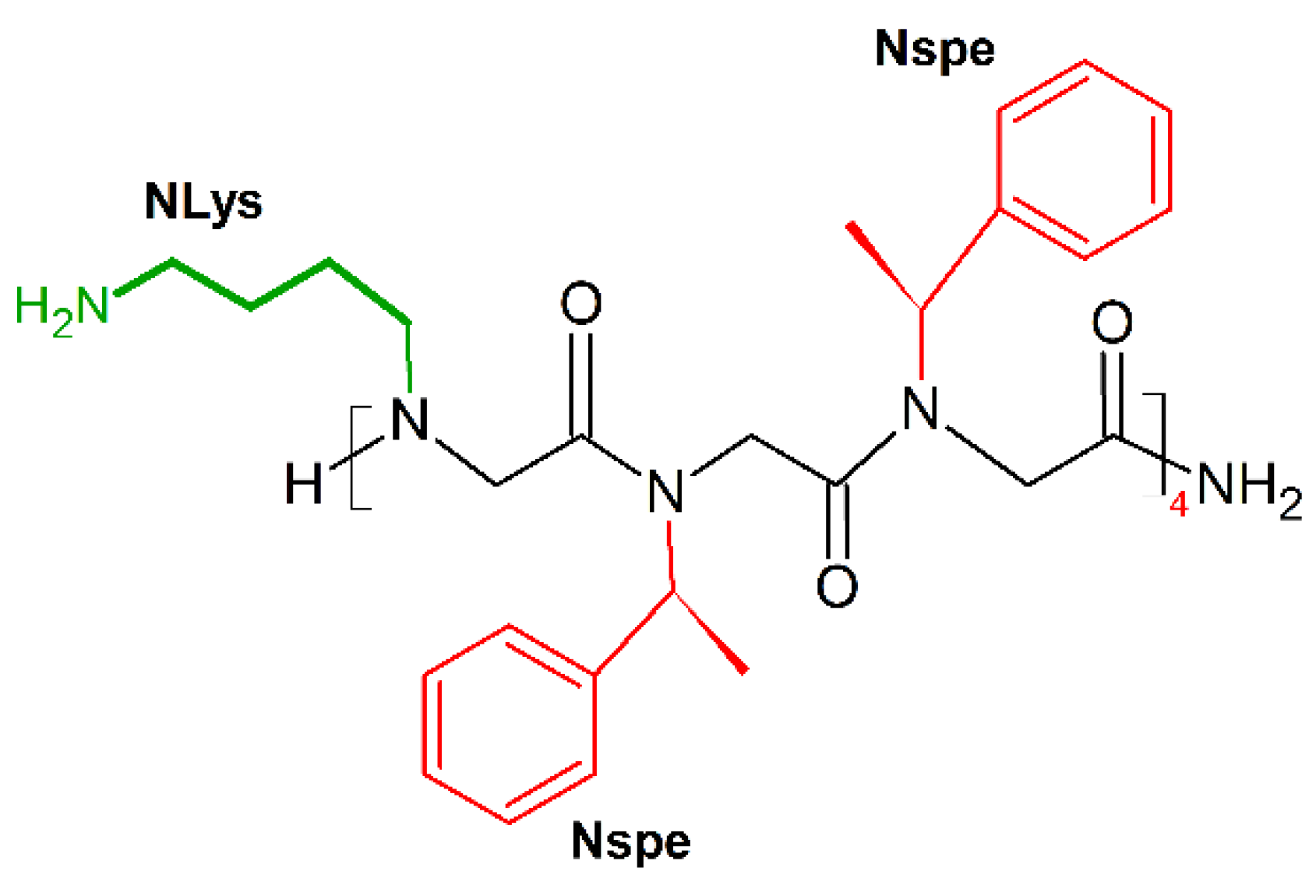
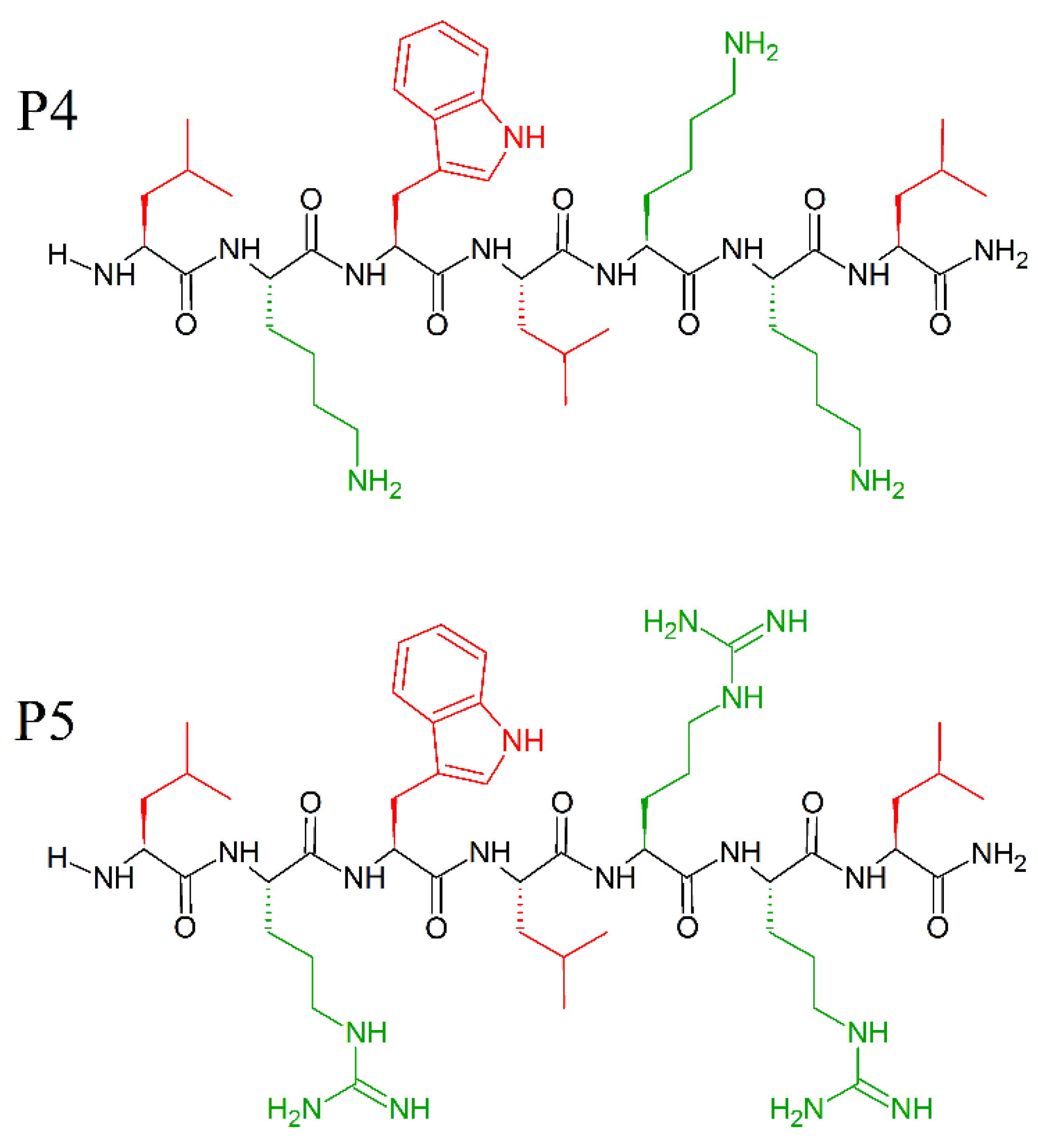
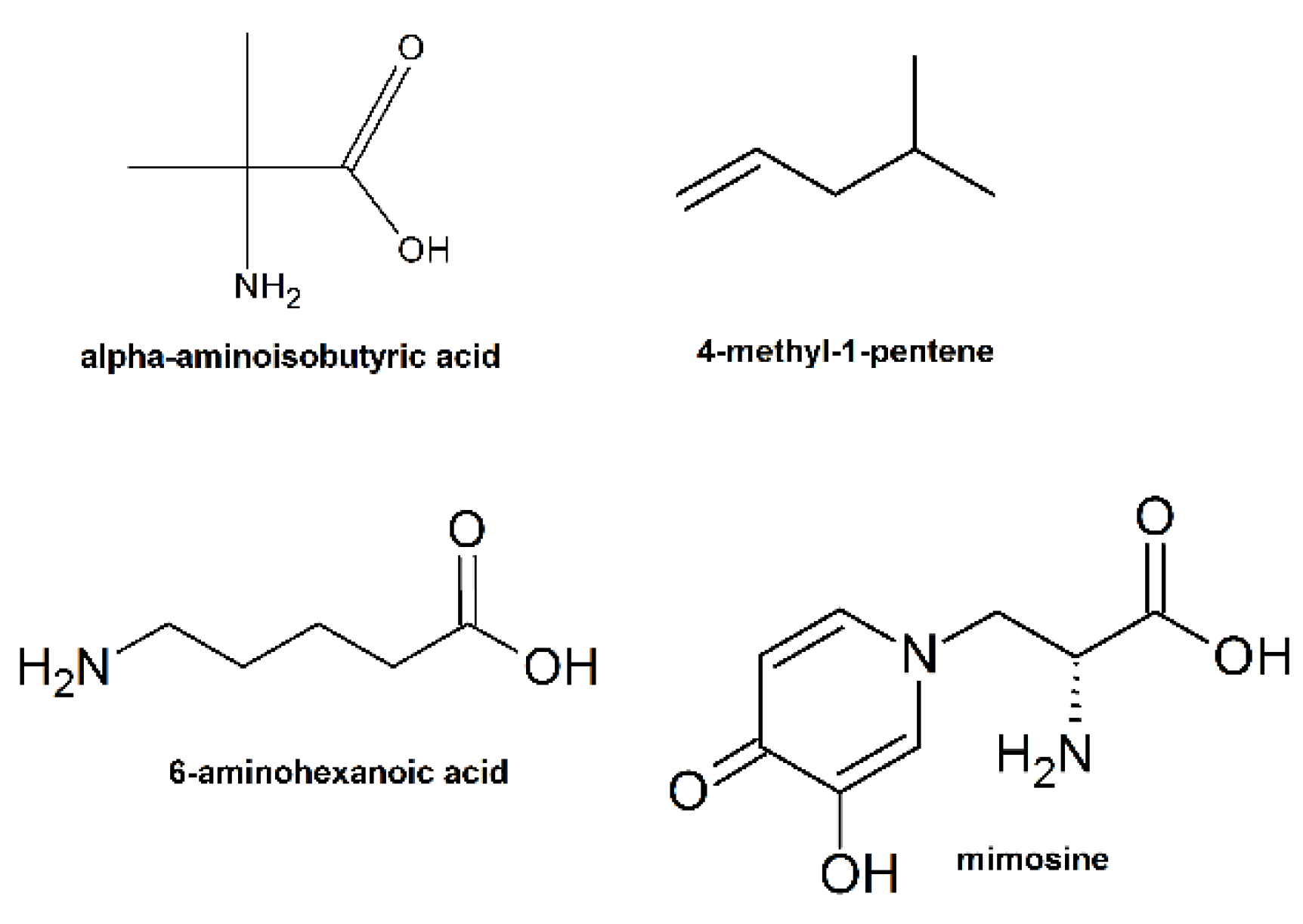
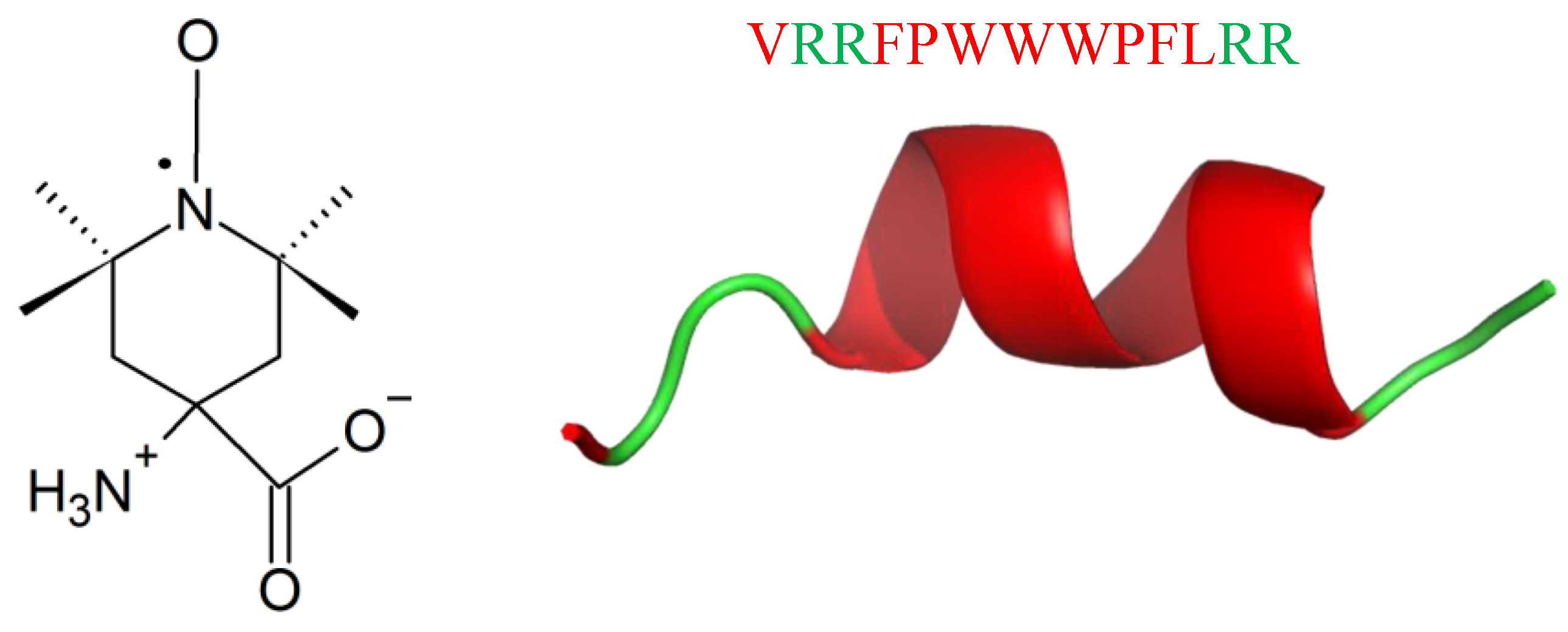
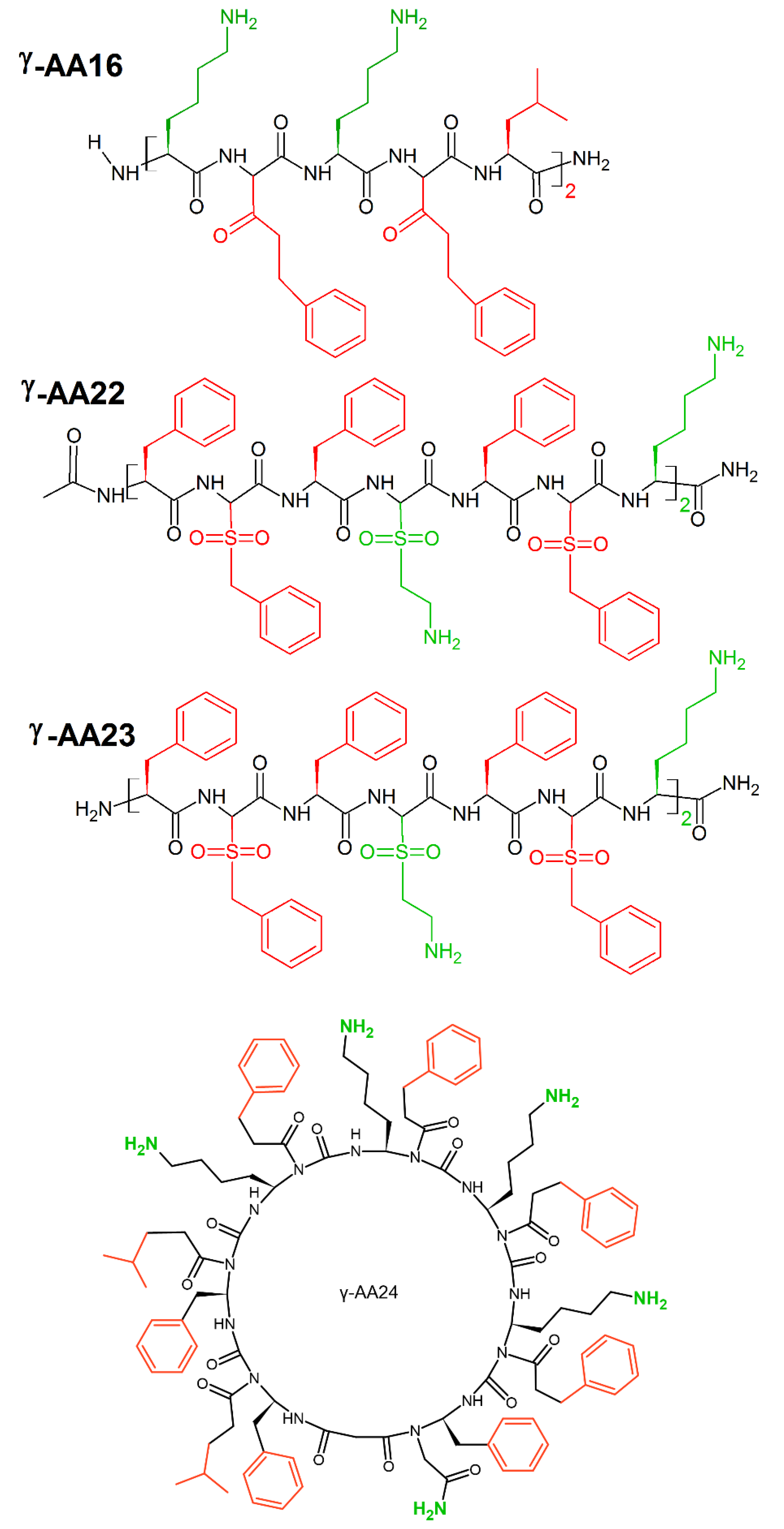
9. Unnatural Amino Acids
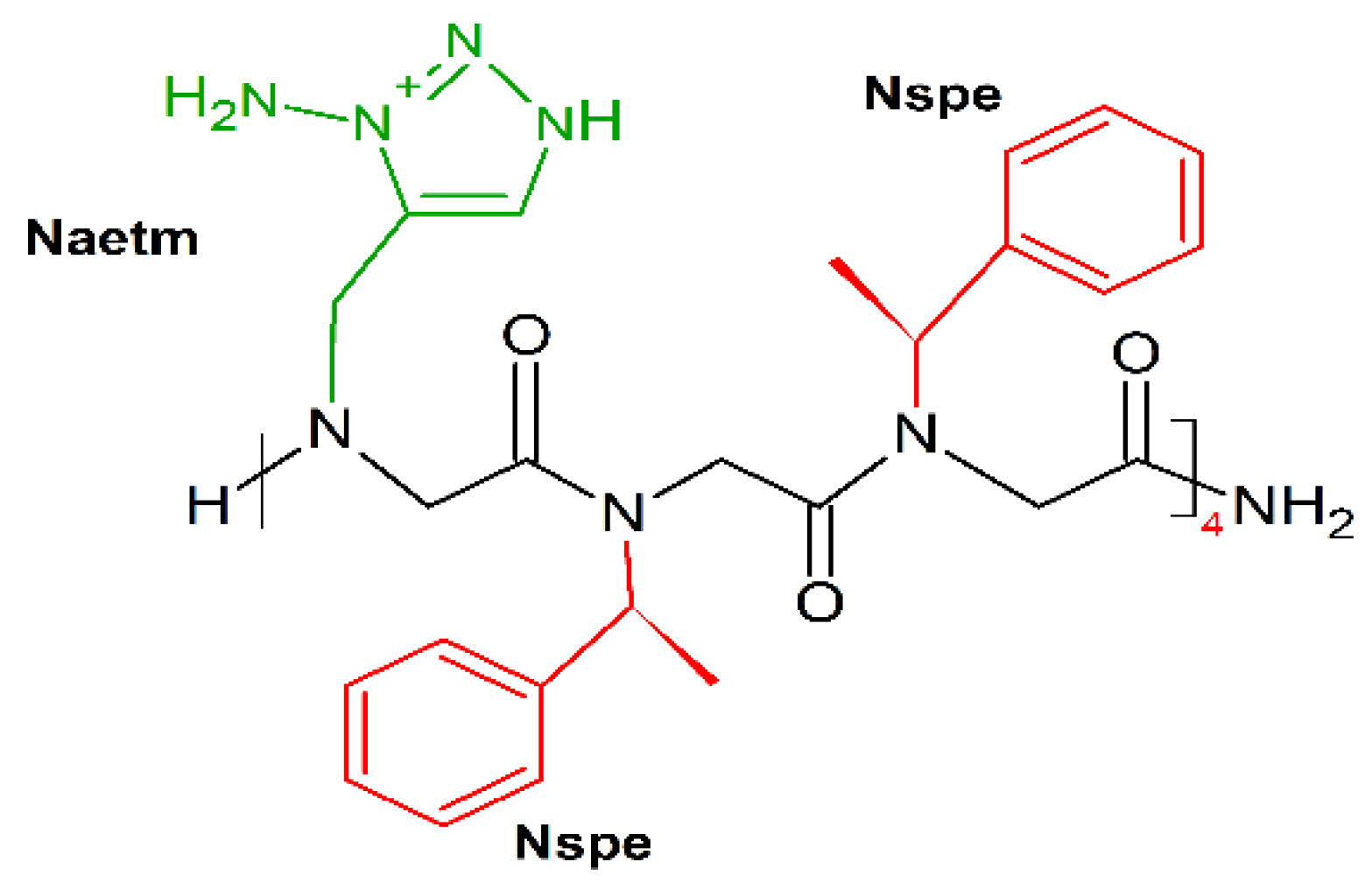
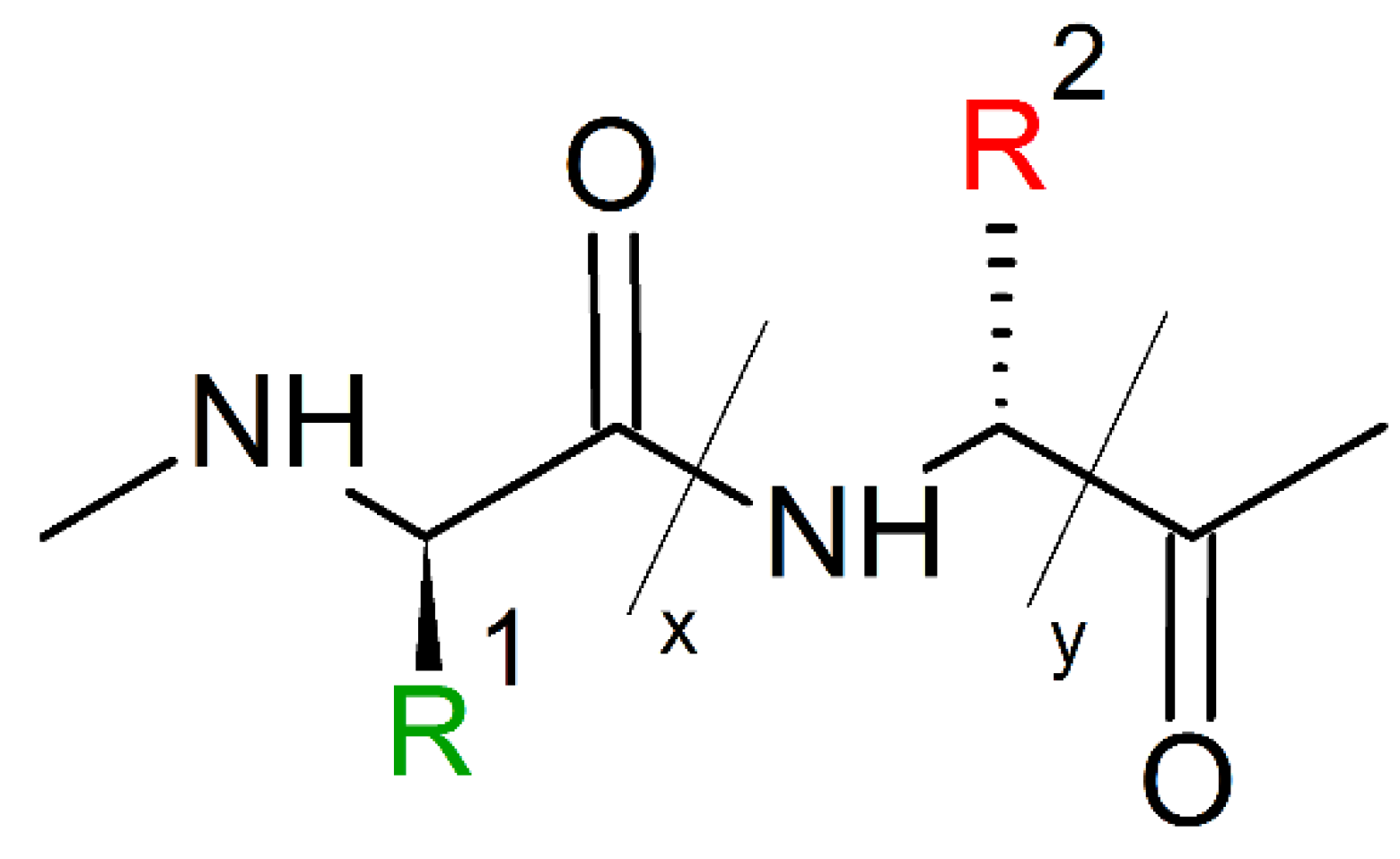
10. Mimicking Peptide Bonds
11. Enhancing AMPs Activity with Metal Ions
12. Bacteriocins
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef] [PubMed]
- Lewies, A.; Du Plessis, L.H.; Wentzel, J.F. Antimicrobial peptides: The Achilles’ heel of antibiotic resistance? Probiotics Antimicrob. Proteins 2019, 11, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Maróti, G.; Kereszt, A.; Kondorosi, E.; Mergaert, P. Natural roles of antimicrobial peptides in microbes, plants and animals. Res. Microbiol. 2011, 162, 363–374. [Google Scholar] [CrossRef]
- Kuppusamy, R.; Willcox, M.; Black, D.S.; Kumar, N. Short cationic peptidomimetic antimicrobials. Antibiotics 2019, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Antibacterial peptides: Key components needed in immunity. Cell 1991, 65, 205–207. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Blaskovich, M.A.; Cooper, M.A. Structure–Activity and Toxicity Relationships of the Antimicrobial Peptide Tachyplesin-1. ACS Infect. Dis. 2017, 3, 917–926. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Lee, E.Y.; Zhang, C.; Di Domizio, J.; Jin, F.; Connell, W.; Hung, M.; Malkoff, N.; Veksler, V.; Gilliet, M.; Ren, P. Helical antimicrobial peptides assemble into protofibril scaffolds that present ordered dsDNA to TLR9. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Ma, Z.; Han, J.; Chang, B.; Gao, L.; Lu, Z.; Lu, F.; Zhao, H.; Zhang, C.; Bie, X. Membrane-active amphipathic peptide WRL3 with in vitro antibiofilm capability and in vivo efficacy in treating methicillin-resistant Staphylococcus aureus burn wound infections. ACS Infect. Dis. 2017, 3, 820–832. [Google Scholar] [CrossRef]
- Mishra, B.; Reiling, S.; Zarena, D.; Wang, G. Host defense antimicrobial peptides as antibiotics: Design and application strategies. Curr. Opin. Chem. Biol. 2017, 38, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Bray, B.L. Large-scale manufacture of peptide therapeutics by chemical synthesis. Nat. Rev. Drug Discov. 2003, 2, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wójcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 2004, 48, 4673–4679. [Google Scholar] [CrossRef]
- Choi, S.; Isaacs, A.; Clements, D.; Liu, D.; Kim, H.; Scott, R.W.; Winkler, J.D.; DeGrado, W.F. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 6968–6973. [Google Scholar] [CrossRef]
- Porter, E.A.; Wang, X.; Lee, H.-S.; Weisblum, B.; Gellman, S.H. Non-haemolytic β-amino-acid oligomers. Nature 2000, 404, 565. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Teng, P.; Sang, P.; She, F.; Wei, L.; Cai, J. γ-AApeptides: Design, structure, and applications. Acc. Chem. Res. 2016, 49, 428–441. [Google Scholar] [CrossRef]
- Wu, H.; Niu, Y.; Padhee, S.; Wang, R.E.; Li, Y.; Qiao, Q.; Bai, G.; Cao, C.; Cai, J. Design and synthesis of unprecedented cyclic γ-AApeptides for antimicrobial development. Chem. Sci. 2012, 3, 2570–2575. [Google Scholar] [CrossRef]
- Bremner, J.B.; Keller, P.A.; Pyne, S.G.; Boyle, T.P.; Brkic, Z.; David, D.M.; Garas, A.; Morgan, J.; Robertson, M.; Somphol, K. Binaphthyl-Based Dicationic Peptoids with Therapeutic Potential. Angew. Chem. Int. Ed. 2010, 49, 537–540. [Google Scholar] [CrossRef]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef]
- Ghosh, C.; Manjunath, G.B.; Akkapeddi, P.; Yarlagadda, V.; Hoque, J.; Uppu, D.S.; Konai, M.M.; Haldar, J. Small molecular antibacterial peptoid mimics: The simpler the better! J. Med. Chem. 2014, 57, 1428–1436. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Ziegler, H.L.; Nielsen, H.M.; Frimodt-Møller, N.; Jaroszewski, J.W.; Franzyk, H. Antimicrobial, Hemolytic, and Cytotoxic Activities of β-Peptoid–Peptide Hybrid Oligomers: Improved Properties Compared to Natural AMPs. ChemBioChem 2010, 11, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Patch, J.A.; Barron, A.E. Helical peptoid mimics of magainin-2 amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Poon, Y.F.; Li, W.; Zhu, H.-Y.; Yeap, S.H.; Cao, Y.; Qi, X.; Zhou, C.; Lamrani, M.; Beuerman, R.W. A polycationic antimicrobial and biocompatible hydrogel with microbe membrane suctioning ability. Nat. Mater. 2011, 10, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Ilker, M.F.; Nüsslein, K.; Tew, G.N.; Coughlin, E.B. Tuning the hemolytic and antibacterial activities of amphiphilic polynorbornene derivatives. J. Am. Chem. Soc. 2004, 126, 15870–15875. [Google Scholar] [CrossRef]
- Chin, W.; Zhong, G.; Pu, Q.; Yang, C.; Lou, W.; De Sessions, P.F.; Periaswamy, B.; Lee, A.; Liang, Z.C.; Ding, X. A macromolecular approach to eradicate multidrug resistant bacterial infections while mitigating drug resistance onset. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Chakraborty, S.; Liu, R.; Hayouka, Z.; Chen, X.; Ehrhardt, J.; Lu, Q.; Burke, E.; Yang, Y.; Weisblum, B.; Wong, G.C. Ternary nylon-3 copolymers as host-defense peptide mimics: Beyond hydrophobic and cationic subunits. J. Am. Chem. Soc. 2014, 136, 14530–14535. [Google Scholar] [CrossRef]
- Mowery, B.P.; Lindner, A.H.; Weisblum, B.; Stahl, S.S.; Gellman, S.H. Structure—activity relationships among random nylon-3 copolymers that mimic antibacterial host-defense peptides. J. Am. Chem. Soc. 2009, 131, 9735–9745. [Google Scholar] [CrossRef]
- Qian, Y.-X.; Zhang, D.-F.; Wu, Y.-M.; Chen, Q.; Liu, R.-H. The design, synthesis and biological activity study of nylon-3 polymers as mimics of host defense peptides. ACTA Polym. Sin. 2016, 10, 1300–1311. [Google Scholar]
- Xiong, M.; Lee, M.W.; Mansbach, R.A.; Song, Z.; Bao, Y.; Peek, R.M.; Yao, C.; Chen, L.-F.; Ferguson, A.L.; Wong, G.C. Helical antimicrobial polypeptides with radial amphiphilicity. Proc. Natl. Acad. Sci. USA 2015, 112, 13155–13160. [Google Scholar] [CrossRef]
- Xiong, M.; Han, Z.; Song, Z.; Yu, J.; Ying, H.; Yin, L.; Cheng, J. Bacteria-assisted activation of antimicrobial polypeptides by a random-coil to helix transition. Angew. Chem. Int. Ed. 2017, 56, 10826–10829. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ma, P.; Xie, J.; Zhang, S.; Xiao, X.; Qiao, Z.; Shao, N.; Zhou, M.; Zhang, W.; Dai, C. Host defense peptide mimicking poly-β-peptides with fast, potent and broad spectrum antibacterial activities. Biomater. Sci. 2019, 7, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-K.; Lam, S.J.; Ho, K.K.; Kumar, N.; Qiao, G.G.; Egan, S.; Boyer, C.; Wong, E.H. Rational design of single-chain polymeric nanoparticles that kill planktonic and biofilm bacteria. ACS Infect. Dis. 2017, 3, 237–248. [Google Scholar] [CrossRef]
- Hartlieb, M.; Williams, E.G.; Kuroki, A.; Perrier, S.; Locock, K.E. Antimicrobial Polymers: Mimicking Amino Acid Functionali ty, Sequence Control and Three-dimensional Structure of Host-defen se Peptides. Curr. Med. Chem. 2017, 24, 2115–2140. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Zhang, J.; Hu, X.; Li, Z.; Fa, K.; Liu, H.; Waigh, T.A.; McBain, A.; Lu, J.R. Hydrophobic Control of the Bioactivity and Cytotoxicity of de Novo-Designed Antimicrobial Peptides. ACS Appl. Mater. Interfaces 2019, 11, 34609–34620. [Google Scholar] [CrossRef]
- Carratalá, J.V.; Serna, N.; Villaverde, A.; Vázquez, E.; Ferrer-Miralles, N. Nanostructured antimicrobial peptides: The last push towards clinics. Biotechnol. Adv. 2020, 44, 107603. [Google Scholar] [CrossRef]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar] [CrossRef]
- Clifton, L.A.; Skoda, M.W.; Le Brun, A.P.; Ciesielski, F.; Kuzmenko, I.; Holt, S.A.; Lakey, J.H. Effect of divalent cation removal on the structure of gram-negative bacterial outer membrane models. Langmuir 2015, 31, 404–412. [Google Scholar] [CrossRef]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolym. Pept. Sci. Sect. 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Lohner, K. Antimicrobial peptides targeting gram-positive bacteria. Pharmaceuticals 2016, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Andreev, K.; Martynowycz, M.W.; Huang, M.L.; Kuzmenko, I.; Bu, W.; Kirshenbaum, K.; Gidalevitz, D. Hydrophobic interactions modulate antimicrobial peptoid selectivity towards anionic lipid membranes. Biochim. Biophys. Acta BBA Biomembr. 2018, 1860, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Houghten, R.A. Design of model amphipathic peptides having potent antimicrobial activities. Biochemistry 1992, 31, 12688–12694. [Google Scholar] [CrossRef]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA 1984, 81, 140–144. [Google Scholar] [CrossRef]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic α helical antimicrobial peptides. A systematic study of the effects of structural and physical properties on biological activity. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Iavicoli, P.; Rossi, F.; Lamarre, B.; Bella, A.; Ryadnov, M.G.; Calzolai, L. Modulating charge-dependent and folding-mediated antimicrobial interactions at peptide—lipid interfaces. Eur. Biophys. J. 2017, 46, 375–382. [Google Scholar] [CrossRef]
- Findlay, B.; Zhanel, G.G.; Schweizer, F. Cationic amphiphiles, a new generation of antimicrobials inspired by the natural antimicrobial peptide scaffold. Antimicrob. Agents Chemother. 2010, 54, 4049–4058. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Wade, J.D. Current challenges in peptide-based drug discovery. Front. Chem. 2014, 2, 62. [Google Scholar] [CrossRef]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.; Silva-Pereira, I.; Kyaw, C. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef]
- Eckert, R. Road to clinical efficacy: Challenges and novel strategies for antimicrobial peptide development. Future Microbiol. 2011, 6, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Bednarska, N.G.; Wren, B.W.; Willcocks, S.J. The importance of the glycosylation of antimicrobial peptides: Natural and synthetic approaches. Drug Discov. Today 2017, 22, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, A.; Frick, I.M.; Andersson, E.; Tapper, H.; Björck, L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 2002, 46, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updates 2016, 26, 43–57. [Google Scholar] [CrossRef]
- Gai, Z.; Samodelov, S.L.; Kullak-Ublick, G.A.; Visentin, M. Molecular mechanisms of colistin-induced nephrotoxicity. Molecules 2019, 24, 653. [Google Scholar] [CrossRef]
- Radermacher, S.; Schoop, V.; Schluesener, H. Bactenecin, a leukocytic antimicrobial peptide, is cytotoxic to neuronal and glial cells. J. Neurosci. Res. 1993, 36, 657–662. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Deptuła, M.; Wardowska, A.; Dzierżyńska, M.; Rodziewicz-Motowidło, S.; Pikuła, M. Antibacterial peptides in dermatology–strategies for evaluation of allergic potential. Molecules 2018, 23, 414. [Google Scholar] [CrossRef]
- Corominas, M.; Gastaminza, G.; Lobera, T. Hypersensitivity reactions to biological drugs. J. Investig. Allergol. Clin. Immunol. 2014, 24, 212–225. [Google Scholar]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of therapeutic protein aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, M.; Bird, C.; Dilger, P.; Gaines-Das, R.; Thorpe, R. Strategies for detection, measurement and characterization of unwanted antibodies induced by therapeutic biologicals. J. Immunol. Methods 2003, 278, 1–17. [Google Scholar] [CrossRef]
- Fathallah, A.M.; Bankert, R.B.; Balu-Iyer, S.V. Immunogenicity of subcutaneously administered therapeutic proteins—a mechanistic perspective. AAPS J. 2013, 15, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Mari, A.; Rasi, C.; Palazzo, P.; Scala, E. Allergen databases: Current status and perspectives. Curr. Allergy Asthma Rep. 2009, 9, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Pikuła, M.; Zieliński, M.; Specjalski, K.; Barańska-Rybak, W.; Dawgul, M.; Langa, P.; Jassem, E.; Kamysz, W.; Trzonkowski, P. In vitro evaluation of the allergic potential of antibacterial peptides: Camel and citropin. Chem. Biol. Drug Des. 2016, 87, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Bobone, S.; Stella, L. Selectivity of Antimicrobial Peptides: A Complex Interplay of Multiple Equilibria. In Antimicrobial Peptides; Springer: Berlin/Heidelberg, Germany, 2019; pp. 175–214. [Google Scholar]
- Maturana, P.; Martinez, M.; Noguera, M.E.; Santos, N.; Disalvo, E.A.; Semorile, L.; Maffia, P.C.; Hollmann, A. Lipid selectivity in novel antimicrobial peptides: Implication on antimicrobial and hemolytic activity. Colloids Surf. B Biointerfaces 2017, 153, 152–159. [Google Scholar] [CrossRef]
- Dörr, T.; Moynihan, P.J.; Mayer, C. Bacterial cell wall structure and dynamics. Front. Microbiol. 2019, 10, 2051. [Google Scholar] [CrossRef]
- Tomasz, A.; McDonnell, M.; Westphal, M.; Zanati, E. Coordinated incorporation of nascent peptidoglycan and teichoic acid into pneumococcal cell walls and conservation of peptidoglycan during growth. J. Biol. Chem. 1975, 250, 337–341. [Google Scholar]
- Bertani, B.; Ruiz, N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 2018. [Google Scholar] [CrossRef]
- Zhang, G.; Meredith, T.C.; Kahne, D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr. Opin. Microbiol. 2013, 16, 779–785. [Google Scholar] [CrossRef]
- Gan, L.; Chen, S.; Jensen, G.J. Molecular organization of Gram-negative peptidoglycan. Proc. Natl. Acad. Sci. USA 2008, 105, 18953–18957. [Google Scholar] [CrossRef] [PubMed]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-κB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Bocchinfuso, G.; Palleschi, A.; Orioni, B.; Grande, G.; Formaggio, F.; Toniolo, C.; Park, Y.; Hahm, K.S.; Stella, L. Different mechanisms of action of antimicrobial peptides: Insights from fluorescence spectroscopy experiments and molecular dynamics simulations. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2009, 15, 550–558. [Google Scholar] [CrossRef]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial peptides in action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef]
- Bogdanova, L.R.; Valiullina, Y.A.; Faizullin, D.A.; Kurbanov, R.K.; Ermakova, E.A. Spectroscopic, zeta potential and molecular dynamics studies of the interaction of antimicrobial peptides with model bacterial membrane. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 242, 118785. [Google Scholar] [CrossRef]
- Lohner, K. Membrane-active Antimicrobial Peptides as Template Structures for Novel Antibiotic Agents. Curr. Top. Med. Chem. 2017, 17, 508–519. [Google Scholar] [CrossRef]
- Aisenbrey, C.; Marquette, A.; Bechinger, B. The Mechanisms of Action of Cationic Antimicrobial Peptides Refined by Novel Concepts from Biophysical Investigations. Adv. Exp. Med. Biol. 2019, 1117, 33–64. [Google Scholar]
- Zeth, K.; Sancho-Vaello, E. The Human Antimicrobial Peptides Dermcidin and LL-37 Show Novel Distinct Pathways in Membrane Interactions. Front. Chem. 2017, 5, 86. [Google Scholar] [CrossRef]
- Yount, N.Y.; Bayer, A.S.; Xiong, Y.Q.; Yeaman, M.R. Advances in antimicrobial peptide immunobiology. Pept. Sci. Orig. Res. Biomol. 2006, 84, 435–458. [Google Scholar] [CrossRef]
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular targeting mechanisms by antimicrobial peptides. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yin, J.; Zhao, J.; Ma, S.R.; Wang, H.R.; Wang, M.; Chen, W.; Wei, W.Q. Microbial Similarity and Preference for Specific Sites in Healthy Oral Cavity and Esophagus. Front. Microbiol. 2018, 9, 1603. [Google Scholar] [CrossRef] [PubMed]
- Kitada, K.; de Toledo, A.; Oho, T. Increase in detectable opportunistic bacteria in the oral cavity of orthodontic patients. Int. J. Dent. Hyg. 2009, 7, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Tets, G.V.; Vikina, D.S.; Vecherkovskaia, M.F.; Domorad, A.A.; Kharlamova, V.V.; Tets, V.V. New approaches to oral cavity opportunistic microbiota study. Stomatologiia 2013, 92, 14–16. [Google Scholar]
- Feazel, L.M.; Baumgartner, L.K.; Peterson, K.L.; Frank, D.N.; Harris, J.K.; Pace, N.R. Opportunistic pathogens enriched in showerhead biofilms. Proc. Natl. Acad. Sci. USA 2009, 106, 16393–16399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Pabst, B.; Klapper, I.; Stewart, P.S. General theory for integrated analysis of growth, gene, and protein expression in biofilms. PLoS ONE 2013, 8, e83626. [Google Scholar] [CrossRef]
- Muras, A.; Mayer, C.; Otero-Casal, P.; Exterkate, R.A.M.; Brandt, B.W.; Crielaard, W.; Otero, A.; Krom, B.P. Short-Chain N-Acylhomoserine Lactone Quorum-Sensing Molecules Promote Periodontal Pathogens in In Vitro Oral Biofilms. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Cho, Y.J.; Song, H.Y.; Ben Amara, H.; Choi, B.K.; Eunju, R.; Cho, Y.A.; Seol, Y.; Lee, Y.; Ku, Y.; Rhyu, I.C.; et al. In Vivo Inhibition of Porphyromonas gingivalis Growth and Prevention of Periodontitis With Quorum-Sensing Inhibitors. J. Periodontol. 2016, 87, 1075–1082. [Google Scholar] [CrossRef]
- Ertugrul, A.S.; Sahin, H.; Dikilitas, A.; Alpaslan, N.Z.; Bozoglan, A.; Tekin, Y. Gingival crevicular fluid levels of human beta-defensin-2 and cathelicidin in smoker and non-smoker patients: A cross-sectional study. J. Periodontal Res. 2014, 49, 282–289. [Google Scholar] [CrossRef]
- Turkoglu, O.; Emingil, G.; Kutukculer, N.; Atilla, G. Gingival crevicular fluid levels of cathelicidin LL-37 and interleukin-18 in patients with chronic periodontitis. J. Periodontol. 2009, 80, 969–976. [Google Scholar] [CrossRef]
- Available online: http://aps.unmc.edu/AP/database/mysql.php (accessed on 27 September 2020).
- Greer, A.; Zenobia, C.; Darveau, R.P. Defensins and LL-37: A review of function in the gingival epithelium. Periodontology 2000 2013, 63, 67–79. [Google Scholar] [CrossRef] [PubMed]
- de Sousa-Pereira, P.; Amado, F.; Abrantes, J.; Ferreira, R.; Esteves, P.J.; Vitorino, R. An evolutionary perspective of mammal salivary peptide families: Cystatins, histatins, statherin and PRPs. Arch. Oral Biol. 2013, 58, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Khurshid, Z.; Naseem, M.; Sheikh, Z.; Najeeb, S.; Shahab, S.; Zafar, M.S. Oral antimicrobial peptides: Types and role in the oral cavity. Saudi Pharm. J. SPJ Off. Publ. Saudi Pharm. Soc. 2016, 24, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Lachowicz, J.I.; Dalla Torre, G.; Cappai, R.; Randaccio, E.; Nurchi, V.M.; Bachor, R.; Szewczuk, Z.; Jaremko, L.; Jaremko, M.; Pisano, M.B.; et al. Metal self-assembly mimosine peptides with enhanced antimicrobial activity: Towards a new generation of multitasking chelating agents. Dalton Trans. 2020, 49, 2862–2879. [Google Scholar] [CrossRef]
- Puklo, M.; Guentsch, A.; Hiemstra, P.S.; Eick, S.; Potempa, J. Analysis of neutrophil-derived antimicrobial peptides in gingival crevicular fluid suggests importance of cathelicidin LL-37 in the innate immune response against periodontogenic bacteria. Oral Microbiol. Immunol. 2008, 23, 328–335. [Google Scholar] [CrossRef]
- Orru, G.; Marini, M.F.; Ciusa, M.L.; Isola, D.; Cotti, M.; Baldoni, M.; Piras, V.; Pisano, E.; Montaldo, C. Usefulness of real time PCR for the differentiation and quantification of 652 and JP2 Actinobacillus actinomycetemcomitans genotypes in dental plaque and saliva. BMC Infect. Dis. 2006, 6, 98. [Google Scholar] [CrossRef]
- Abriani, A.; Hamad, C. Activated antimicrobial peptides due to periodontal bacteria in synovial fluid - The link between psoriatic arthritis and periodontitis? Med. Hypotheses 2020, 144, 109967. [Google Scholar] [CrossRef]
- Jourdain, M.L.; Velard, F.; Pierrard, L.; Sergheraert, J.; Gangloff, S.C.; Braux, J. Cationic antimicrobial peptides and periodontal physiopathology: A systematic review. J. Periodontal Res. 2019, 54, 589–600. [Google Scholar] [CrossRef]
- Li, S.; Schmalz, G.; Schmidt, J.; Krause, F.; Haak, R.; Ziebolz, D. Antimicrobial peptides as a possible interlink between periodontal diseases and its risk factors: A systematic review. J. Periodontal Res. 2018, 53, 145–155. [Google Scholar] [CrossRef]
- Turkoglu, O.; Gurkan, A.; Emingil, G.; Afacan, B.; Toz, H.; Kutukculer, N.; Atilla, G. Are antimicrobial peptides related to cyclosporine A-induced gingival overgrowth? Arch. Oral Biol. 2015, 60, 508–515. [Google Scholar] [CrossRef]
- Hussain, M.; Stover, C.M.; Dupont, A. P. gingivalis in Periodontal Disease and Atherosclerosis—Scenes of Action for Antimicrobial Peptides and Complement. Front. Immunol. 2015, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Guncu, G.N.; Yilmaz, D.; Kononen, E.; Gursoy, U.K. Salivary Antimicrobial Peptides in Early Detection of Periodontitis. Front. Cell. Infect. Microbiol. 2015, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Dommisch, H.; Jepsen, S. Diverse functions of defensins and other antimicrobial peptides in periodontal tissues. Periodontology 2000 2015, 69, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Bedran, T.B.; Mayer, M.P.; Spolidorio, D.P.; Grenier, D. Synergistic anti-inflammatory activity of the antimicrobial peptides human beta-defensin-3 (hBD-3) and cathelicidin (LL-37) in a three-dimensional co-culture model of gingival epithelial cells and fibroblasts. PLoS ONE 2014, 9, e106766. [Google Scholar] [CrossRef]
- Abbassi, F.; Lequin, O.; Piesse, C.; Goasdoué, N.; Foulon, T.; Nicolas, P.; Ladram, A. Temporin-SHf, a new type of phe-rich and hydrophobic ultrashort antimicrobial peptide. J. Biol. Chem. 2010, 285, 16880–16892. [Google Scholar] [CrossRef]
- Mishra, B.; Lushnikova, T.; Golla, R.M.; Wang, X.; Wang, G. Design and surface immobilization of short anti-biofilm peptides. Acta Biomater. 2017, 49, 316–328. [Google Scholar] [CrossRef]
- Bozelli, J.C.; Yune, J.; Dang, X.; Narayana, J.L.; Wang, G.; Epand, R.M. Membrane activity of two short Trp-rich amphipathic peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183280. [Google Scholar] [CrossRef]
- Manzo, G.; Ferguson, P.M.; Hind, C.K.; Clifford, M.; Gustilo, V.B.; Ali, H.; Bansal, S.S.; Bui, T.T.; Drake, A.F.; Atkinson, R.A.; et al. Temporin L and aurein 2.5 have identical conformations but subtly distinct membrane and antibacterial activities. Sci. Rep. 2019, 9, 10934. [Google Scholar] [CrossRef]
- Suh, J.-Y.; Lee, K.-H.; Chi, S.-W.; Hong, S.-Y.; Choi, B.-W.; Moon, H.-M.; Choi, B.-S. Unusually stable helical kink in the antimicrobial peptide—A derivative of gaegurin. FEBS Lett. 1996, 392, 309–312. [Google Scholar] [CrossRef]
- Hossain, M.A.; Guilhaudis, L.; Sonnevend, A.; Attoub, S.; Van Lierop, B.J.; Robinson, A.J.; Wade, J.D.; Conlon, J.M. Synthesis, conformational analysis and biological properties of a dicarba derivative of the antimicrobial peptide, brevinin-1BYa. Eur. Biophys. J. 2011, 40, 555–564. [Google Scholar] [CrossRef]
- Timmons, P.B.; O’Flynn, D.; Conlon, J.M.; Hewage, C.M. Structural and positional studies of the antimicrobial peptide brevinin-1BYa in membrane-mimetic environments. J. Pept. Sci. 2019, 25. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins Struct. Funct. Bioinform. 2009, 77, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Yang, J.; Zhang, Y. COFACTOR: An accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res. 2012, 40, W471–W477. [Google Scholar] [CrossRef] [PubMed]
- Timmons, P.B.; O’Flynn, D.; Conlon, J.M.; Hewage, C.M. Insights into conformation and membrane interactions of the acyclic and dicarba-bridged brevinin-1BYa antimicrobial peptides. Eur. Biophys. J. 2019, 48, 701–710. [Google Scholar] [CrossRef]
- Pál, T.; Abraham, B.; Sonnevend, Á.; Jumaa, P.; Conlon, J.M. Brevinin-1BYa: A naturally occurring peptide from frog skin with broad-spectrum antibacterial and antifungal properties. Int. J. Antimicrob. Agents 2006, 27, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.-Y.; Hong, S.-Y.; Lee, K.-H. Structure-activity analysis of brevinin 1E amide, an antimicrobial peptide from Rana esculenta. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzymol. 1998, 1387, 239–248. [Google Scholar] [CrossRef]
- Almeida, J.R.; Mendes, B.; Lancellotti, M.; Marangoni, S.; Vale, N.; Passos, Ó.; Ramos, M.J.; Fernandes, P.A.; Gomes, P.; Da Silva, S.L. A novel synthetic peptide inspired on Lys49 phospholipase A2 from Crotalus oreganus abyssus snake venom active against multidrug-resistant clinical isolates. Eur. J. Med. Chem. 2018, 149, 248–256. [Google Scholar] [CrossRef]
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713. [Google Scholar]
- Kawano, K.; Yoneya, T.; Miyata, T.; Yoshikawa, K.; Tokunaga, F.; Terada, Y.; Iwanaga, S. Antimicrobial peptide, tachyplesin I, isolated from hemocytes of the horseshoe crab (Tachypleus tridentatus). NMR determination of the beta-sheet structure. J. Biol. Chem. 1990, 265, 15365–15367. [Google Scholar]
- Kushibiki, T.; Kamiya, M.; Aizawa, T.; Kumaki, Y.; Kikukawa, T.; Mizuguchi, M.; Demura, M.; Kawabata, S.-I.; Kawano, K. Interaction between tachyplesin I, an antimicrobial peptide derived from horseshoe crab, and lipopolysaccharide. Biochim. Biophys. Acta BBA Proteins Proteom. 2014, 1844, 527–534. [Google Scholar] [CrossRef]
- Tamamura, H.; Ikoma, R.; Niwa, M.; Funakoshi, S.; Murakami, T.; Fujii, N. Antimicrobial activity and conformation of tachyplesin I and its analogs. Chem. Pharm. Bull. 1993, 41, 978–980. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Hu, J.; Ke, F. Experimental induction of bacterial resistance to the antimicrobial peptide tachyplesin I and investigation of the resistance mechanisms. Antimicrob. Agents Chemother. 2016, 60, 6067–6075. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Guan, W.; Jin, G.; Zhao, H.; Jiang, X.; Dai, J. Mechanism of tachyplesin I injury to bacterial membranes and intracellular enzymes, determined by laser confocal scanning microscopy and flow cytometry. Microbiol. Res. 2015, 170, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Brogden, N.K.; Brogden, K.A. Will new generations of modified antimicrobial peptides improve their potential as pharmaceuticals? Int. J. Antimicrob. Agents 2011, 38, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Dai, J.; Guan, W.; Jin, G.; Huang, Z.L.; Zhang, L.; Dang, J.Z.; Zhang, Y. Tachyplesin I induce drug resistance in bacteria in vitro. J. Anim. Vet. Adv. 2012, 11, 939–945. [Google Scholar]
- Varnava, K.G.; Mohid, S.A.; Calligari, P.; Stella, L.; Reynison, J.; Bhunia, A.; Sarojini, V. Design, synthesis, antibacterial potential, and structural characterization of N-acylated derivatives of the human autophagy 16 polypeptide. Bioconjugate Chem. 2019, 30, 1998–2010. [Google Scholar] [CrossRef]
- Tsubery, H.; Ofek, I.; Cohen, S.; Fridkin, M. N-terminal modifications of polymyxin B nonapeptide and their effect on antibacterial activity. Peptides 2001, 22, 1675–1681. [Google Scholar] [CrossRef]
- Wakabayashi, H.; Matsumoto, H.; Hashimoto, K.; Teraguchi, S.; Takase, M.; Hayasawa, H. N-Acylated and D enantiomer derivatives of a nonamer core peptide of lactoferricin B showing improved antimicrobial activity. Antimicrob. Agents Chemother. 1999, 43, 1267–1269. [Google Scholar] [CrossRef] [PubMed]
- Majerle, A.; Kidrič, J.; Jerala, R. Enhancement of antibacterial and lipopolysaccharide binding activities of a human lactoferrin peptide fragment by the addition of acyl chain. J. Antimicrob. Chemother. 2003, 51, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Chu-Kung, A.F.; Nguyen, R.; Bozzelli, K.N.; Tirrell, M. Chain length dependence of antimicrobial peptide–fatty acid conjugate activity. J. Colloid Interface Sci. 2010, 345, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Chu-Kung, A.F.; Bozzelli, K.N.; Lockwood, N.A.; Haseman, J.R.; Mayo, K.H.; Tirrell, M.V. Promotion of peptide antimicrobial activity by fatty acid conjugation. Bioconjugate Chem. 2004, 15, 530–535. [Google Scholar] [CrossRef] [PubMed]
- De Zoysa, G.H.; Cameron, A.J.; Hegde, V.V.; Raghothama, S.; Sarojini, V. Antimicrobial peptides with potential for biofilm eradication: Synthesis and structure activity relationship studies of battacin peptides. J. Med. Chem. 2015, 58, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Moussouni, M.; Nogaret, P.; Garai, P.; Ize, B.; Vivès, E.; Blanc-Potard, A.B. Activity of a synthetic peptide targeting MgtC on Pseudomonas aeruginosa intramacrophage survival and biofilm formation. Front. Cell. Infect. Microbiol. 2019, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Kanaoka, S.; Sato, T.; Aoshima, S.; Kuroda, K. Block versus random amphiphilic copolymers as antibacterial agents. Biomacromolecules 2011, 12, 3581–3591. [Google Scholar] [CrossRef]
- Engler, A.C.; Wiradharma, N.; Ong, Z.Y.; Coady, D.J.; Hedrick, J.L.; Yang, Y.-Y. Emerging trends in macromolecular antimicrobials to fight multi-drug-resistant infections. Nano Today 2012, 7, 201–222. [Google Scholar] [CrossRef]
- Ergene, C.; Yasuhara, K.; Palermo, E.F. Biomimetic antimicrobial polymers: Recent advances in molecular design. Polym. Chem. 2018, 9, 2407–2427. [Google Scholar] [CrossRef]
- Mowery, B.P.; Lee, S.E.; Kissounko, D.A.; Epand, R.F.; Epand, R.M.; Weisblum, B.; Stahl, S.S.; Gellman, S.H. Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc. 2007, 129, 15474–15476. [Google Scholar] [CrossRef]
- Palermo, E.F.; Lee, D.-K.; Ramamoorthy, A.; Kuroda, K. Role of cationic group structure in membrane binding and disruption by amphiphilic copolymers. J. Phys. Chem. B 2011, 115, 366–375. [Google Scholar] [CrossRef]
- Avery, C.W.; Palermo, E.F.; McLaughlin, A.; Kuroda, K.; Chen, Z. Investigations of the interactions between synthetic antimicrobial polymers and substrate-supported lipid bilayers using sum frequency generation vibrational spectroscopy. Anal. Chem. 2011, 83, 1342–1349. [Google Scholar] [CrossRef]
- Kuroda, K.; DeGrado, W.F. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J. Am. Chem. Soc. 2005, 127, 4128–4129. [Google Scholar] [CrossRef]
- Kuroda, K.; Caputo, G.A.; DeGrado, W.F. The role of hydrophobicity in the antimicrobial and hemolytic activities of polymethacrylate derivatives. Chem. A Eur. J. 2009, 15, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Punia, A.; He, E.; Lee, K.; Banerjee, P.; Yang, N.-L. Cationic amphiphilic non-hemolytic polyacrylates with superior antibacterial activity. Chem. Commun. 2014, 50, 7071–7074. [Google Scholar] [CrossRef] [PubMed]
- Punia, A.; Debata, P.R.; Banerjee, P.; Yang, N.-L. Structure–property relationships of antibacterial amphiphilic polymers derived from 2-aminoethyl acrylate. RSC Adv. 2015, 5, 95300–95306. [Google Scholar] [CrossRef]
- Sgolastra, F.; Deronde, B.M.; Sarapas, J.M.; Som, A.; Tew, G.N. Designing mimics of membrane active proteins. Acc. Chem. Res. 2013, 46, 2977–2987. [Google Scholar] [CrossRef]
- Al-Badri, Z.M.; Som, A.; Lyon, S.; Nelson, C.F.; Nüsslein, K.; Tew, G.N. Investigating the effect of increasing charge density on the hemolytic activity of synthetic antimicrobial polymers. Biomacromolecules 2008, 9, 2805–2810. [Google Scholar] [CrossRef]
- Uppu, D.; Konai, M.; Baul, U.; Singh, P.; Siersma, T.; Samaddar, S.; Vemparala, S.; Hamoen, L.; Narayana, C.; Haldar, J. Isosteric substitution in cationic-amphiphilic polymers reveals an important role for hydrogen bonding in bacterial membrane interactions. Chem. Sci. 2016, 7, 4613–4623. [Google Scholar] [CrossRef]
- Uppu, D.S.; Samaddar, S.; Hoque, J.; Konai, M.M.; Krishnamoorthy, P.; Shome, B.R.; Haldar, J. Side chain degradable cationic–amphiphilic polymers with tunable hydrophobicity show in vivo activity. Biomacromolecules 2016, 17, 3094–3102. [Google Scholar] [CrossRef]
- Nimmagadda, A.; Liu, X.; Teng, P.; Su, M.; Li, Y.; Qiao, Q.; Khadka, N.K.; Sun, X.; Pan, J.; Xu, H. Polycarbonates with potent and selective antimicrobial activity toward gram-positive bacteria. Biomacromolecules 2017, 18, 87–95. [Google Scholar] [CrossRef]
- Yang, C.; Lou, W.; Zhong, G.; Lee, A.; Leong, J.; Chin, W.; Ding, B.; Bao, C.; Tan, J.P.; Pu, Q. Degradable antimicrobial polycarbonates with unexpected activity and selectivity for treating multidrug-resistant Klebsiella pneumoniae lung infection in mice. Acta Biomater. 2019, 94, 268–280. [Google Scholar] [CrossRef]
- Kenawy, E.-R.; Worley, S.; Broughton, R. The chemistry and applications of antimicrobial polymers: A state-of-the-art review. Biomacromolecules 2007, 8, 1359–1384. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, J.; Zhang, Y.; Yan, H.; Liu, K. Antimicrobial and hemolytic activities of copolymers with cationic and hydrophobic groups: A comparison of block and random copolymers. Macromol. Biosci. 2011, 11, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Gelman, M.A.; Weisblum, B.; Lynn, D.M.; Gellman, S.H. Biocidal activity of polystyrenes that are cationic by virtue of protonation. Org. Lett. 2004, 6, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Lienkamp, K.; Madkour, A.E.; Musante, A.; Nelson, C.F.; Nusslein, K.; Tew, G.N. Antimicrobial polymers prepared by ROMP with unprecedented selectivity: A molecular construction kit approach. J. Am. Chem. Soc. 2008, 130, 9836–9843. [Google Scholar] [CrossRef]
- Ikeda, T.; Yamaguchi, H.; Tazuke, S. New polymeric biocides: Synthesis and antibacterial activities of polycations with pendant biguanide groups. Antimicrob. Agents Chemother. 1984, 26, 139–144. [Google Scholar] [CrossRef]
- Palermo, E.F.; Kuroda, K. Chemical structure of cationic groups in amphiphilic polymethacrylates modulates the antimicrobial and hemolytic activities. Biomacromolecules 2009, 10, 1416–1428. [Google Scholar] [CrossRef]
- Epand, R.F.; Mowery, B.P.; Lee, S.E.; Stahl, S.S.; Lehrer, R.I.; Gellman, S.H.; Epand, R.M. Dual mechanism of bacterial lethality for a cationic sequence-random copolymer that mimics host-defense antimicrobial peptides. J. Mol. Biol. 2008, 379, 38–50. [Google Scholar] [CrossRef]
- Gabriel, G.J.; Madkour, A.E.; Dabkowski, J.M.; Nelson, C.F.; Nüsslein, K.; Tew, G.N. Synthetic mimic of antimicrobial peptide with nonmembrane-disrupting antibacterial properties. Biomacromolecules 2008, 9, 2980–2983. [Google Scholar] [CrossRef] [PubMed]
- Palermo, E.F.; Sovadinova, I.; Kuroda, K. Structural determinants of antimicrobial activity and biocompatibility in membrane-disrupting methacrylamide random copolymers. Biomacromolecules 2009, 10, 3098–3107. [Google Scholar] [CrossRef]
- Exley, S.E.; Paslay, L.C.; Sahukhal, G.S.; Abel, B.A.; Brown, T.D.; McCormick, C.L.; Heinhorst, S.; Koul, V.; Choudhary, V.; Elasri, M.O.; et al. Antimicrobial Peptide Mimicking Primary Amine and Guanidine Containing Methacrylamide Copolymers Prepared by Raft Polymerization. Biomacromolecules 2015, 16, 3845–3852. [Google Scholar] [CrossRef]
- Brittin, J.; Fry, M.R.; Punia, A.; Johnson, K.A.; Sengupta, A. Antibacterial and hemolytic properties of acrylate-based random ternary copolymers comprised of same center cationic, ethyl and poly(oligoethylene glycol) side chains. Eur. Polym. J. 2020, 132. [Google Scholar] [CrossRef]
- Porter, E.A.; Weisblum, B.; Gellman, S.H. Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial β-peptides. J. Am. Chem. Soc. 2002, 124, 7324–7330. [Google Scholar] [CrossRef] [PubMed]
- Frackenpohl, J.; Arvidsson, P.I.; Schreiber, J.V.; Seebach, D. The outstanding biological stability of β-and γ-peptides toward proteolytic enzymes: An in vitro investigation with fifteen peptidases. ChemBioChem 2001, 2, 445–455. [Google Scholar] [CrossRef]
- Zhou, X.; He, J.; Zhou, C. Strategies from nature: Polycaprolactone-based mimetic antimicrobial peptide block copolymers with low cytotoxicity and excellent antibacterial efficiency. Polym. Chem. 2019, 10, 945–953. [Google Scholar] [CrossRef]
- Sinha, V.; Bansal, K.; Kaushik, R.; Kumria, R.; Trehan, A. Poly-ϵ-caprolactone microspheres and nanospheres: An overview. Int. J. Pharm. 2004, 278, 1–23. [Google Scholar] [CrossRef]
- Wei, X.; Gong, C.; Gou, M.; Fu, S.; Guo, Q.; Shi, S.; Luo, F.; Guo, G.; Qiu, L.; Qian, Z. Biodegradable poly (ɛ-caprolactone)—poly (ethylene glycol) copolymers as drug delivery system. Int. J. Pharm. 2009, 381, 1–18. [Google Scholar] [CrossRef]
- Zhou, S.; Deng, X.; Yang, H. Biodegradable poly (ε-caprolactone)-poly (ethylene glycol) block copolymers: Characterization and their use as drug carriers for a controlled delivery system. Biomaterials 2003, 24, 3563–3570. [Google Scholar] [CrossRef]
- Qian, Y.; Zhou, X.; He, J.; Zhou, C. Polycaprolactone-based mimetic antimicrobial peptide copolymers vesicles as an effective drug-carrier for cancer therapy. Polymers 2019, 11, 1783. [Google Scholar] [CrossRef]
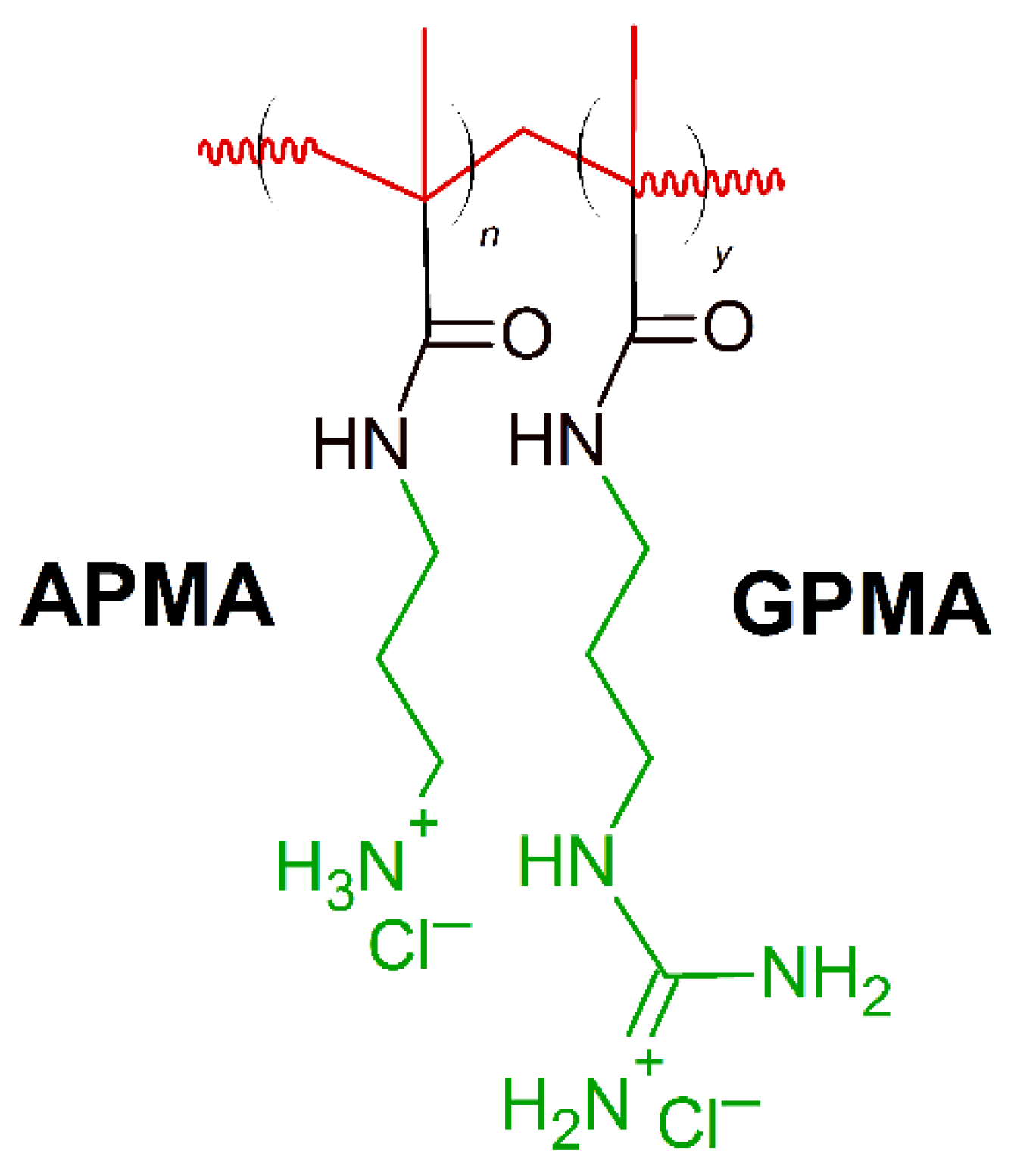
- Barman, S.; Konai, M.M.; Samaddar, S.; Haldar, J. Amino Acid Conjugated Polymers: Antibacterial Agents Effective against Drug-Resistant Acinetobacter baumannii with No Detectable Resistance. ACS Appl. Mater. Interfaces 2019, 11, 33559–33572. [Google Scholar] [CrossRef]
- Costa, F.; Gomes, P.; Martins, M.C.L. Peptides and Proteins as Biomaterials for Tissue Regeneration and Repair; Woodhead Publishing: Cambridge, UK, 2018; pp. 329–345. [Google Scholar]
- Acosta, S.; Ibañez-Fonseca, A.; Aparicio, C.; Rodríguez-Cabello, J.C. Antibiofilm coatings based on protein-engineered polymers and antimicrobial peptides for preventing implant-associated infections. Biomater. Sci. 2020, 8, 2866–2877. [Google Scholar] [CrossRef]
- Mojsoska, B.; Zuckermann, R.N.; Jenssen, H. Structure-activity relationship study of novel peptoids that mimic the structure of antimicrobial peptides. Antimicrob. Agents Chemother. 2015, 59, 4112–4120. [Google Scholar] [CrossRef]
- Thakkar, A.; Cohen, A.S.; Connolly, M.D.; Zuckermann, R.N.; Pei, D. High-throughput sequencing of peptoids and peptide—peptoid hybrids by partial Edman degradation and mass spectrometry. J. Comb. Chem. 2009, 11, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Szleifer, I. Structural and dynamical characteristics of peptoid oligomers with achiral aliphatic side chains studied by molecular dynamics simulation. J. Phys. Chem. B 2011, 115, 10967–10975. [Google Scholar] [CrossRef] [PubMed]
- Mirijanian, D.T.; Mannige, R.V.; Zuckermann, R.N.; Whitelam, S. Development and use of an atomistic CHARMM-based forcefield for peptoid simulation. J. Comput. Chem. 2014, 35, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Baer, M.D.; Mundy, C.J.; Pfaendtner, J. Peptoid backbone flexibilility dictates its interaction with water and surfaces: A molecular dynamics investigation. Biomacromolecules 2018, 19, 1006–1015. [Google Scholar] [CrossRef]
- Andreev, K.; Martynowycz, M.W.; Gidalevitz, D. Peptoid drug discovery and optimization via surface X-ray scattering. Biopolymers 2019, 110. [Google Scholar] [CrossRef]
- Andreev, K.; Bianchi, C.; Laursen, J.S.; Citterio, L.; Hein-Kristensen, L.; Gram, L.; Kuzmenko, I.; Olsen, C.A.; Gidalevitz, D. Guanidino groups greatly enhance the action of antimicrobial peptidomimetics against bacterial cytoplasmic membranes. Biochim. Biophys. Acta BBA Biomembr. 2014, 1838, 2492–2502. [Google Scholar] [CrossRef]
- Huang, M.L.; Shin, S.B.Y.; Benson, M.A.; Torres, V.J.; Kirshenbaum, K. A comparison of linear and cyclic peptoid oligomers as potent antimicrobial agents. ChemMedChem 2012, 7, 114–122. [Google Scholar] [CrossRef]
- Gellman, S.H. Foldamers: A manifesto. Acc. Chem. Res. 1998, 31, 173–180. [Google Scholar] [CrossRef]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev. Res. 1995, 35, 20–32. [Google Scholar] [CrossRef]
- Ibrahim, O.O. Classification of Antimicrobial Peptides Bacteriocins, and the Nature of Some Bacteriocins with Potential Applications in Food Safety and Bio-Pharmaceuticals. EC Microbiol. 2019, 15, 591–608. [Google Scholar]
- Olsen, C.A.; Bonke, G.; Vedel, L.; Adsersen, A.; Witt, M.; Franzyk, H.; Jaroszewski, J.W. α-peptide/β-peptoid chimeras. Org. Lett. 2007, 9, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Ren, G.; Liu, H.; Miao, Z.; Park, M.; Wang, Y.; Miller, T.M.; Barron, A.E.; Cheng, Z. In vivo biodistribution and small animal PET of 64Cu-Labeled antimicrobial peptoids. Bioconjugate Chem. 2012, 23, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Turkett, J.A.; Corson, A.E.; Bicker, K.L. Peptoid Library Agar Diffusion (PLAD) assay for the high-throughput identification of antimicrobial peptoids. ACS Comb. Sci. 2016, 18, 287–291. [Google Scholar] [CrossRef]
- Mojsoska, B.; Carretero, G.; Larsen, S.; Mateiu, R.V.; Jenssen, H. Peptoids successfully inhibit the growth of gram negative E. coli causing substantial membrane damage. Sci. Rep. 2017, 7, 42332. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, K.; Barron, A.E.; Goldsmith, R.A.; Armand, P.; Bradley, E.K.; Truong, K.T.; Dill, K.A.; Cohen, F.E.; Zuckermann, R.N. Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. USA 1998, 95, 4303–4308. [Google Scholar] [CrossRef]
- Huang, W.; Seo, J.; Willingham, S.B.; Czyzewski, A.M.; Gonzalgo, M.L.; Weissman, I.L.; Barron, A.E. Learning from host-defense peptides: Cationic, amphipathic peptoids with potent anticancer activity. PLoS ONE 2014, 9, e90397. [Google Scholar] [CrossRef]
- Shyam, R.; Charbonnel, N.; Job, A.; Blavignac, C.; Forestier, C.; Taillefumier, C.; Faure, S. 1,2,3-Triazolium-Based Cationic Amphipathic Peptoid Oligomers Mimicking Antimicrobial Helical Peptides. ChemMedChem 2018, 13, 1513–1516. [Google Scholar] [CrossRef]
- Chou, P.Y.; Fasman, G.D. Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry 1974, 13, 211–222. [Google Scholar] [CrossRef]
- Béven, L.; Castano, S.; Dufourcq, J.; Wieslander, Å.; Wróblewski, H. The antibiotic activity of cationic linear amphipathic peptides: Lessons from the action of leucine/lysine copolymers on bacteria of the class Mollicutes. Eur. J. Biochem. 2003, 270, 2207–2217. [Google Scholar] [CrossRef]
- Monroc, S.; Badosa, E.; Feliu, L.; Planas, M.; Montesinos, E.; Bardají, E. De novo designed cyclic cationic peptides as inhibitors of plant pathogenic bacteria. Peptides 2006, 27, 2567–2574. [Google Scholar] [CrossRef]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta Bba Biomembr. 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Won, H.S.; Park, S.H.; Kim, H.E.; Hyun, B.; Kim, M.; Lee, B.J.; Lee, B.J. Effects of a tryptophanyl substitution on the structure and antimicrobial activity of C-terminally truncated gaegurin 4. Eur. J. Biochem. 2002, 269, 4367–4374. [Google Scholar] [CrossRef] [PubMed]
- Pandit, G.; Ilyas, H.; Ghosh, S.; Bidkar, A.P.; Mohid, S.A.; Bhunia, A.; Satpati, P.; Chatterjee, S. Insights into the Mechanism of Antimicrobial Activity of Seven-Residue Peptides. J. Med. Chem. 2018, 61, 7614–7629. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; He, P.; Xiao, C.; Chen, X. From Antimicrobial Peptides to Antimicrobial Poly(α-amino acid)s. Adv. Healthc. Mater. 2018, 7, e1800354. [Google Scholar] [CrossRef] [PubMed]
- Wyrsta, M.D.; Cogen, A.L.; Deming, T.J. A parallel synthetic approach for the analysis of membrane interactive copolypeptides. J. Am. Chem. Soc. 2001, 123, 12919–12920. [Google Scholar] [CrossRef]
- Borase, T.; Heise, A. Hybrid Nanomaterials by Surface Grafting of Synthetic Polypeptides Using N-Carboxyanhydride (NCA) Polymerization. Adv. Mater. 2016, 28, 5725–5731. [Google Scholar] [CrossRef]
- Cloutier, M.; Mantovani, D.; Rosei, F. Antibacterial coatings: Challenges, perspectives, and opportunities. Trends Biotechnol. 2015, 33, 637–652. [Google Scholar] [CrossRef]
- Dong, N.; Chou, S.; Li, J.; Xue, C.; Li, X.; Cheng, B.; Shan, A.; Xu, L. Short symmetric-end antimicrobial peptides centered on β-turn amino acids unit improve selectivity and stability. Front. Microbiol. 2018, 9, 2832. [Google Scholar] [CrossRef]
- Fjell, C.D.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.; Cherkasov, A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar] [CrossRef]
- Fjell, C.D.; Jenssen, H.; Cheung, W.A.; Hancock, R.E.; Cherkasov, A. Optimization of antibacterial peptides by genetic algorithms and cheminformatics. Chem. Biol. Drug Des. 2011, 77, 48–56. [Google Scholar] [CrossRef]
- Godballe, T.; Mojsoska, B.; Nielsen, H.M.; Jenssen, H. Antimicrobial activity of GN peptides and their mode of action. Biopolymers 2016, 106, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.; Liu, L.-P.; Deber, C.M. Cationic hydrophobic peptides with antimicrobial activity. Antimicrob. Agents Chemother. 2002, 46, 3585–3590. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef]
- Hu, J.; Chen, C.; Zhang, S.; Zhao, X.; Xu, H.; Zhao, X.; Lu, J.R. Designed antimicrobial and antitumor peptides with high selectivity. Biomacromolecules 2011, 12, 3839–3843. [Google Scholar] [CrossRef]
- Chen, C.; Hu, J.; Yang, C.; Zhang, Y.; Wang, F.; Mu, Q.; Pan, F.; Xu, H.; Lu, J.R. Amino acid side chains affect the bioactivity of designed short peptide amphiphiles. J. Mater. Chem. B 2016, 4, 2359–2368. [Google Scholar] [CrossRef]
- Chen, C.; Yang, C.; Chen, Y.; Wang, F.; Mu, Q.; Zhang, J.; Li, Z.; Pan, F.; Xu, H.; Lu, J.R. Surface physical activity and hydrophobicity of designed helical peptide amphiphiles control their bioactivity and cell selectivity. ACS Appl. Mater. Interfaces 2016, 8, 26501–26510. [Google Scholar] [CrossRef]
- Grassi, L.; Batoni, G.; Ostyn, L.; Rigole, P.; Van Den Bossche, S.; Rinaldi, A.C.; Maisetta, G.; Esin, S.; Coenye, T.; Crabbé, A. The antimicrobial peptide lin-SB056-1 and its dendrimeric derivative prevent pseudomonas aeruginosabiofilm formation in physiologically relevant models of chronic infections. Front. Microbiol. 2019, 10, 198. [Google Scholar] [CrossRef]
- Meng, H.; Kumar, K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J. Am. Chem. Soc. 2007, 129, 15615–15622. [Google Scholar] [CrossRef]
- Zikou, S.; Koukkou, A.I.; Mastora, P.; Sakarellos-Daitsiotis, M.; Sakarellos, C.; Drainas, C.; Panou-Pomonis, E. Design and synthesis of cationic Aib-containing antimicrobial peptides: Conformational and biological studies. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2007, 13, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, X.; Falk, S.P.; Masters, K.S.; Weisblum, B.; Gellman, S.H. Nylon-3 polymers active against drug-resistant Candida albicans biofilms. J. Am. Chem. Soc. 2015, 137, 2183–2186. [Google Scholar] [CrossRef] [PubMed]
- Szkudlarek, M.; Heine, E.; Keul, H.; Beginn, U.; Möller, M. Synthesis, characterization, and antimicrobial properties of peptides mimicking copolymers of maleic anhydride and 4-methyl-1-pentene. Int. J. Mol. Sci. 2018, 19, 2617. [Google Scholar] [CrossRef] [PubMed]
- Bozelli, J.C.; Salay, L.C.; Arcisio-Miranda, M.; Procopio, J.; Riciluca, K.C.T.; Silva Junior, P.I.; Nakaie, C.R.; Schreier, S. A comparison of activity, toxicity, and conformation of tritrpticin and two TOAC-labeled analogues. Effects on the mechanism of action. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183110. [Google Scholar] [CrossRef]
- Arias, M.; Jensen, K.V.; Nguyen, L.T.; Storey, D.G.; Vogel, H.J. Hydroxy-tryptophan containing derivatives of tritrpticin: Modification of antimicrobial activity and membrane interactions. Biochim. Biophys. Acta Bba Biomembr. 2015, 1848, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Cirioni, O.; Giacometti, A.; Silvestri, C.; Della Vittoria, A.; Licci, A.; Riva, A.; Scalise, G. In vitro activities of tritrpticin alone and in combination with other antimicrobial agents against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 3923–3925. [Google Scholar] [CrossRef]
- Lawyer, C.; Pai, S.; Watabe, M.; Borgia, P.; Mashimo, T.; Eagleton, L.; Watabe, K. Antimicrobial activity of a 13 amino acid tryptophan-rich peptide derived from a putative porcine precursor protein of a novel family of antibacterial peptides. FEBS Lett. 1996, 390, 95–98. [Google Scholar] [CrossRef]
- Nguyen, L.T.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Investigating the cationic side chains of the antimicrobial peptide tritrpticin: Hydrogen bonding properties govern its membrane-disruptive activities. Biochim. Biophys. Acta BBA Biomembr. 2011, 1808, 2297–2303. [Google Scholar] [CrossRef]
- Schibli, D.J.; Nguyen, L.T.; Kernaghan, S.D.; Rekdal, Ø.; Vogel, H.J. Structure-function analysis of tritrpticin analogs: Potential relationships between antimicrobial activities, model membrane interactions, and their micelle-bound NMR structures. Biophys. J. 2006, 91, 4413–4426. [Google Scholar] [CrossRef]
- Yang, S.-T.; Shin, S.Y.; Kim, Y.-C.; Kim, Y.; Hahm, K.-S.; Kim, J.I. Conformation-dependent antibiotic activity of tritrpticin, a cathelicidin-derived antimicrobial peptide. Biochem. Biophys. Res. Commun. 2002, 296, 1044–1050. [Google Scholar] [CrossRef]
- Schibli, D.J.; Epand, R.F.; Vogel, H.J.; Epand, R.M. Tryptophan-rich antimicrobial peptides: Comparative properties and membrane interactions. Biochem. Cell Biol. 2002, 80, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Salay, L.C.; Procopio, J.; Oliveira, E.; Nakaie, C.R.; Schreier, S. Ion channel-like activity of the antimicrobial peptide tritrpticin in planar lipid bilayers. FEBS Lett. 2004, 565, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Ageitos, J.; Sánchez-Pérez, A.; Calo-Mata, P.; Villa, T. Antimicrobial peptides (AMPs): Ancient compounds that represent novel weapons in the fight against bacteria. Biochem. Pharmacol. 2017, 133, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Oliva, R.; Chino, M.; Pane, K.; Pistorio, V.; De Santis, A.; Pizzo, E.; D’Errico, G.; Pavone, V.; Lombardi, A.; Del Vecchio, P.; et al. Exploring the role of unnatural amino acids in antimicrobial peptides. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Zhu, R.; Zhao, Y.; An, X.; Jia, F.; Peng, J.; Ma, Z.; Zhu, Y.; Wang, J.; Su, J.; et al. Antimicrobial activity and stability of protonectin with D-amino acid substitutions. J. Pept. Sci. 2017, 23, 392–402. [Google Scholar] [CrossRef]
- Mendes, M.A.; de Souza, B.M.; Marques, M.R.; Palma, M.S. Structural and biological characterization of two novel peptides from the venom of the neotropical social wasp Agelaia pallipes pallipes. Toxicon 2004, 44, 67–74. [Google Scholar] [CrossRef]
- Wang, K.; Dang, W.; Yan, J.; Chen, R.; Liu, X.; Yan, W.; Zhang, B.; Xie, J.; Zhang, J.; Wang, R. Membrane perturbation action mode and structure-activity relationships of Protonectin, a novel antimicrobial peptide from the venom of the neotropical social wasp Agelaia pallipes pallipes. Antimicrob. Agents Chemother. 2013, 57, 4632–4639. [Google Scholar] [CrossRef]
- Wang, K.; Dang, W.; Xie, J.; Zhu, R.; Sun, M.; Jia, F.; Zhao, Y.; An, X.; Qiu, S.; Li, X. Antimicrobial peptide protonectin disturbs the membrane integrity and induces ROS production in yeast cells. Biochim. Biophys. Acta Bba Biomembr. 2015, 1848, 2365–2373. [Google Scholar] [CrossRef]
- Patch, J.A.; Barron, A.E. Mimicry of bioactive peptides via non-natural, sequence-specific peptidomimetic oligomers. Curr. Opin. Chem. Biol. 2002, 6, 872–877. [Google Scholar] [CrossRef]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef]
- Laursen, J.S.; Engel-Andreasen, J.; Olsen, C.A. β-Peptoid Foldamers at Last. Acc. Chem. Res. 2015, 48, 2696–2704. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.-D.; Yang, D. α-Aminoxy acids: New possibilities from foldamers to anion receptors and channels. Acc. Chem. Res. 2008, 41, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Johnson, L.M.; Ketas, T.J.; Klasse, P.J.; Lu, M.; Moore, J.P.; Gellman, S.H. Structural and biological mimicry of protein surface recognition by α/β-peptide foldamers. Proc. Natl. Acad. Sci. USA 2009, 106, 14751–14756. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Song, J.-W.; Choi, Y.-S.; Park, H.-M.; Lee, K.-B. A theoretical study of conformational properties of N-methyl azapeptide derivatives. J. Am. Chem. Soc. 2002, 124, 11881–11893. [Google Scholar] [CrossRef]
- Claudon, P.; Violette, A.; Lamour, K.; Decossas, M.; Fournel, S.; Heurtault, B.; Godet, J.; Mély, Y.; Jamart-Grégoire, B.; Averlant-Petit, M.C. Consequences of isostructural main-chain modifications for the design of antimicrobial foldamers: Helical mimics of host-defense peptides based on a heterogeneous amide/urea backbone. Angew. Chem. Int. Ed. 2010, 49, 333–336. [Google Scholar] [CrossRef]
- Chandramouli, N.; Ferrand, Y.; Lautrette, G.; Kauffmann, B.; Mackereth, C.D.; Laguerre, M.; Dubreuil, D.; Huc, I. Iterative design of a helically folded aromatic oligoamide sequence for the selective encapsulation of fructose. Nat. Chem. 2015, 7, 334–341. [Google Scholar] [CrossRef]
- Wu, H.; Qiao, Q.; Hu, Y.; Teng, P.; Gao, W.; Zuo, X.; Wojtas, L.; Larsen, R.W.; Ma, S.; Cai, J. Sulfono-γ-AApeptides as a New Class of Nonnatural Helical Foldamer. Chem. A Eur. J. 2015, 21, 2501–2507. [Google Scholar] [CrossRef]
- Li, Y.; Wu, H.; Teng, P.; Bai, G.; Lin, X.; Zuo, X.; Cao, C.; Cai, J. Helical antimicrobial sulfono-γ-AApeptides. J. Med. Chem. 2015, 58, 4802–4811. [Google Scholar] [CrossRef]
- Anacona, J.R.; Rodriguez, A. Synthesis and antibacterial activity of ceftriaxone metal complexes. Transit. Met. Chem. 2005, 30, 897–901. [Google Scholar] [CrossRef]
- Chohan, Z.H.; Supuran, C.T.; Scozzafava, A. Metalloantibiotics: Synthesis and antibacterial activity of cobalt (II), copper (II), nickel (II) and zinc (II) complexes of kefzol. J. Enzym. Inhib. Med. Chem. 2004, 19, 79–84. [Google Scholar] [CrossRef]
- Anacona, J.R.; Osorio, I. Synthesis and antibacterial activity of copper (II) complexes with sulphathiazole and cephalosporin ligands. Transit. Met. Chem. 2008, 33, 517–521. [Google Scholar] [CrossRef]
- Möhler, J.S.; Kolmar, T.; Synnatschke, K.; Hergert, M.; Wilson, L.A.; Ramu, S.; Elliott, A.G.; Blaskovich, M.A.; Sidjabat, H.E.; Paterson, D.L. Enhancement of antibiotic-activity through complexation with metal ions-Combined ITC, NMR, enzymatic and biological studies. J. Inorg. Biochem. 2017, 167, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Medici, S.; Peana, M.; Crisponi, G.; Nurchi, V.M.; Lachowicz, J.I.; Remelli, M.; Zoroddu, M.A. Silver coordination compounds: A new horizon in medicine. Coord. Chem. Rev. 2016, 327, 349–359. [Google Scholar] [CrossRef]
- Waldron, K.J.; Robinson, N.J. How do bacterial cells ensure that metalloproteins get the correct metal? Nat. Rev. Microbiol. 2009, 7, 25–35. [Google Scholar] [CrossRef]
- Łoboda, D.; Kozłowski, H.; Rowińska-Żyrek, M. Antimicrobial peptide–metal ion interactions–a potential way of activity enhancement. New J. Chem. 2018, 42, 7560–7568. [Google Scholar] [CrossRef]
- Silva, F.D.; Rezende, C.A.; Rossi, D.C.; Esteves, E.; Dyszy, F.H.; Schreier, S.; Gueiros-Filho, F.; Campos, C.B.; Pires, J.R.; Daffre, S. Structure and mode of action of microplusin, a copper II-chelating antimicrobial peptide from the cattle tick Rhipicephalus (Boophilus) microplus. J. Biol. Chem. 2009, 284, 34735–34746. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Song, Y.-K.; Cho, J.-H. Risk assessment of ciprofloxacin, flavomycin, olaquindox and colistin sulfate based on microbiological impact on human gut biota. Regul. Toxicol. Pharmacol. 2009, 53, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Kim, Y.-H.; Tahk, S.; Hong, T.; Weis, P.; Waring, A.J.; Ganz, T. Calcitermin, a novel antimicrobial peptide isolated from human airway secretions. FEBS Lett. 2001, 504, 5–10. [Google Scholar] [CrossRef]
- Wrzesinski, J.; Błaszczyk, L.; Wrońska, M.; Kasprowicz, A.; Stokowa-Sołtys, K.; Nagaj, J.; Szafraniec, M.; Kulinski, T.; Jeżowska-Bojczuk, M.; Ciesiołka, J. Mapping the interactions of selected antibiotics and their C u2+ complexes with the antigenomic delta ribozyme. FEBS J. 2013, 280, 2652–2664. [Google Scholar] [CrossRef]
- Oppenheim, F.; Xu, T.; McMillian, F.; Levitz, S.; Diamond, R.; Offner, G.; Troxler, R. Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans. J. Biol. Chem. 1988, 263, 7472–7477. [Google Scholar]
- Al-Harthi, S.; Lachowicz, J.I.; Nowakowski, M.E.; Jaremko, M.; Jaremko, Ł. Towards the functional high-resolution coordination chemistry of blood plasma human serum albumin. J. Inorg. Biochem. 2019, 198, 110716. [Google Scholar] [CrossRef] [PubMed]
- Jeżowska-Bojczuk, M.; Stokowa-Sołtys, K. Peptides having antimicrobial activity and their complexes with transition metal ions. Eur. J. Med. Chem. 2018, 143, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Agbale, C.M.; Sarfo, J.K.; Galyuon, I.K.; Juliano, S.A.; Silva, G.G.; Buccini, D.F.; Cardoso, M.H.; Torres, M.D.; Angeles-Boza, A.M.; de la Fuente-Nunez, C. Antimicrobial and Antibiofilm Activities of Helical Antimicrobial Peptide Sequences Incorporating Metal-Binding Motifs. Biochemistry 2019, 58, 3802–3812. [Google Scholar] [CrossRef] [PubMed]
- Harford, C.; Sarkar, B. Amino terminal Cu (II)-and Ni (II)-binding (ATCUN) motif of proteins and peptides: Metal binding, DNA cleavage, and other properties. Acc. Chem. Res. 1997, 30, 123–130. [Google Scholar] [CrossRef]
- Mujika, J.; Dalla Torre, G.; Lachowicz, J.; Lopez, X. In silico design of mimosine containing peptides as new efficient chelators of aluminum. RSC Adv. 2019, 9, 7688–7697. [Google Scholar] [CrossRef]
- Perez, R.H.; Zendo, T.; Sonomoto, K. Circular and leaderless bacteriocins: Biosynthesis, mode of action, applications, and prospects. Front. Microbiol. 2018, 9, 2085. [Google Scholar] [CrossRef]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Drider, D.; Bendali, F.; Naghmouchi, K.; Chikindas, M.L. Bacteriocins: Not only antibacterial agents. Probiotics Antimicrob. Proteins 2016, 8, 177–182. [Google Scholar] [CrossRef]
- Sahl, H.-G.; Bierbaum, G. Lantibiotics: Biosynthesis and biological activities of uniquely modified peptides from gram-positive bacteria. Annu. Rev. Microbiol. 1998, 52, 41–79. [Google Scholar] [CrossRef]
- Cotter, P.D.; Hill, C.; Ross, R.P. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005, 3, 777–788. [Google Scholar] [CrossRef]
- Cleveland, J.; Montville, T.J.; Nes, I.F.; Chikindas, M.L. Bacteriocins: Safe, natural antimicrobials for food preservation. Int. J. Food Microbiol. 2001, 71, 1–20. [Google Scholar] [CrossRef]
- Héchard, Y.; Sahl, H.-G. Mode of action of modified and unmodified bacteriocins from Gram-positive bacteria. Biochimie 2002, 84, 545–557. [Google Scholar] [CrossRef]
- Klaenhammer, T.R. Genetics of bacteriocins produced by lactic acid bacteria. FEMS Microbiol. Rev. 1993, 12, 39–85. [Google Scholar] [CrossRef]
- Carolissen-Mackay, V.; Arendse, G.; Hastings, J.W. Purification of bacteriocins of lactic acid bacteria: Problems and pointers. Int. J. Food Microbiol. 1997, 34, 1–16. [Google Scholar] [CrossRef]
- Galvez, A.; Burgos, M.; Lopez, R.; Pulido, R. Natural antimicrobials for food biopreservation. In Food Biopreservation; Springer: New York, NY, USA; Heidelberg, Germany; Dordrecht, The Netherland; London, UK, 2014; pp. 3–11. [Google Scholar]
- Prudêncio, C.V.; Dos Santos, M.T.; Vanetti, M.C.D. Strategies for the use of bacteriocins in Gram-negative bacteria: Relevance in food microbiology. J. Food Sci. Technol. 2015, 52, 5408–5417. [Google Scholar] [CrossRef]
- Pisano, M.B.; Fadda, M.E.; Melis, R.; Ciusa, M.L.; Viale, S.; Deplano, M.; Cosentino, S. Molecular identification of bacteriocins produced by Lactococcus lactis dairy strains and their technological and genotypic characterization. Food Control 2015, 51, 1–8. [Google Scholar] [CrossRef]































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
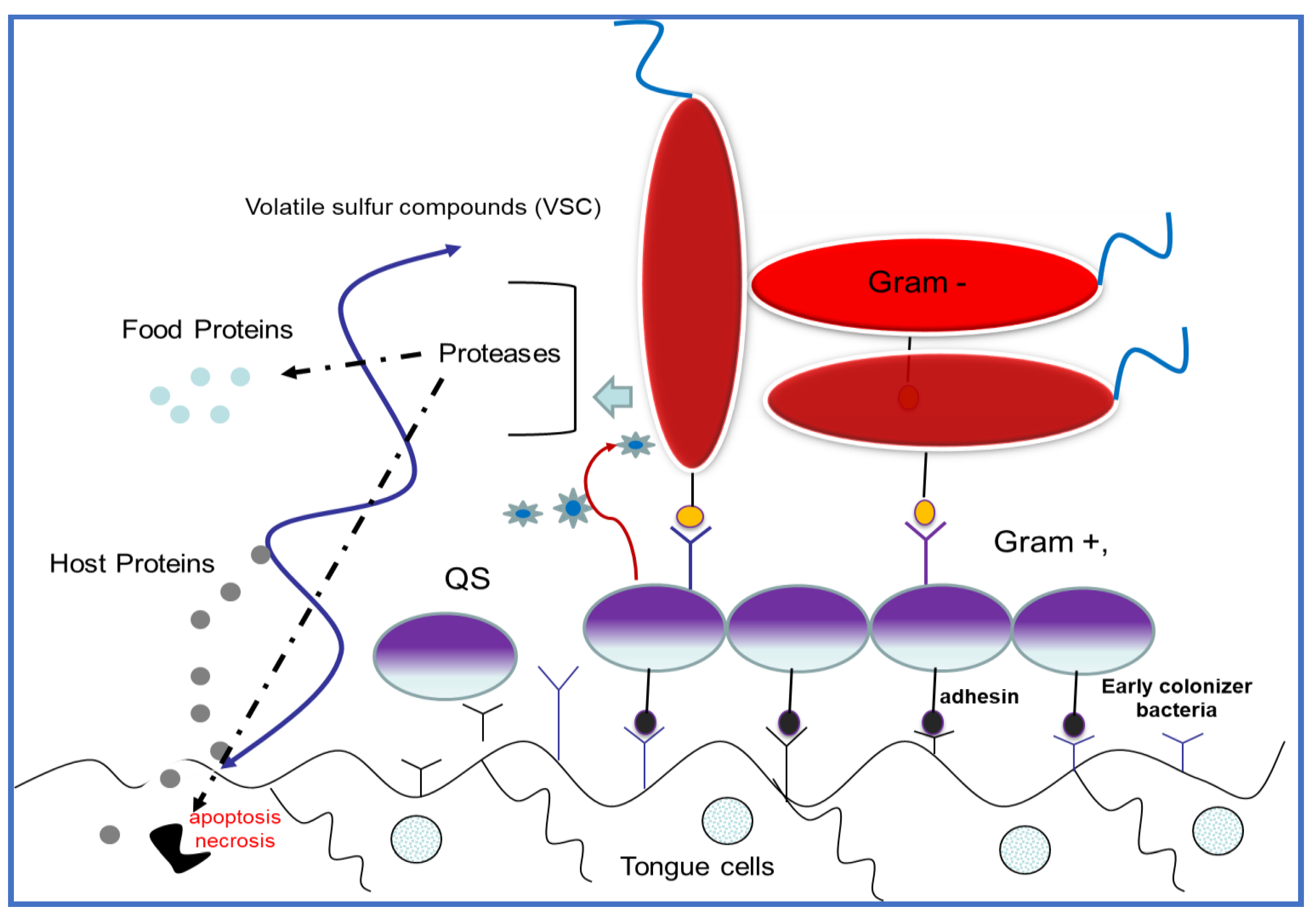
| Killing Mechanisms | Bacterial Membrane Type of Interaction | References |
|---|---|---|
| Barrel-stave model | AMPs are perpendicularly inserted into the membrane, promoting peptide–peptide lateral interactions. | [75,76] |
| Carpet model | Binding of AMPs to the outer surface (phospholipids) of cell membrane. | [77,78] |
| Toroidal model | Attached AMPs start aggregation and force the lipid monolayer to bend incessantly through the pores | [79,80] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lachowicz, J.I.; Szczepski, K.; Scano, A.; Casu, C.; Fais, S.; Orrù, G.; Pisano, B.; Piras, M.; Jaremko, M. The Best Peptidomimetic Strategies to Undercover Antibacterial Peptides. Int. J. Mol. Sci. 2020, 21, 7349. https://doi.org/10.3390/ijms21197349
Lachowicz JI, Szczepski K, Scano A, Casu C, Fais S, Orrù G, Pisano B, Piras M, Jaremko M. The Best Peptidomimetic Strategies to Undercover Antibacterial Peptides. International Journal of Molecular Sciences. 2020; 21(19):7349. https://doi.org/10.3390/ijms21197349
Chicago/Turabian StyleLachowicz, Joanna Izabela, Kacper Szczepski, Alessandra Scano, Cinzia Casu, Sara Fais, Germano Orrù, Barbara Pisano, Monica Piras, and Mariusz Jaremko. 2020. "The Best Peptidomimetic Strategies to Undercover Antibacterial Peptides" International Journal of Molecular Sciences 21, no. 19: 7349. https://doi.org/10.3390/ijms21197349
APA StyleLachowicz, J. I., Szczepski, K., Scano, A., Casu, C., Fais, S., Orrù, G., Pisano, B., Piras, M., & Jaremko, M. (2020). The Best Peptidomimetic Strategies to Undercover Antibacterial Peptides. International Journal of Molecular Sciences, 21(19), 7349. https://doi.org/10.3390/ijms21197349