CaMKIIβ in Neuronal Development and Plasticity: An Emerging Candidate in Brain Diseases


Abstract
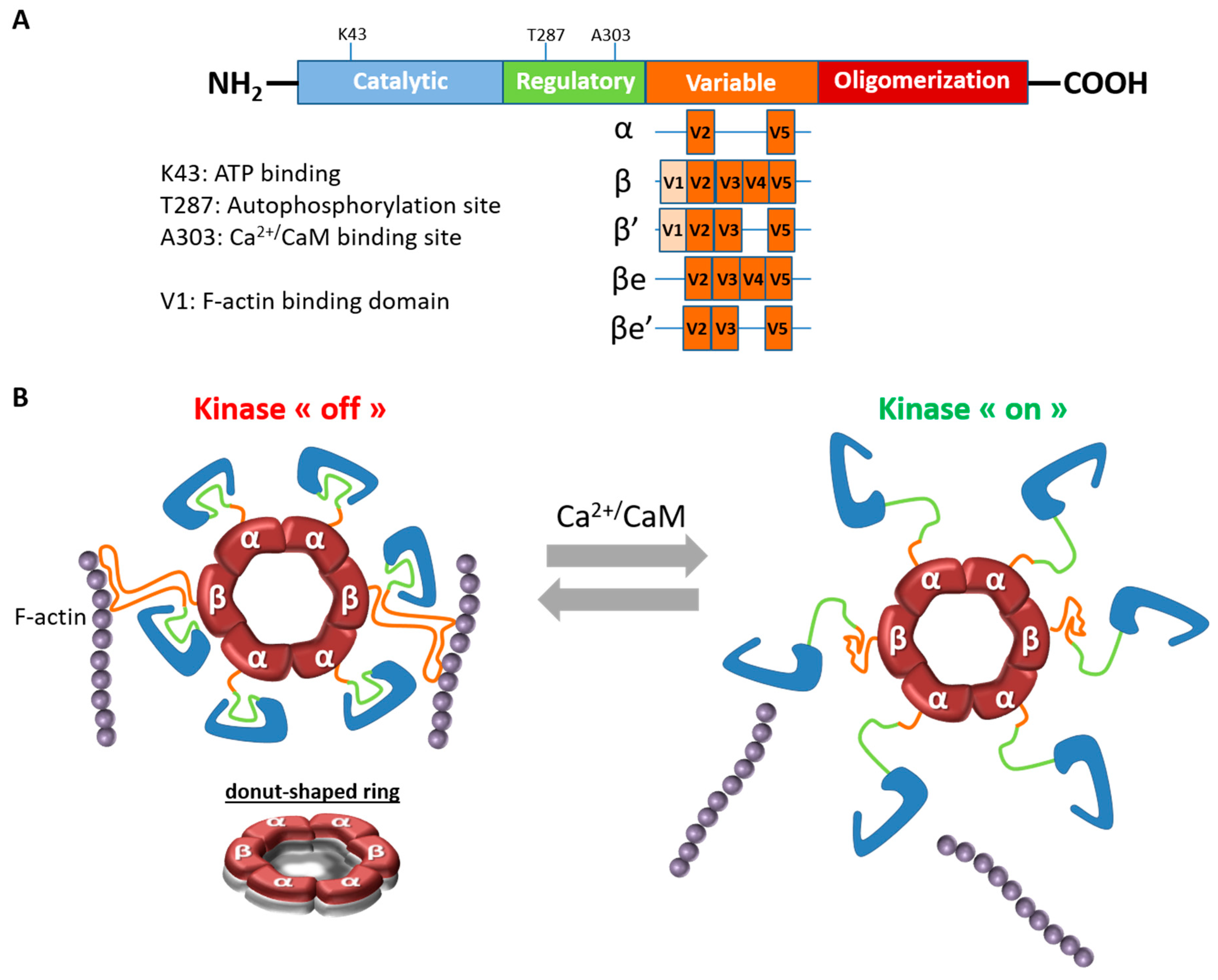
1. Features of CaMKIIβ
1.1. CaMKIIβ Structure and Properties
1.2. CaMKIIβ Expression in the Nervous System
2. CaMKIIβ in Neuronal Development
2.1. CaMKIIβ and Neuronal Migration
2.2. CaMKIIβ and Dendrite Formation/Pruning
2.3. CaMKIIβ and Spine/Synapse Formation
3. CaMKIIβ in Neuronal Plasticity
3.1. Molecular Mechanisms of CaMKIIβ in Synaptic Plasticity
3.2. Role of CaMKIIβ in LTP (Hippocampal and Cerebellar)
3.3. CaMKIIβ and Memory
4. CaMKIIβ and Brain Disorders
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| Ca2+/CaM | Ca2+-bound calmodulin |
| CaMKII | calcium/calmodulin-dependent protein kinase II |
| Cdc20-APC | cell division cycle 20–anaphase promoting complex |
| COX-2 | cyclo-oxygenase-2 |
| CP | cortical plate |
| CTS | centrosomal targeting sequence |
| DG | dentate gyrus |
| DKO | deletion of both CaMKIIα and CaMKIIβ |
| FABD | F-actin binding domain |
| F-actin | filamentous-actin |
| IZ | intermediate zone |
| LTD | long-term depression |
| LTP | long-term potentiation |
| mGluR1 | group I metabotropic glutamate receptor |
| NMDA | N-Methyl-D-aspartate |
| PCM1 | pericentriolar material 1 |
| PGE2 | prostaglandin E2 |
| PKC | protein kinase C |
| PP2B | protein phosphatase 2B |
| PRP | plasticity-related proteins |
| PSD | post-synaptic density |
| VZ | ventricular zone |
| WM | white matter |
References
- Erondu, N.E.; Kennedy, M.B. Regional distribution of type II Ca2+/calmodulin-dependent protein kinase in rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1985, 5, 3270–3277. [Google Scholar] [CrossRef]
- Tombes, R.M.; Faison, M.O.; Turbeville, J.M. Organization and evolution of multifunctional Ca(2+)/CaM-dependent protein kinase genes. Gene 2003, 322, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Bayer, K.U.; Lohler, J.; Schulman, H.; Harbers, K. Developmental expression of the CaM kinase II isoforms: Ubiquitous gamma- and delta-CaM kinase II are the early isoforms and most abundant in the developing nervous system. Brain Res. Mol. Brain Res. 1999, 70, 147–154. [Google Scholar] [CrossRef]
- Brocke, L.; Chiang, L.W.; Wagner, P.D.; Schulman, H. Functional implications of the subunit composition of neuronal CaM kinase II. J. Biol. Chem. 1999, 274, 22713–22722. [Google Scholar] [CrossRef] [PubMed]
- Bayer, K.U.; Schulman, H. CaM Kinase: Still Inspiring at 40. Neuron 2019, 103, 380–394. [Google Scholar] [CrossRef]
- Hell, J.W. CaMKII: Claiming center stage in postsynaptic function and organization. Neuron 2014, 81, 249–265. [Google Scholar] [CrossRef]
- Tobimatsu, T.; Fujisawa, H. Tissue-specific expression of four types of rat calmodulin-dependent protein kinase II mRNAs. J. Biol. Chem. 1989, 264, 17907–17912. [Google Scholar]
- Shen, K.; Teruel, M.N.; Subramanian, K.; Meyer, T. CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 1998, 21, 593–606. [Google Scholar] [CrossRef]
- Okamoto, K.; Narayanan, R.; Lee, S.H.; Murata, K.; Hayashi, Y. The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl. Acad. Sci. USA 2007, 104, 6418–6423. [Google Scholar] [CrossRef]
- Brocke, L.; Srinivasan, M.; Schulman, H. Developmental and regional expression of multifunctional Ca2+/calmodulin-dependent protein kinase isoforms in rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 6797–6808. [Google Scholar] [CrossRef]
- Cook, S.G.; Bourke, A.M.; O’Leary, H.; Zaegel, V.; Lasda, E.; Mize-Berge, J.; Quillinan, N.; Tucker, C.L.; Coultrap, S.J.; Herson, P.S.; et al. Analysis of the CaMKIIalpha and beta splice-variant distribution among brain regions reveals isoform-specific differences in holoenzyme formation. Sci. Rep. 2018, 8, 5448. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, H.; Lasda, E.; Bayer, K.U. CaMKIIbeta association with the actin cytoskeleton is regulated by alternative splicing. Mol. Biol. Cell 2006, 17, 4656–4665. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Redmond, L.; Xu, C.; Kuang, J.; Liao, W. Alternative splicing in the variable domain of CaMKIIbeta affects the level of F-actin association in developing neurons. Int. J. Clin. Exp. Pathol. 2014, 7, 2963–2975. [Google Scholar]
- Okuno, H.; Akashi, K.; Ishii, Y.; Yagishita-Kyo, N.; Suzuki, K.; Nonaka, M.; Kawashima, T.; Fujii, H.; Takemoto-Kimura, S.; Abe, M.; et al. Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIbeta. Cell 2012, 149, 886–898. [Google Scholar] [CrossRef]
- Puram, S.V.; Kim, A.H.; Ikeuchi, Y.; Wilson-Grady, J.T.; Merdes, A.; Gygi, S.P.; Bonni, A. A CaMKIIbeta signaling pathway at the centrosome regulates dendrite patterning in the brain. Nat. Neurosci. 2011, 14, 973–983. [Google Scholar] [CrossRef]
- Gaertner, T.R.; Kolodziej, S.J.; Wang, D.; Kobayashi, R.; Koomen, J.M.; Stoops, J.K.; Waxham, M.N. Comparative analyses of the three-dimensional structures and enzymatic properties of alpha, beta, gamma and delta isoforms of Ca2+-calmodulin-dependent protein kinase II. J. Biol. Chem. 2004, 279, 12484–12494. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Redmond, L. CaMKIIbeta binding to stable F-actin in vivo regulates F-actin filament stability. Proc. Natl. Acad. Sci. USA 2008, 105, 15791–15796. [Google Scholar] [CrossRef] [PubMed]
- Nicole, O.; Bell, D.M.; Leste-Lasserre, T.; Doat, H.; Guillemot, F.; Pacary, E. A novel role for CAMKIIbeta in the regulation of cortical neuron migration: Implications for neurodevelopmental disorders. Mol. Psychiatry 2018, 23, 2209–2226. [Google Scholar] [CrossRef]
- Miller, S.G.; Kennedy, M.B. Distinct forebrain and cerebellar isozymes of type II Ca2+/calmodulin-dependent protein kinase associate differently with the postsynaptic density fraction. J. Biol. Chem. 1985, 260, 9039–9046. [Google Scholar]
- van Woerden, G.M.; Hoebeek, F.E.; Gao, Z.; Nagaraja, R.Y.; Hoogenraad, C.C.; Kushner, S.A.; Hansel, C.; De Zeeuw, C.I.; Elgersma, Y. betaCaMKII controls the direction of plasticity at parallel fiber-Purkinje cell synapses. Nat. Neurosci. 2009, 12, 823–825. [Google Scholar] [CrossRef]
- Liu, X.B.; Jones, E.G. Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not gamma-aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc. Natl. Acad. Sci. USA 1996, 93, 7332–7336. [Google Scholar] [CrossRef] [PubMed]
- Sik, A.; Hajos, N.; Gulacsi, A.; Mody, I.; Freund, T.F. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc. Natl. Acad. Sci. USA 1998, 95, 3245–3250. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, T.C.; Piedras-Renteria, E.S.; Tsien, R.W. alpha- and betaCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 2002, 36, 1103–1114. [Google Scholar] [CrossRef]
- Conlee, J.W.; Shapiro, S.M.; Churn, S.B. Expression of the alpha and beta subunits of Ca2+/calmodulin kinase II in the cerebellum of jaundiced Gunn rats during development: A quantitative light microscopic analysis. Acta Neuropathol. 2000, 99, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Waggener, C.T.; Dupree, J.L.; Elgersma, Y.; Fuss, B. CaMKIIbeta regulates oligodendrocyte maturation and CNS myelination. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 10453–10458. [Google Scholar] [CrossRef]
- Bennett, M.K.; Erondu, N.E.; Kennedy, M.B. Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J. Biol. Chem. 1983, 258, 12735–12744. [Google Scholar]
- Fink, C.C.; Bayer, K.U.; Myers, J.W.; Ferrell, J.E., Jr.; Schulman, H.; Meyer, T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 2003, 39, 283–297. [Google Scholar] [CrossRef]
- Shen, K.; Meyer, T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 1999, 284, 162–166. [Google Scholar] [CrossRef]
- Kim, K.; Lakhanpal, G.; Lu, H.E.; Khan, M.; Suzuki, A.; Hayashi, M.K.; Narayanan, R.; Luyben, T.T.; Matsuda, T.; Nagai, T.; et al. A Temporary Gating of Actin Remodeling during Synaptic Plasticity Consists of the Interplay between the Kinase and Structural Functions of CaMKII. Neuron 2015, 87, 813–826. [Google Scholar] [CrossRef]
- Gao, Z.; van Woerden, G.M.; Elgersma, Y.; De Zeeuw, C.I.; Hoebeek, F.E. Distinct roles of alpha- and betaCaMKII in controlling long-term potentiation of GABAA-receptor mediated transmission in murine Purkinje cells. Front. Cell. Neurosci. 2014, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Bachstetter, A.D.; Webster, S.J.; Tu, T.; Goulding, D.S.; Haiech, J.; Watterson, D.M.; Van Eldik, L.J. Generation and behavior characterization of CaMKIIbeta knockout mice. PLoS ONE 2014, 9, e105191. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.J.; van de Bree, J.E.; Bodde, H.E.; Elgersma, Y.; van Woerden, G.M. The molecular, temporal and region-specific requirements of the beta isoform of Calcium/Calmodulin-dependent protein kinase type 2 (CAMK2B) in mouse locomotion. Sci. Rep. 2016, 6, 26989. [Google Scholar] [CrossRef] [PubMed]
- Borgesius, N.Z.; van Woerden, G.M.; Buitendijk, G.H.; Keijzer, N.; Jaarsma, D.; Hoogenraad, C.C.; Elgersma, Y. betaCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting alphaCaMKII to synapses. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 10141–10148. [Google Scholar] [CrossRef]
- Kim, K.; Suzuki, A.; Kojima, H.; Kawamura, M.; Miya, K.; Abe, M.; Yamada, I.; Furuse, T.; Wakana, S.; Sakimura, K.; et al. Autophosphorylation of F-actin binding domain of CaMKIIbeta is required for fear learning. Neurobiol. Learn. Mem. 2019, 157, 86–95. [Google Scholar] [CrossRef]
- Elgersma, Y.; Fedorov, N.B.; Ikonen, S.; Choi, E.S.; Elgersma, M.; Carvalho, O.M.; Giese, K.P.; Silva, A.J. Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 2002, 36, 493–505. [Google Scholar] [CrossRef]
- Kool, M.J.; Proietti Onori, M.; Borgesius, N.Z.; van de Bree, J.E.; Elgersma-Hooisma, M.; Nio, E.; Bezstarosti, K.; Buitendijk, G.H.S.; Aghadavoud Jolfaei, M.; Demmers, J.A.A.; et al. CAMK2-Dependent Signaling in Neurons Is Essential for Survival. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 5424–5439. [Google Scholar] [CrossRef]
- Kury, S.; van Woerden, G.M.; Besnard, T.; Proietti Onori, M.; Latypova, X.; Towne, M.C.; Cho, M.T.; Prescott, T.E.; Ploeg, M.A.; Sanders, S.; et al. De Novo Mutations in Protein Kinase Genes CAMK2A and CAMK2B Cause Intellectual Disability. Am. J. Hum. Genet. 2017, 101, 768–788. [Google Scholar] [CrossRef]
- Azzarelli, R.; Guillemot, F.; Pacary, E. Function and regulation of Rnd proteins in cortical projection neuron migration. Front. Neurosci. 2015, 9, 19. [Google Scholar] [CrossRef]
- Komuro, H.; Rakic, P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron 1996, 17, 275–285. [Google Scholar] [CrossRef]
- Rash, B.G.; Ackman, J.B.; Rakic, P. Bidirectional radial Ca(2+) activity regulates neurogenesis and migration during early cortical column formation. Sci. Adv. 2016, 2, e1501733. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Bonni, A. Cell-intrinsic drivers of dendrite morphogenesis. Development 2013, 140, 4657–4671. [Google Scholar] [CrossRef] [PubMed]
- Hering, H.; Sheng, M. Dendritic spines: Structure, dynamics and regulation. Nat. Rev. Neurosci. 2001, 2, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Hlushchenko, I.; Koskinen, M.; Hotulainen, P. Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton 2016, 73, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Hisatsune, C.; Miyamoto, H.; Ogawa, N.; Mikoshiba, K. Regulation of spinogenesis in mature Purkinje cells via mGluR/PKC-mediated phosphorylation of CaMKIIbeta. Proc. Natl. Acad. Sci. USA 2017, 114, E5256–E5265. [Google Scholar] [CrossRef]
- Sweatt, J.D. Neural plasticity and behavior—sixty years of conceptual advances. J. Neurochem. 2016, 139 Suppl. 2, 179–199. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev.. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef]
- Okamoto, K.; Nagai, T.; Miyawaki, A.; Hayashi, Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 2004, 7, 1104–1112. [Google Scholar] [CrossRef]
- Bosch, M.; Hayashi, Y. Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 2012, 22, 383–388. [Google Scholar] [CrossRef]
- Redondo, R.L.; Morris, R.G. Making memories last: The synaptic tagging and capture hypothesis. Nat. Rev.. Neurosci. 2011, 12, 17–30. [Google Scholar] [CrossRef]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Bosch, M.; Hayashi, Y. The roles of CaMKII and F-actin in the structural plasticity of dendritic spines: A potential molecular identity of a synaptic tag? Physiology 2009, 24, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Burgin, K.E.; Waxham, M.N.; Rickling, S.; Westgate, S.A.; Mobley, W.C.; Kelly, P.T. In situ hybridization histochemistry of Ca2+/calmodulin-dependent protein kinase in developing rat brain. J. Neurosci. Off. J. Soc. Neurosci. 1990, 10, 1788–1798. [Google Scholar] [CrossRef]
- Mayford, M.; Bach, M.E.; Huang, Y.Y.; Wang, L.; Hawkins, R.D.; Kandel, E.R. Control of memory formation through regulated expression of a CaMKII transgene. Science 1996, 274, 1678–1683. [Google Scholar] [CrossRef]
- Bhattacharyya, M.; Stratton, M.M.; Going, C.C.; McSpadden, E.D.; Huang, Y.; Susa, A.C.; Elleman, A.; Cao, Y.M.; Pappireddi, N.; Burkhardt, P.; et al. Molecular mechanism of activation-triggered subunit exchange in Ca(2+)/calmodulin-dependent protein kinase II. eLife 2016, 5, e13405. [Google Scholar] [CrossRef]
- Stratton, M.; Lee, I.H.; Bhattacharyya, M.; Christensen, S.M.; Chao, L.H.; Schulman, H.; Groves, J.T.; Kuriyan, J. Activation-triggered subunit exchange between CaMKII holoenzymes facilitates the spread of kinase activity. eLife 2014, 3, e01610. [Google Scholar] [CrossRef]
- Cho, M.H.; Cao, X.; Wang, D.; Tsien, J.Z. Dentate gyrus-specific manipulation of beta-Ca2+/calmodulin-dependent kinase II disrupts memory consolidation. Proc. Natl. Acad. Sci. USA 2007, 104, 16317–16322. [Google Scholar] [CrossRef]
- Incontro, S.; Diaz-Alonso, J.; Iafrati, J.; Vieira, M.; Asensio, C.S.; Sohal, V.S.; Roche, K.W.; Bender, K.J.; Nicoll, R.A. The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat. Commun. 2018, 9, 2069. [Google Scholar] [CrossRef]
- Hansel, C.; de Jeu, M.; Belmeguenai, A.; Houtman, S.H.; Buitendijk, G.H.; Andreev, D.; De Zeeuw, C.I.; Elgersma, Y. alphaCaMKII Is essential for cerebellar LTD and motor learning. Neuron 2006, 51, 835–843. [Google Scholar] [CrossRef]
- Pinto, T.M.; Schilstra, M.J.; Roque, A.C.; Steuber, V. Binding of Filamentous Actin to CaMKII as Potential Regulation Mechanism of Bidirectional Synaptic Plasticity by beta CaMKII in Cerebellar Purkinje Cells. Sci. Rep. 2020, 10, 9019. [Google Scholar] [CrossRef]
- Yin, P.; Xu, H.; Wang, Q.; Wang, J.; Yin, L.; Xu, M.; Xie, Z.; Liu, W.; Cao, X. Overexpression of betaCaMKII impairs behavioral flexibility and NMDAR-dependent long-term depression in the dentate gyrus. Neuropharmacology 2017, 116, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Frankland, P.W.; Bontempi, B.; Talton, L.E.; Kaczmarek, L.; Silva, A.J. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science 2004, 304, 881–883. [Google Scholar] [CrossRef] [PubMed]
- Akita, T.; Aoto, K.; Kato, M.; Shiina, M.; Mutoh, H.; Nakashima, M.; Kuki, I.; Okazaki, S.; Magara, S.; Shiihara, T.; et al. De novo variants in CAMK2A and CAMK2B cause neurodevelopmental disorders. Ann. Clin. Transl. Neurol. 2018, 5, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Seeman, P.; Tallerico, T. Schizophrenia: Elevated mRNA for calcium-calmodulin-dependent protein kinase IIbeta in frontal cortex. Brain Res. Mol. Brain Res. 2000, 82, 95–100. [Google Scholar] [CrossRef]
- Novak, G.; Seeman, P.; Tallerico, T. Increased expression of calcium/calmodulin-dependent protein kinase IIbeta in frontal cortex in schizophrenia and depression. Synapse 2006, 59, 61–68. [Google Scholar] [CrossRef]
- Novak, G.; Fan, T.; O’Dowd, B.F.; George, S.R. Postnatal maternal deprivation and pubertal stress have additive effects on dopamine D2 receptor and CaMKII beta expression in the striatum. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2013, 31, 189–195. [Google Scholar] [CrossRef]
- Greenstein, R.; Novak, G.; Seeman, P. Amphetamine sensitization elevates CaMKIIbeta mRNA. Synapse 2007, 61, 827–834. [Google Scholar] [CrossRef]
- Li, K.; Zhou, T.; Liao, L.; Yang, Z.; Wong, C.; Henn, F.; Malinow, R.; Yates, J.R., Jr.; Hu, H. betaCaMKII in lateral habenula mediates core symptoms of depression. Science 2013, 341, 1016–1020. [Google Scholar] [CrossRef]
- Song, Q.; Fan, C.; Wang, P.; Li, Y.; Yang, M.; Yu, S.Y. Hippocampal CA1 betaCaMKII mediates neuroinflammatory responses via COX-2/PGE2 signaling pathways in depression. J. Neuroinflammation 2018, 15, 338. [Google Scholar] [CrossRef]
- Hoffman, L.; Farley, M.M.; Waxham, M.N. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry 2013, 52, 1198–1207. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| CaMKIIα | CaMKIIβ | ||
|---|---|---|---|
| Temporal expression (total brain) | Embryonic life | − 2 | + 3 |
| Post-natal | + | ++ | |
| Adult | +++ | ++ | |
| Regional expression (adult brain) | Hippocampus | +++ | + |
| Cerebral cortex | +++ | + | |
| Cerebellum | + | ++++ | |
| Cellular expression | Excitatory pyramidal neurons 1 | + | + |
| Inhibitory interneurons 1 | − | + | |
| Purkinje cells | + | + | |
| Cerebellar granule neurons | − | + |
| Name | Description | References |
|---|---|---|
| CaMKIIβ-T287D | Constitutively active mutant | [12,17,28,29] |
| CaMKIIβ-A303R | Ca2+/CaM binding-deficient mutant | [12,17,28,29] |
| CaMKIIβ-K43R | Impaired for ATP binding, kinase inactive mutant | [9,12,28,29] |
| CaMKIIβ285-542 CaMKIIβ344-542 | Mutants lacking kinase domain but containing F-actin binding and association domains | [9] |
| CaMKIIβ-ΔFABD | Mutant without F-actin binding domain | [15,18] |
| CaMKIIβ1-401 | Mutant without association domain | [9] |
| CaMKIIβ-Δasso | Mutant without association domain | [15,18] |
| CaMKIIβ285-401 | Mutant without kinase and association domains | [9] |
| CaMKIIβ-ΔCTS | Mutant without centrosomal targeting sequence | [15,18] |
| CaMKIIβ-All A | Phosphoblock All A mutant (all S and T residues within the FABD are changed to alanine) | [18,30] |
| CaMKIIβ-All D | Phosphomimetic All D mutant (all S and T residues within the FABD are changed to aspartic acid), loss of F-actin-binding activity | [18,30] |
| Name | Description | References |
|---|---|---|
| Camk2b−/− | Deletion exon 11 | [20] |
| Camk2b−/− | Deletion exon 2 | [31] |
| Camk2b−/− | Camk2bf/f (loxP sites flanking exons 7–8) crossed with a CMV-Cre mouse | [32] |
| Camk2bf/f | LoxP sites flanking exon 2 | [33] |
| Camk2bA303R/A303R | Mutant which cannot bind Ca2+/CaM (mutation prevents CaMKIIβ enzymatic activation, while preserving its ability to bind to actin) | [34] |
| Camk2bT287A/T287A | Autophosphorylation-deficient mutant (mutation blocks CaMKIIβ autonomous activity) | [33] |
| Camk2b exon 13:TS/A knock-in mouse | Mouse carrying phosphoblock mutations in the actin binding domain (phosphorylation sites of this region are critical for CaMKII detachment from F-actin) | [35] |
| Camk2a−/−;Camk2b−/− | Camk2a−/− [36] and Camk2b−/− [20] were used to generate double mutants | [37] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicole, O.; Pacary, E. CaMKIIβ in Neuronal Development and Plasticity: An Emerging Candidate in Brain Diseases. Int. J. Mol. Sci. 2020, 21, 7272. https://doi.org/10.3390/ijms21197272
Nicole O, Pacary E. CaMKIIβ in Neuronal Development and Plasticity: An Emerging Candidate in Brain Diseases. International Journal of Molecular Sciences. 2020; 21(19):7272. https://doi.org/10.3390/ijms21197272
Chicago/Turabian StyleNicole, Olivier, and Emilie Pacary. 2020. "CaMKIIβ in Neuronal Development and Plasticity: An Emerging Candidate in Brain Diseases" International Journal of Molecular Sciences 21, no. 19: 7272. https://doi.org/10.3390/ijms21197272
APA StyleNicole, O., & Pacary, E. (2020). CaMKIIβ in Neuronal Development and Plasticity: An Emerging Candidate in Brain Diseases. International Journal of Molecular Sciences, 21(19), 7272. https://doi.org/10.3390/ijms21197272