Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies



Abstract
1. Introduction
2. UV-Induced DNA Damage
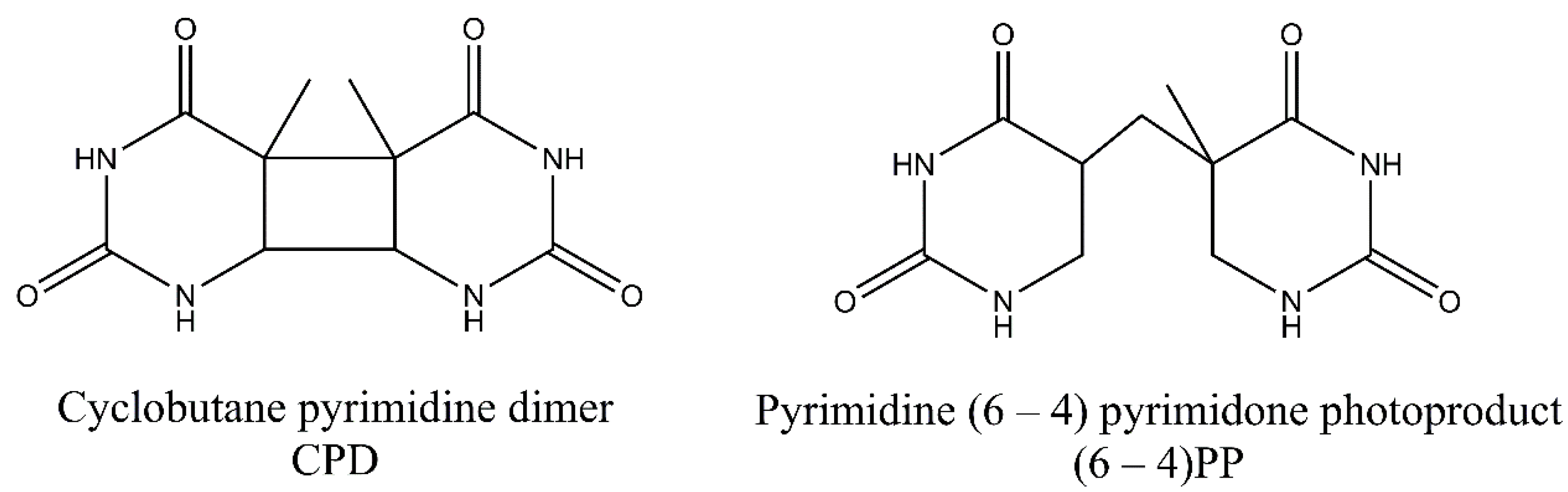
3. Pyrimidine Photoproducts
4. Purine Photoproducts
5. UV-Associated ROS Damage
6. Factors That Affect Formation of UV Damage
7. Cellular Response to UV Radiation
7.1. Kinase Signaling Pathways
7.2. Transcriptional Response to UV Radiation
7.3. Canonical DNA Damage Response and UV Radiation
7.4. Cell Cycle Regulation in Response to UV-Induced Bulky DNA Adducts
8. UV Damage Repair
8.1. Direct Reversal of Damage (DR)
8.2. UVDE-mediated Repair (UVER)
8.3. Nucleotide Excision Repair (NER)
8.4. Base Excision Repair (BER)
8.5. Double Strand Break Repair (DSBR)
8.6. Translesion Synthesis (TLS)
8.7. DNA Repair in Mitochondria
9. Disease Relevance and Future Therapeutic Strategies
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 53BP1 | TP53-binding protein 1 |
| 6-4PPs | 6-4 photoproducts |
| 8-AIA | 8-(5-aminoimidazol-4-yl)adenine |
| 8-HDF | 8-hydroxy-5-deazaflavin |
| AEJ | Alternative end-joining |
| AMD1 | S-adenosylmethionine decarboxylase proenzyme |
| AMP | Adenosine monophosphate |
| ANP32E | Acidic leucine-rich nuclear phosphoprotein 32 family member E |
| ANP32E | Acidic leucine-rich nuclear phosphoprotein 32 family member E |
| AP1/2 | Activating protein 1/2 |
| APE | AP-endonulcease |
| aPKCs | Atypical protein kinase C |
| APLF | Aprataxin and PNK-like factor |
| APTX | Aprataxin |
| ATM | Ataxia telangiectasia mutated |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| ATRIP | ATR-interacting protein |
| BAK | Bcl-2 homologous antagonist/killer |
| BAX | Apoptosis regulator BAX |
| BCL2 | Apoptosis regulator BCL2 |
| BER | Base excision repair |
| BIRC3 | Baculoviral IAP repeat-containing protein 3 |
| BLM | Bloom syndrome protein |
| BRCA1 | Breast cancer type 1 susceptibility protein |
| BSC | Basal cell carcinoma |
| BTG1 | BTG anti-proliferation factor 1 |
| CAF-1 | Chromatin assembly factor 1 |
| CAT | Catalase |
| CCNG1 | Cyclin G1 |
| CDC25A/C | M-phase inducer phosphatases |
| CDKs | Cyclin dependent kinases |
| CDT1 | DNA replication factor Cdt1 |
| CENT2 | Centrin 2 |
| CHK1/2 | Serine/threonine-protein kinase CHK1/2 |
| Cho | Excinuclease Cho |
| COFS | Cerebro-oculo-facioskeletal syndrome |
| COP9 | Constitutive photomorphogenesis 9 |
| CPDs | Cyclobutane-pyrimidine dimers |
| CS | Cockayne syndrome |
| CSA/B | Cockayne syndrome protein A/B |
| CSN | Constitutive photomorphogenesis 9 (COP9) signalosome |
| CtlP | CtBP-interacting protein |
| DDB2 | Damaged DNA-binding protein 2 |
| DDR | DNA damage response |
| DDR1 | Epithelial discoidin domain-containing receptor 1 |
| DGPY | 4,6-diamino-5-guanidinopyrimidine |
| DNA2 | DNA replication ATP-dependent helicase/nuclease DNA2 |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DP1 | Transcription factor Dp-1 |
| DR | Direct reversal of damage |
| dRP | 5’ deoxyribosephosphate |
| DSBR | Double strand break repair |
| DSBs | Double strand breaks |
| DUSP5 | Dual specificity protein phosphatase 5 |
| EGFR | Epidermal growth factor receptor |
| ERKs | Extracellular signal-regulated kinases |
| EXO1 | Exonuclease 1 |
| FACT | Facilitates chromatin transcription |
| FADH | Reduced flavin adenine dinucleotide |
| FEN-1 | Flap endonuclease 1 |
| GADD45 | Growth arrest and DNA damage-inducible protein GADD45 alpha |
| GG-NER | Global genomic NER |
| GPX | Glutathione peroxidase |
| GR | Glutathione reductase |
| HIRA | Histone regulator A |
| HMGN1 | High mobility group nucleosome−binding domain−containing protein 1 |
| HR | Homologous recombination |
| IER3 | Radiation-inducible immediate-early gene IEX-1 |
| INO80 | Chromatin-remodeling ATPase |
| JNKs | C-Jun NH2-terminal kinases |
| KAP1 | Nuclear corepressor KAP-1 |
| KU70/80 | X-ray repair cross-complementing protein 5/6 |
| MAPK | Mitogen-activated protein kinase |
| MCM6 | Minichromosome maintenance complex component 6 |
| MCM7 | DNA replication licensing factor 7 |
| MDC1 | Mediator of DNA damage checkpoint protein 1 |
| MDM2 | E3 ubiquitin-protein ligase MDM2 |
| MM | Malignant melanoma |
| MMEJ | Microhomology-mediated end-joining |
| MRN | MRE11-RAD50-NBS1 complexes |
| mtDNA | Mitochondrial DNA |
| MTHF | 5,10-methyl tetrahydrofolate |
| NAD+ | Nicotinamide adenine dinucleotide |
| NBS1 | Nibrin |
| NER | Nucleotide excision repair |
| NFAT | Nuclear factor of activated T cells |
| NFκB | Nuclear factor kappa B |
| NHEJ | Non-homologous end joining |
| NOLC1 | Nucleolar and coiled-body phosphoprotein 1 |
| ODC | Ornithine decarboxylase |
| P21 | Cyclin-dependent kinase inhibitor 1A |
| PAR | Poly-ADP-ribose |
| PARG | Poly (ADP-ribose) glycohydrolase |
| PARPs | Poly [ADP-ribose] polymerases |
| PCNA | Proliferating cell nuclear antigen |
| PEA15 | Astrocytic Phosphoprotein PEA-15 |
| PI3K | Phosphoinositide 3-kinase |
| Pol | Polymerase |
| POLE3 | DNA polymerase epsilon subunit 3 |
| pRB | Retinoblastoma-associated protein |
| PTMA | Prothymosin alpha |
| PUMA | Bcl-2-binding component 3 |
| RBX | Ubiquitin-protein ligase RBX1 |
| RAD23B | UV excision repair protein RAD23 homolog B |
| RAD51 | DNA repair protein RAD51 |
| RAD9-RAD1-HUS1 | Cell cycle checkpoint control protein complex |
| RARRES | Retinoic acid receptor responder 1 |
| RecA | Recombinase A |
| RFC | Replication factor C |
| RNAPII | RNA polymerase II |
| RNF8/20/40/168 | E3 ubiquitin-protein ligases RNF8/20/40/168 |
| ROS | Reactive oxygen species |
| RPA | Replication protein A |
| RUNX3 | RUNX family transcription factor 3 |
| S100A11 | S100 calcium binding protein A11 |
| SCC | Squamous cell carcinoma |
| SOD | Superoxide dismutase |
| SRM | Spermidine synthase |
| SSBR | Single strand break repair |
| SSBs | Single strand breaks |
| ssDNA | Single strand DNA |
| STATs | Signal transducers and activators of transcription |
| SWI/SNF | Chromatin remodeling complexes |
| TAX1BP1 | Tax1-binding protein 1 |
| TC-NER | Transcription-coupled NER |
| TDP-1 | Tyrosyl-DNA phosphodiesterase 1 |
| TF | Transcription factor |
| TFIIH | Transcription factor II |
| TLS | Translesion synthesis |
| TNF-α | Tumor necrosis factor α |
| TOB1 | Transducer of erbB-2 1 |
| TOPBP1 | DNA topoisomerase 2-binding protein 1 |
| TP53 | Cellular tumor antigen p53 |
| TRAF1 | TNF receptor-associated factor 1 |
| TRCF | Transcriptional-repair coupling factor |
| TRX | Thioredoxin reductase |
| TTD | Trichothiodystrophy |
| USP7 | Ubiquitin−specific−processing protease 7 |
| TTDA | General transcription factor 2H subunit 5 |
| TTDN1 | Trichothiodystrophy non-phothosensitive 1 |
| UVDE | UV damage endonuclease |
| UVER/UVDR | UVDE-mediated excision repair |
| UvrA/B/C | UvrABC system protein |
| UV-SS | UV-sensitive syndrome |
| WRN | Werner syndrome ATP-dependent helicase |
| XAB2 | XPA−binding protein 2 |
| XLF | Cernunos |
| XP | Xeroderma pigmentosum |
| XPC | Xeroderma pigmentosum group C |
| XRCC1/4 | X-ray repair cross-complementing protein 1/4 |
References
- Sinha, R.P.; Häder, D.-P. UV-induced DNA damage and repair: A review. Photochem. Photobiol. Sci. 2002, 1, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Pfeifer, G.P. UV damage and repair mechanisms in mammalian cells. BioEssays 1996, 18, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA Damage Response. Cold Spring Harb. Perspect. Biol. 2010, 3, a000745. [Google Scholar] [CrossRef]
- Douki, T.; Sage, E. Dewar valence isomers, the third type of environmentally relevant DNA photoproducts induced by solar radiation. Photochem. Photobiol. Sci. 2016, 15, 24–30. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J. Nucleic Acids 2010, 2010, 592980. [Google Scholar] [CrossRef]
- Latonen, L.; Laiho, M. Cellular UV damage responses—Functions of tumor suppressor p53. Biochim. Biophys. Acta (BBA)-Bioenerg. 2005, 1755, 71–89. [Google Scholar] [CrossRef]
- Perrier, S.; Hau, J.; Gasparutto, D.; Cadet, J.; Favier, A.; Ravanat, J.-L. Characterization of Lysine−Guanine Cross-Links upon One-Electron Oxidation of a Guanine-Containing Oligonucleotide in the Presence of a Trilysine Peptide. J. Am. Chem. Soc. 2006, 128, 5703–5710. [Google Scholar] [CrossRef]
- Silerme, S.; Bobyk, L.; Taverna-Porro, M.; Cuier, C.; Saint-Pierre, C.; Ravanat, J.-L. DNA-Polyamine Cross-Links Generated upon One Electron Oxidation of DNA. Chem. Res. Toxicol. 2014, 27, 1011–1018. [Google Scholar] [CrossRef]
- Cadet, J.; Ravanat, J.-L.; TavernaPorro, M.; Menoni, H.; Angelov, D. Oxidatively generated complex DNA damage: Tandem and clustered lesions. Cancer Lett. 2012, 327, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Crean, C.; Uvaydov, Y.; Geacintov, N.E.; Shafirovich, V. Oxidation of single-stranded oligonucleotides by carbonate radical anions: Generating intrastrand cross-links between guanine and thymine bases separated by cytosines. Nucleic Acids Res. 2007, 36, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Sczepanski, J.T.; Jacobs, A.C.; Majumdar, A.; Greenberg, M.M. Scope and Mechanism of Interstrand Cross-Link Formation by the C4′-Oxidized Abasic Site. J. Am. Chem. Soc. 2009, 131, 11132–11139. [Google Scholar] [CrossRef] [PubMed]
- De Lima-Bessa, K.M.; Armelini, M.G.; Chiganças, V.; Jacysyn, J.F.; Amarante-Mendes, G.P.; Sarasin, A.; Menck, C.F.M. CPDs and 6-4PPs play different roles in UV-induced cell death in normal and NER-deficient human cells. DNA Repair 2008, 7, 303–312. [Google Scholar] [CrossRef]
- Cadet, J.; Douki, T. Formation of UV-induced DNA damage contributing to skin cancer development. Photochem. Photobiol. Sci. 2018, 17, 1816–1841. [Google Scholar] [CrossRef]
- Douki, T. The variety of UV-induced pyrimidine dimeric photoproducts in DNA as shown by chromatographic quantification methods. Photochem. Photobiol. Sci. 2013, 12, 1286–1302. [Google Scholar] [CrossRef]
- Sassa, A.; Kanemaru, Y.; Kamoshita, N.; Honma, M.; Yasui, M. Mutagenic consequences of cytosine alterations site-specifically embedded in the human genome. Genes Environ. 2016, 38, 17. [Google Scholar] [CrossRef]
- Su, D.G.T.; Taylor, J.-S.; Gross, M.L. A New Photoproduct of 5-Methylcytosine and Adenine Characterized by High-Performance Liquid Chromatography and Mass Spectrometry. Chem. Res. Toxicol. 2010, 23, 474–479. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Jen, J.; Cleaver, J.E. Relative induction of cyclobutane dimers and cytosine photohydrates in DNA irradiated in vitro and in vivo with ultraviolet-C and ultraviolet-B light. Photochem. Photobiol. 1991, 54, 741–746. [Google Scholar] [CrossRef]
- Wierzchowski, K.L.; Shugar, D. Photochemistry of cytosine nucleosides and nucleotides. II. Acta Biochim. Pol. 1961, 8, 219–234. [Google Scholar]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively generated base damage to cellular DNA. Free. Radic. Biol. Med. 2010, 49, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Bauer, N.C.; Corbett, A.H.; Doetsch, P.W. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res. 2015, 43, 10083–10101. [Google Scholar] [CrossRef]
- Strumberg, D.; Pilon, A.A.; Smith, M.; Hickey, R.; Malkas, L.; Pommier, Y.G. Conversion of Topoisomerase I Cleavage Complexes on the Leading Strand of Ribosomal DNA into 5′-Phosphorylated DNA Double-Strand Breaks by Replication Runoff. Mol. Cell. Biol. 2000, 20, 3977–3987. [Google Scholar] [CrossRef]
- Ohnishi, T.; Mori, E.; Takahashi, A. DNA double-strand breaks: Their production, recognition, and repair in eukaryotes. Mutat. Res. Mol. Mech. Mutagen. 2009, 669, 8–12. [Google Scholar] [CrossRef]
- Banáth, J.; Olive, P.L. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003, 63, 4347–4350. [Google Scholar]
- Cooke, M.S.; Evans, M.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Morales, V.; Giamarchi, C.; Chailleux, C.; Moro, F.; Marsaud, V.; Le Ricousse, S.; Richard-Foy, H. Chromatin structure and dynamics: Functional implications. Biochimie 2001, 83, 1029–1039. [Google Scholar] [CrossRef]
- Woodcock, C.L.; Ghosh, R.P. Chromatin Higher-order Structure and Dynamics. Cold Spring Harb. Perspect. Biol. 2010, 2, a000596. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Reinberg, D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011, 21, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Widom, J. Role of DNA sequence in nucleosome stability and dynamics. Q. Rev. Biophys. 2001, 34, 269–324. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Swygert, S.G.; Peterson, C.L. Chromatin dynamics: Interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta (BBA)-Bioenerg. 2014, 1839, 728–736. [Google Scholar] [CrossRef]
- Segal, E.; Widom, J. What controls nucleosome positions? Trends Genet. 2009, 25, 335–343. [Google Scholar] [CrossRef]
- Li, G.; Levitus, M.; Bustamante, C.; Widom, J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2004, 12, 46–53. [Google Scholar] [CrossRef]
- Mao, P.; Smerdon, M.J.; Roberts, S.A.; Wyrick, J.J. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 9057–9062. [Google Scholar] [CrossRef]
- Mao, P.; Wyrick, J.J.; Roberts, S.A.; Smerdon, M.J. UV-Induced DNA Damage and Mutagenesis in Chromatin. Photochem. Photobiol. 2016, 93, 216–228. [Google Scholar] [CrossRef]
- Hu, J.; Adebali, O.; Adar, S.; Sancar, A. Dynamic maps of UV damage formation and repair for the human genome. Proc. Natl. Acad. Sci. USA 2017, 114, 6758–6763. [Google Scholar] [CrossRef]
- Piquet, S.; Le Parc, F.; Bai, S.-K.; Chevallier, O.; Adam, S.; Polo, S.E. The Histone Chaperone FACT Coordinates H2A.X-Dependent Signaling and Repair of DNA Damage. Mol. Cell 2018, 72, 888–901.e7. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Downs, J.A.; Gasser, S.M. Chromatin modifiers and remodellers in DNA repair and signalling. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160279. [Google Scholar] [CrossRef] [PubMed]
- Hauer, M.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [PubMed]
- Luijsterburg, M.S.; Van Attikum, H. Chromatin and the DNA damage response: The cancer connection. Mol. Oncol. 2011, 5, 349–367. [Google Scholar] [CrossRef]
- Schick, S.; Fournier, D.; Thakurela, S.; Sahu, S.K.; Garding, A.; Tiwari, V.K. Dynamics of chromatin accessibility and epigenetic state in response to UV damage. J. Cell Sci. 2015, 128, 4380–4394. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Mitogen-Activated Protein Kinase Activation in UV-Induced Signal Transduction. Sci. Signal. 2003, 2003, re2. [Google Scholar] [CrossRef]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFκB and TNFα signal transduction pathways. Arch. Dermatol. Res. 2009, 302, 5–17. [Google Scholar] [CrossRef]
- Chen, H.; Weng, Q.Y.; Fisher, D.E. UV signaling pathways within the skin. J. Investig. Dermatol. 2014, 134, 2080–2085. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef]
- Sachsenmaier, C.; Radler-Pohl, A.; Müller, A.; Herrlich, P.; Rahmsdorf, H.J. Damage to DNA by UV light and activation of transcription factors. Biochem. Pharmacol. 1994, 47, 129–136. [Google Scholar] [CrossRef]
- Marais, T.L.D.; Kluz, T.; Xu, D.; Zhang, X.; Gesumaria, L.; Matsui, M.S.; Costa, M.; Sun, H. Transcription factors and stress response gene alterations in human keratinocytes following Solar Simulated Ultra Violet Radiation. Sci. Rep. 2017, 7, 13622. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Latonen, L.; Laiho, M. Cell cycle arrest and apoptosis provoked by UV radiation-induced DNA damage are transcriptionally highly divergent responses. Nucleic Acids Res. 2003, 31, 4779–4790. [Google Scholar] [CrossRef] [PubMed]
- Rubbi, C.P.; Milner, J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003, 22, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Rieger, K.E.; Chu, G. Portrait of transcriptional responses to ultraviolet and ionizing radiation in human cells. Nucleic Acids Res. 2004, 32, 4786–4803. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; Cortez, D. DNA Damage Response: Three Levels of DNA Repair Regulation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012724. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.T.; Karlseder, J. DNA damage associated with mitosis and cytokinesis failure. Oncogene 2013, 32, 4593–4601. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM activation additional sites add to the complexity. J. Biol. Chem. 2010, 286, 9107–9119. [Google Scholar] [CrossRef]
- Harper, W.; Elledge, S.J. The DNA Damage Response: Ten Years After. Mol. Cell 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Dasika, G.K.; Lin, S.-C.J.; Zhao, S.; Sung, P.; Tomkinson, A.; Lee, E.Y.-H.P. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene 1999, 18, 7883–7899. [Google Scholar] [CrossRef]
- Raleigh, D.R.; Haas-Kogan, D.A. Molecular targets and mechanisms of radiosensitization using DNA damage response pathways. Future Oncol. 2013, 9, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Ribezzo, F.; Shiloh, Y.; Schumacher, B. Systemic DNA damage responses in aging and diseases. Semin. Cancer Biol. 2016, 37–38, 26–35. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Antoniono, R.J.; Redpath, J.L.; Stanbridge, E.J. DNA damage and p53-mediated cell cycle arrest: A reevaluation. Proc. Natl. Acad. Sci. USA 1996, 93, 15209–15214. [Google Scholar] [CrossRef] [PubMed]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.M.; Newbold, A.; Johnstone, R.W. Intrinsic and Extrinsic Apoptotic Pathway Signaling as Determinants of Histone Deacetylase Inhibitor Antitumor Activity. Adv. Cancer Res. 2012, 116, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Asp. Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [PubMed]
- Purohit, N.K.; Robu, M.; Shah, R.G.; Geacintov, N.E.; Shah, G.M. Characterization of the interactions of PARP-1 with UV-damaged DNA in vivo and in vitro. Sci. Rep. 2016, 6, 19020. [Google Scholar] [CrossRef]
- Qin, X.-J.; Liu, W.; Li, Y.-N.; Sun, X.; Hai, C.-X.; Hudson, L.G.; Liu, K.J. Poly(ADP-Ribose) Polymerase-1 Inhibition by Arsenite Promotes the Survival of Cells With Unrepaired DNA Lesions Induced by UV Exposure. Toxicol. Sci. 2012, 127, 120–129. [Google Scholar] [CrossRef]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef]
- Stokes, M.; Comb, M. A wide-ranging cellular response to UV damage of DNA. Cell Cycle 2008, 7, 2097–2099. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pavey, S.; Russell, T.; Gabrielli, B. G2 phase cell cycle arrest in human skin following UV irradiation. Oncogene 2001, 20, 6103–6110. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K. Xeroderma pigmentosum genes: Functions inside and outside DNA repair. Carcinogenesis 2008, 29, 455–465. [Google Scholar] [CrossRef]
- Liu, O.-G.; Xiong, X.-Y.; Li, C.-M.; Zhou, X.-S.; Li, S.-S. Role of Xeroderma Pigmentosum Group D in Cell Cycle and Apoptosis in Cutaneous Squamous Cell Carcinoma A431 Cells. Med. Sci. Monit. 2018, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Musich, P.R.; Li, Z.; Zou, Y. Xeroderma Pigmentosa Group A (XPA), Nucleotide Excision Repair and Regulation by ATR in Response to Ultraviolet Irradiation. Adv. Exp. Med. Biol. 2017, 996, 41–54. [Google Scholar] [CrossRef]
- Morino, M.; Nukina, K.; Sakaguchi, H.; Maeda, T.; Takahara, M.; Shiomi, Y.; Nishitani, H. Mitotic UV Irradiation Induces a DNA Replication-Licensing Defect that Potentiates G1 Arrest Response. PLoS ONE 2015, 10, e0120553. [Google Scholar] [CrossRef]
- Yi, C.; He, C. DNA Repair by Reversal of DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012575. [Google Scholar] [CrossRef]
- Sancar, A. Structure and Function of Photolyase andin VivoEnzymology: 50th Anniversary. J. Biol. Chem. 2008, 283, 32153–32157. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, L.; Zhong, D. Photolyase: Dynamics and electron-transfer mechanisms of DNA repair. Arch. Biochem. Biophys. 2017, 632, 158–174. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, L.; Zhong, D. Dynamics and mechanisms of DNA repair by photolyase. Phys. Chem. Chem. Phys. 2015, 17, 11933–11949. [Google Scholar] [CrossRef]
- Essen, L.-O.; Klar, T. Light-driven DNA repair by photolyases. Cell. Mol. Life Sci. 2006, 63, 1266–1277. [Google Scholar] [CrossRef]
- You, Y.-H.; Lee, D.-H.; Yoon, J.-H.; Nakajima, S.; Yasui, A.; Pfeifer, G.P. Cyclobutane Pyrimidine Dimers Are Responsible for the Vast Majority of Mutations Induced by UVB Irradiation in Mammalian Cells. J. Biol. Chem. 2001, 276, 44688–44694. [Google Scholar] [CrossRef]
- Schul, W.; Jans, J.; Rijksen, Y.M.; Klemann, K.H.; Eker, A.P.; De Wit, J.; Nikaido, O.; Nakajima, S.; Yasui, A.; Hoeijmakers, J.H.; et al. Enhanced repair of cyclobutane pyrimidine dimers and improved UV resistance in photolyase transgenic mice. EMBO J. 2002, 21, 4719–4729. [Google Scholar] [CrossRef]
- Jans, J.; Schul, W.; Sert, Y.-G.; Rijksen, Y.; Rebel, H.; Eker, A.P.; Nakajima, S.; Van Steeg, H.; De Gruijl, F.R.; Yasui, A.; et al. Powerful Skin Cancer Protection by a CPD-Photolyase Transgene. Curr. Biol. 2005, 15, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Asagoshi, K.; Liu, Y.; Masaoka, A.; Lan, L.; Prasad, R.; Horton, J.K.; Brown, A.R.; Wang, X.-H.; Bdour, H.M.; Sobol, R.W.; et al. DNA polymerase β-dependent long patch base excision repair in living cells. DNA Repair 2010, 9, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Yasui, A. Alternative Excision Repair Pathways. Cold Spring Harb. Perspect. Biol. 2013, 5, a012617. [Google Scholar] [CrossRef] [PubMed]
- Sha, Y.; Vartanian, V.; Owen, N.; Koon, S.J.M.; Calkins, M.J.; Thompson, C.S.; MirAfzali, Z.; Mir, S.; Goldsmith, L.E.; He, H.; et al. Modulation of UVB-induced Carcinogenesis by Activation of Alternative DNA Repair Pathways. Sci. Rep. 2018, 8, 705. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef]
- Park, J.-M.; Kang, T.H. Transcriptional and Posttranslational Regulation of Nucleotide Excision Repair: The Guardian of the Genome against Ultraviolet Radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef]
- Kisker, C.; Kuper, J.; Van Houten, B. Prokaryotic Nucleotide Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012591. [Google Scholar] [CrossRef]
- Truglio, J.J.; Croteau, D.L.; Van Houten, B.; Kisker, C. Prokaryotic Nucleotide Excision Repair: The UvrABC System. Chem. Rev. 2006, 106, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Selby, C.P.; Adar, S.; Adebali, O.; Sancar, A. Molecular mechanisms and genomic maps of DNA excision repair inEscherichia coliand humans. J. Biol. Chem. 2017, 292, 15588–15597. [Google Scholar] [CrossRef] [PubMed]
- Czaja, W.; Mao, P.; Smerdon, M.J. The Emerging Roles of ATP-Dependent Chromatin Remodeling Enzymes in Nucleotide Excision Repair. Int. J. Mol. Sci. 2012, 13, 11954–11973. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Polo, S.E. Chromatin Dynamics during Nucleotide Excision Repair: Histones on the Move. Int. J. Mol. Sci. 2012, 13, 11895–11911. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef]
- Vermeulen, W.; Fousteri, M.I. Mammalian Transcription-Coupled Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012625. [Google Scholar] [CrossRef]
- Spivak, G. Transcription-coupled repair: An update. Arch. Toxicol. 2016, 90, 2583–2594. [Google Scholar] [CrossRef]
- Fousteri, M.I.; Mullenders, L.H. Transcription-coupled nucleotide excision repair in mammalian cells: Molecular mechanisms and biological effects. Cell Res. 2008, 18, 73–84. [Google Scholar] [CrossRef]
- Sarasin, A. UVSSA and USP7: New players regulating transcription-coupled nucleotide excision repair in human cells. Genome Med. 2012, 4, 44. [Google Scholar] [CrossRef]
- Schwertman, P.; Vermeulen, W.; Marteijn, J.A. UVSSA and USP7, a new couple in transcription-coupled DNA repair. Chromosoma 2013, 122, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Valles, G.J.; Bezsonova, I.; Woodgate, R.; Ashton, N.W. USP7 Is a Master Regulator of Genome Stability. Front. Cell Dev. Biol. 2020, 8, 717. [Google Scholar] [CrossRef] [PubMed]
- Pozhidaeva, A.; Bezsonova, I. USP7: Structure, substrate specificity, and inhibition. DNA Repair 2019, 76, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.W.; Raams, A.; Van Der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.J.; Demmers, J.A.A.; Fousteri, M.I.; Vermeulen, W.; et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef]
- Reardon, J.T.; Sancar, A. Nucleotide Excision Repair. Prog. Nucleic Acid Res. Mol. Biol. 2005, 79, 183–235. [Google Scholar] [CrossRef]
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Gong, F.; Kwon, Y.; Smerdon, M.J. Nucleotide excision repair in chromatin and the right of entry. DNA Repair 2005, 4, 884–896. [Google Scholar] [CrossRef]
- Rechkunova, N.I.; Maltseva, E.A.; Lavrik, O.I. Post-translational Modifications of Nucleotide Excision Repair Proteins and Their Role in the DNA Repair. Biochem. (Mosc.) 2019, 84, 1008–1020. [Google Scholar] [CrossRef]
- Kurthkoti, K.; Kumar, P.; Sang, P.B.; Varshney, U. Base excision repair pathways of bacteria: New promise for an old problem. Future Med. Chem. 2020, 12, 339–355. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, A.L.; Schär, P. DNA glycosylases: In DNA repair and beyond. Chromosoma 2011, 121, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef]
- Ignatov, A.V.; Bondarenko, K.A.; Makarova, A.V. Non-bulky Lesions in Human DNA: The Ways of Formation, Repair, and Replication. Acta Naturae 2017, 9, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, J.L.; Dunn, J.; Rees, A.; Rünger, T.M.; Rünger, T.M. No Formation of DNA Double-Strand Breaks and No Activation of Recombination Repair with UVA. J. Investig. Dermatol. 2011, 131, 1139–1148. [Google Scholar] [CrossRef]
- Rolfsmeier, M.L.; Laughery, M.F.; Haseltine, C. Repair of DNA Double-Strand Breaks following UV Damage in Three Sulfolobussolfataricus Strains. J. Bacteriol. 2010, 192, 4954–4962. [Google Scholar] [CrossRef]
- Greinert, R.; Volkmer, B.; Henning, S.; Breitbart, E.W.; Greulich, K.O.; Cardoso, M.C.; Rapp, A. UVA-induced DNA double-strand breaks result from the repair of clustered oxidative DNA damages. Nucleic Acids Res. 2012, 40, 10263–10273. [Google Scholar] [CrossRef]
- Pennisi, R.; Ascenzi, P.; Di Masi, A. Hsp90: A New Player in DNA Repair? Biomolecules 2015, 5, 2589–2618. [Google Scholar] [CrossRef]
- Helleday, T.; Lo, J.; Van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Barnes, D.E. Non-homologous end joining as a mechanism of DNA repair. Curr. Biol. 2001, 11, R455–R457. [Google Scholar] [CrossRef]
- Yang, K.; Guo, R.; Xu, D. Non-homologous end joining: Advances and frontiers. Acta Biochim. Biophys. Sin. 2016, 48, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2017, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E. Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Waters, L.S.; Minesinger, B.K.; Wiltrout, M.E.; D’Souza, S.; Woodruff, R.V.; Walker, G.C. Eukaryotic Translesion Polymerases and Their Roles and Regulation in DNA Damage Tolerance. Microbiol. Mol. Biol. Rev. 2009, 73, 134–154. [Google Scholar] [CrossRef]
- Bertok, D. Žgur DNA Damage Repair and Bacterial Pathogens. PLoS Pathog. 2013, 9, e1003711. [Google Scholar] [CrossRef]
- Sertic, S.; Mollica, A.; Campus, I.; Roma, S.; Tumini, E.; Aguilera, A.; Muzi-Falconi, M. Coordinated Activity of Y Family TLS Polymerases and EXO1 Protects Non-S Phase Cells from UV-Induced Cytotoxic Lesions. Mol. Cell 2018, 70, 34–47. [Google Scholar] [CrossRef]
- Hendel, A.; Krijger, P.H.; Diamant, N.; Goren, Z.; Langerak, P.; Kim, J.; Reißner, T.; Lee, K.-Y.; Geacintov, N.E.; Carell, T.; et al. PCNA Ubiquitination Is Important, But Not Essential for Translesion DNA Synthesis in Mammalian Cells. PLoS Genet. 2011, 7, e1002262. [Google Scholar] [CrossRef]
- Prakash, S.; Prakash, L. Translesion DNA synthesis in eukaryotes: A one- or two-polymerase affair. Genes Dev. 2002, 16, 1872–1883. [Google Scholar] [CrossRef] [PubMed]
- Hedglin, M.; Pandey, B.; Benkovic, S.J. Characterization of human translesion DNA synthesis across a UV-induced DNA lesion. eLife 2016, 5, e19788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Washington, M.T. Translesion Synthesis: Insights into the Selection and Switching of DNA Polymerases. Genes 2017, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Powers, K.T.; Washington, M.T. Eukaryotic translesion synthesis: Choosing the right tool for the job. DNA Repair 2018, 71, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Gao, Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu. Rev. Biochem. 2018, 87, 239–261. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef]
- Zafar, M.K.; Eoff, R.L. Translesion DNA Synthesis in Cancer: Molecular Mechanisms and Therapeutic Opportunities. Chem. Res. Toxicol. 2017, 30, 1942–1955. [Google Scholar] [CrossRef]
- Fuchs, R.P.; Fujii, S. Translesion DNA Synthesis and Mutagenesis in Prokaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012682. [Google Scholar] [CrossRef]
- Ma, X.; Tang, T.; Guo, C. Regulation of translesion DNA synthesis in mammalian cells. Environ. Mol. Mutagen. 2020. [Google Scholar] [CrossRef]
- Brand, R.M.; Wipf, P.; Durham, A.; Epperly, M.W.; Greenberger, J.S.; Falo, L.D.J. Targeting Mitochondrial Oxidative Stress to Mitigate UV-Induced Skin Damage. Front. Pharmacol. 2018, 9, 920. [Google Scholar] [CrossRef]
- Birch-Machin, M.A.; Swalwell, H. How mitochondria record the effects of UV exposure and oxidative stress using human skin as a model tissue. Mutagenesis 2010, 25, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Sia, E.A. Mitochondrial DNA repair and damage tolerance. Front. Biosci. 2017, 22, 920–943. [Google Scholar] [CrossRef]
- Torregrosa-Muñumer, R.; Goffart, S.; Haikonen, J.A.; Pohjoismäki, J. Low doses of ultraviolet radiation and oxidative damage induce dramatic accumulation of mitochondrial DNA replication intermediates, fork regression, and replication initiation shift. Mol. Biol. Cell 2015, 26, 4197–4208. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, B.; Versteegh, A.; Van Hoffen, A.; Van Zeeland, A.; Mullenders, L.; Dogliotti, E. DNA repair of UV photoproducts and mutagenesis in human mitochondrial DNA. J. Mol. Biol. 1997, 273, 417–427. [Google Scholar] [CrossRef]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Chrzanowska-Lightowlers, Z.M.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta (BBA)-Bioenerg. 2011, 1813, 186–200. [Google Scholar] [CrossRef]
- O’Driscoll, M. Diseases Associated with Defective Responses to DNA Damage. Cold Spring Harb. Perspect. Biol. 2012, 4, a012773. [Google Scholar] [CrossRef]
- Sáez, G.T. DNA Damage and Repair in Degenerative Diseases 2016. Int. J. Mol. Sci. 2017, 18, 166. [Google Scholar] [CrossRef]
- Tiwari, V.; Wilson, D.M. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef]
- Black, J.O. Xeroderma Pigmentosum. Head Neck Pathol. 2016, 10, 139–144. [Google Scholar] [CrossRef]
- Lehmann, A.R.; McGibbon, D.; Stefanini, M. Xeroderma pigmentosum. Orphanet J. Rare Dis. 2011, 6, 70. [Google Scholar] [CrossRef]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Rapin, I.; Weidenheim, K.; Lindenbaum, Y.; Rosenbaum, P.; Merchant, S.N.; Krishna, S.; Dickson, D.W. Cockayne syndrome in adults: Review with clinical and pathologic study of a new case. J. Child Neurol. 2006, 21, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Lambert, W.C.; Gagna, C.E.; Lambert, M.W. Trichothiodystrophy: Photosensitive, TTD-P, TTD, Tay syndrome. Adv. Exp. Med. Biol. 2010, 685, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, M.; Botta, E.; Lanzafame, M.; Orioli, D. Trichothiodystrophy: From basic mechanisms to clinical implications. DNA Repair 2010, 9, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Egly, J.-M. Trichothiodystrophy view from the molecular basis of DNA repair/transcription factor TFIIH. Hum. Mol. Genet. 2009, 18, R224–R230. [Google Scholar] [CrossRef]
- Faghri, S.; Tamura, D.; Kraemer, K.H.; DiGiovanna, J.J. Trichothiodystrophy: A systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J. Med Genet. 2008, 45, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Yew, Y.W.; Giordano, C.N.; Spivak, G.; Lim, H.W. Understanding photodermatoses associated with defective DNA repair: Photosensitive syndromes without associated cancer predisposition. J. Am. Acad. Dermatol. 2016, 75, 873–882. [Google Scholar] [CrossRef]
- Weidenheim, K.M.; Dickson, D.W.; Rapin, I. Neuropathology of Cockayne syndrome: Evidence for impaired development, premature aging, and neurodegeneration. Mech. Ageing Dev. 2009, 130, 619–636. [Google Scholar] [CrossRef]
- Rapin, I. Disorders of nucleotide excision repair. Handb. Clin. Neurol. 2013, 113, 1637–1650. [Google Scholar] [CrossRef]
- Moriwaki, S. Hereditary Disorders with Defective Repair of UV-Induced DNA Damage. Jpn. Clin. Med. 2013, 4, 29–35. [Google Scholar] [CrossRef]
- Leibeling, D.; Laspe, P.; Emmert, S. Nucleotide excision repair and cancer. J. Mol. Histol. 2006, 37, 225–238. [Google Scholar] [CrossRef]
- Bergoglio, V.; Magnaldo, T. Nucleotide excision repair and related human diseases. In Genome and Disease; Karger Publishers: Basel, Switzerland, 2006; Volume 1, pp. 35–52. [Google Scholar] [CrossRef]
- Lambert, W.C.; Lambert, M.W. Development of Effective Skin Cancer Treatment and Prevention in Xeroderma Pigmentosum. Photochem. Photobiol. 2015, 91, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Magnaldo, T. Xeroderma pigmentosum: From genetics to hopes and realities of cutaneous gene therapy. Expert Opin. Biol. Ther. 2004, 4, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Maturo, M.G.; Di Nardo, L.; Ciciarelli, V.; García-Rodrigo, C.G.; Fargnoli, M.C. Understanding the Molecular Genetics of Basal Cell Carcinoma. Int. J. Mol. Sci. 2017, 18, 2485. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, L.; Farshchian, M.; Riihilä, P.; Kähäri, V.-M. New perspectives on role of tumor microenvironment in progression of cutaneous squamous cell carcinoma. Cell Tissue Res. 2016, 365, 691–702. [Google Scholar] [CrossRef]
- Rodust, P.M.; Stockfleth, E.; Ulrich, C.; Leverkus, M.; Eberle, J. UV-induced squamous cell carcinoma–A role for antiapoptotic signalling pathways. Br. J. Dermatol. 2009, 161 (Suppl. 3), 107–115. [Google Scholar] [CrossRef]
- Feller, L.; Khammissa, R.A.G.; Kramer, B.; Altini, M.; Lemmer, J. Basal cell carcinoma, squamous cell carcinoma and melanoma of the head and face. Head Face Med. 2016, 12, 11. [Google Scholar] [CrossRef]
- Liu-Smith, F.; Jia, J.; Zheng, Y. UV-Induced Molecular Signaling Differences in Melanoma and Non-melanoma Skin Cancer. Adv. Exp. Med. Biol. 2017, 996, 27–40. [Google Scholar] [CrossRef]
- Brenner, M.; Hearing, V.J. The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 2008, 84, 539–549. [Google Scholar] [CrossRef]
- Amaro-Ortiz, A.; Yan, B.; D’Orazio, J.A. Ultraviolet Radiation, Aging and the Skin: Prevention of Damage by Topical cAMP Manipulation. Molecules 2014, 19, 6202–6219. [Google Scholar] [CrossRef]
- Latha, M.S.; Martis, J.; Shobha, V.; Sham Shinde, R.; Bangera, S.; Krishnankutty, B.; Bellary, S.; Varughese, S.; Rao, P.; Naveen Kumar, B.R. Sunscreening agents: A review. J. Clin. Aesthetic Dermatol. 2013, 6, 16–26. [Google Scholar]
- Dunaway, S.; Odin, R.; Zhou, L.; Ji, L.; Zhang, Y.; Kadekaro, A.L. Natural Antioxidants: Multiple Mechanisms to Protect Skin From Solar Radiation. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.F.; Ramírez, M.L.G.; Campmany, A.C.C.; Martínez, A.R.; Naveros, B.C. In vivo and in vitro evaluation of the use of a newly developed melatonin loaded emulsion combined with UV filters as a protective agent against skin irradiation. J. Dermatol. Sci. 2013, 69, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.P. Ensuring the Safety of Sunscreens, and Their Efficacy in Preventing Skin Cancers: Challenges and Controversies for Clinicians, Formulators, and Regulators. Front. Med. 2019, 6, 195. [Google Scholar] [CrossRef]
- Gupta, A.; Avci, P.; Dai, T.; Huang, Y.-Y.; Hamblin, M.R. Ultraviolet Radiation in Wound Care: Sterilization and Stimulation. Adv. Wound Care 2013, 2, 422–437. [Google Scholar] [CrossRef]
- Yin, R.; Dai, T.; Avci, P.; Jorge, A.E.S.; De Melo, W.; Vecchio, D.; Huang, Y.-Y.; Gupta, A.; Hamblin, M.R. Light based anti-infectives: Ultraviolet C irradiation, photodynamic therapy, blue light, and beyond. Curr. Opin. Pharmacol. 2013, 13, 731–762. [Google Scholar] [CrossRef]
- Reed, N.G. The History of Ultraviolet Germicidal Irradiation for Air Disinfection. Public Health Rep. 2010, 125, 15–27. [Google Scholar] [CrossRef]
- Vatansever, F.; Ferraresi, C.; De Sousa, M.V.P.; Yin, R.; Rineh, A.; Sharma, S.K.; Hamblin, M.R. Can biowarfare agents be defeated with light? Virulence 2013, 4, 796–825. [Google Scholar] [CrossRef]
- Lytle, C.D.; Sagripanti, J.-L. Predicted Inactivation of Viruses of Relevance to Biodefense by Solar Radiation. J. Virol. 2005, 79, 14244–14252. [Google Scholar] [CrossRef]
- Yarosh, D.; Klein, J.; O’Connor, A.; Hawk, J.; Rafal, E.; Wolf, P. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: A randomised study. Xeroderma Pigmentosum Study Group. Lancet 2001, 357, 926–929. [Google Scholar] [CrossRef]
- Lloyd, R.S. Investigations of pyrimidine dimer glycosylases—A paradigm for DNA base excision repair enzymology. Mutat. Res. Mol. Mech. Mutagen. 2005, 577, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Stege, H.; Roza, L.; Vink, A.A.; Grewe, M.; Ruzicka, T.; Grether-Beck, S.; Krutmann, J. Enzyme plus light therapy to repair DNA damage in ultraviolet-B-irradiated human skin. Proc. Natl. Acad. Sci. USA 2000, 97, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Stege, H. Effect of xenogenic repair enzymes on photoimmunology and photocarcinogenesis. J. Photochem. Photobiol. B Biol. 2001, 65, 105–108. [Google Scholar] [CrossRef]
- Leccia, M.T.; Lebbe, C.; Claudel, J.-P.; Narda, M.; Basset-Seguin, N. New Vision in Photoprotection and Photorepair. Dermatol. Ther. 2019, 9, 103–115. [Google Scholar] [CrossRef]
- Yarosh, D.B.; Rosenthal, A.; Moy, R. Six critical questions for DNA repair enzymes in skincare products: A review in dialog. Clin. Cosmet. Investig. Dermatol. 2019, 12, 617–624. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. coli Protein | Function | H. sapiens Protein | Function |
|---|---|---|---|
| UvrA | Damage recognition, molecular matchmaker | DDB1, XPC, XPE, RAD23B | Damage binding, recruitment of other NER factors |
| UvrB | Damage recognition, unwinding and bending of DNA molecule | XPB XPD | 3′-5′ helicase 5′-3′ helicase DNA unwinding |
| UvrC | 3′ and 5′ excision | XPG XPF ERCC1 | Endonuclease/3′ incision Endonuclease/5′ incision |
| UvrD | Helicase | XPA | Damage verification, RPA and ERCC1 recruitment |
| TRCF (Mfd) | Transcription coupled repair | CSA/CSB complex | Transcription coupled repair |
| Feature | Homologous Recombination (HR) | Non-Homologous End Joining (NHEJ) |
|---|---|---|
| Type of lesion repaired | DSBs, stalled replication forks, inter-strand DNA cross-links and DSBs resulting from abortive topoisomerase II action | Radiation- or chemically induced DSBs, V(D)J recombination |
| Key components | RAD51 and RAD51-related proteins (XRCC2, XRCC3), RAD52, BRCA2, RPA, FEN1, DNA polymerase and associated factors. Promoted by MRN, CtIP, BRCA1, and the ATM signaling pathway | Ku and DNA-PKcs, XRCC4, XLF/Cernunnos ligase IV. MRE11-RAD50-NBS1 complex, Artemis nuclease, PNK, Aprataxin and polymerases μ and λ |
| Mechanism | Repair involves invasion of the homologous strand on the DNA duplex | Repair involves formation of DNA-protein complexes with broken ends for efficient repair |
| Template | Requires sister chromatid as a template during repair process | Does not require sister chromatid as a template during repair process |
| Cell cycle | Is active only in S and G2 phase of the cell cycle | Is active throughout most of the cell cycle |
| Fidelity | Repair is generally error-free | Repair is error-prone |
| Phenotype | Genes Mutated (Prevalent) | Genes Mutated (Others) | Features of Classical Variant of the Disease | Ref. |
|---|---|---|---|---|
| XP | XPC, XPE | XPA, XPB/ERCC3, XPG, XPF, XPV | extreme sensitivity to sunlight, hypopigmentation, hyperpigmentation, predisposition to cancer | [149,150] |
| CS | CSA, CSB | XPG, XPB, XPD | hypersensitivity to sunlight and various mutagens, neurological symptoms, dwarfing, microcephaly, mental retardation, dysmyelination, retinal degeneration | [151,152] |
| TTD | XPB, XPD | TTDA, TTDN1 | sensitivity to sunlight, fragile hair and nails, and scaly skin | [153,154,155,156] |
| COFS | CSB | XPD, XPG | Depend on the subtype of the disease: neurodegeneration microcephaly, congenital cataracts, severe mental retardation, facial dysmorphism, and arthrogryposis | [107,157,158,159] |
| UV-SS | UVSSA | CSA, CSB | sun sensitivity |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kciuk, M.; Marciniak, B.; Mojzych, M.; Kontek, R. Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies. Int. J. Mol. Sci. 2020, 21, 7264. https://doi.org/10.3390/ijms21197264
Kciuk M, Marciniak B, Mojzych M, Kontek R. Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies. International Journal of Molecular Sciences. 2020; 21(19):7264. https://doi.org/10.3390/ijms21197264
Chicago/Turabian StyleKciuk, Mateusz, Beata Marciniak, Mariusz Mojzych, and Renata Kontek. 2020. "Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies" International Journal of Molecular Sciences 21, no. 19: 7264. https://doi.org/10.3390/ijms21197264
APA StyleKciuk, M., Marciniak, B., Mojzych, M., & Kontek, R. (2020). Focus on UV-Induced DNA Damage and Repair—Disease Relevance and Protective Strategies. International Journal of Molecular Sciences, 21(19), 7264. https://doi.org/10.3390/ijms21197264