Constitutive Activation and Inactivation of Mutations Inducing Cell Surface Loss of Receptor and Impairing of Signal Transduction of Agonist-Stimulated Eel Follicle-Stimulating Hormone Receptor


Abstract
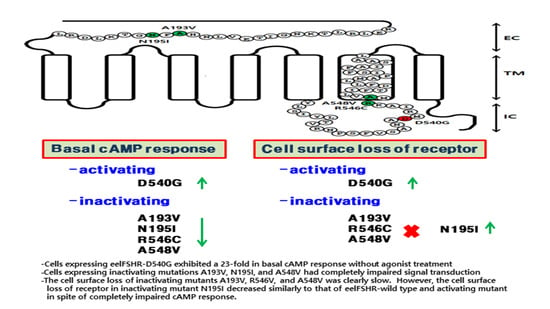
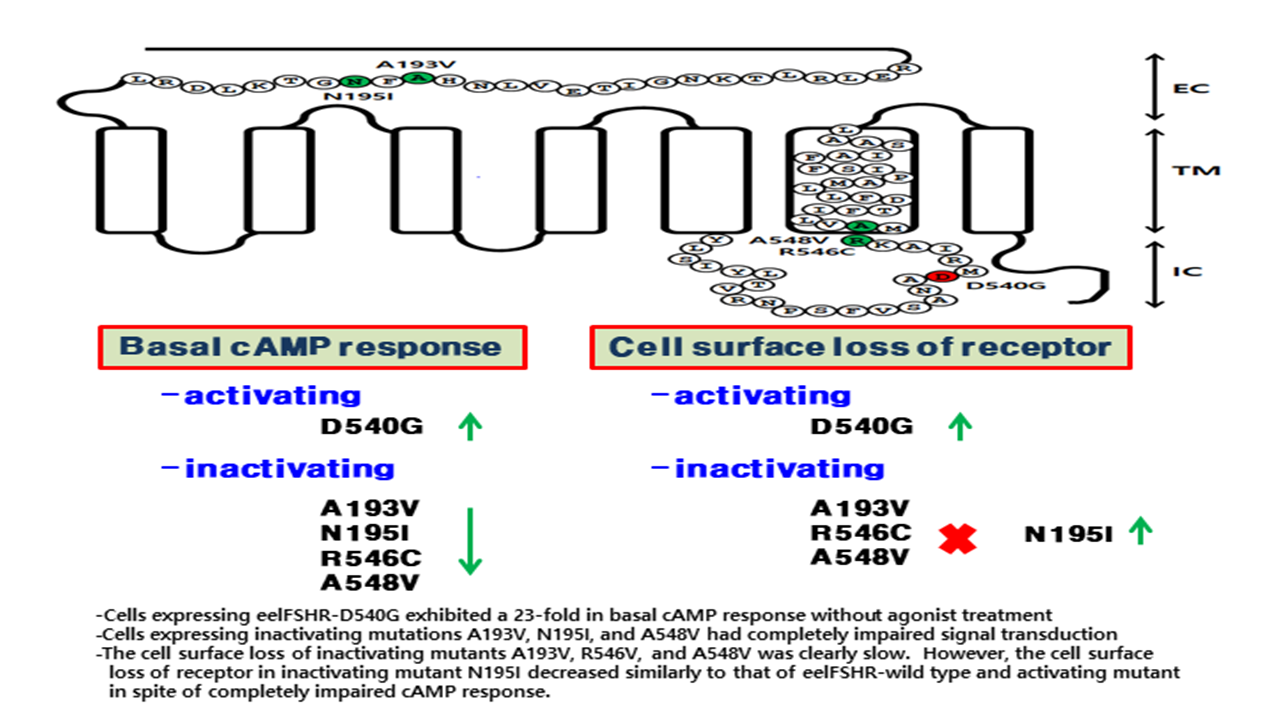
1. Introduction
2. Results
2.1. Preparation and Cell Surface Expression of Wild-Type eelFSHR and the Mutant Receptors
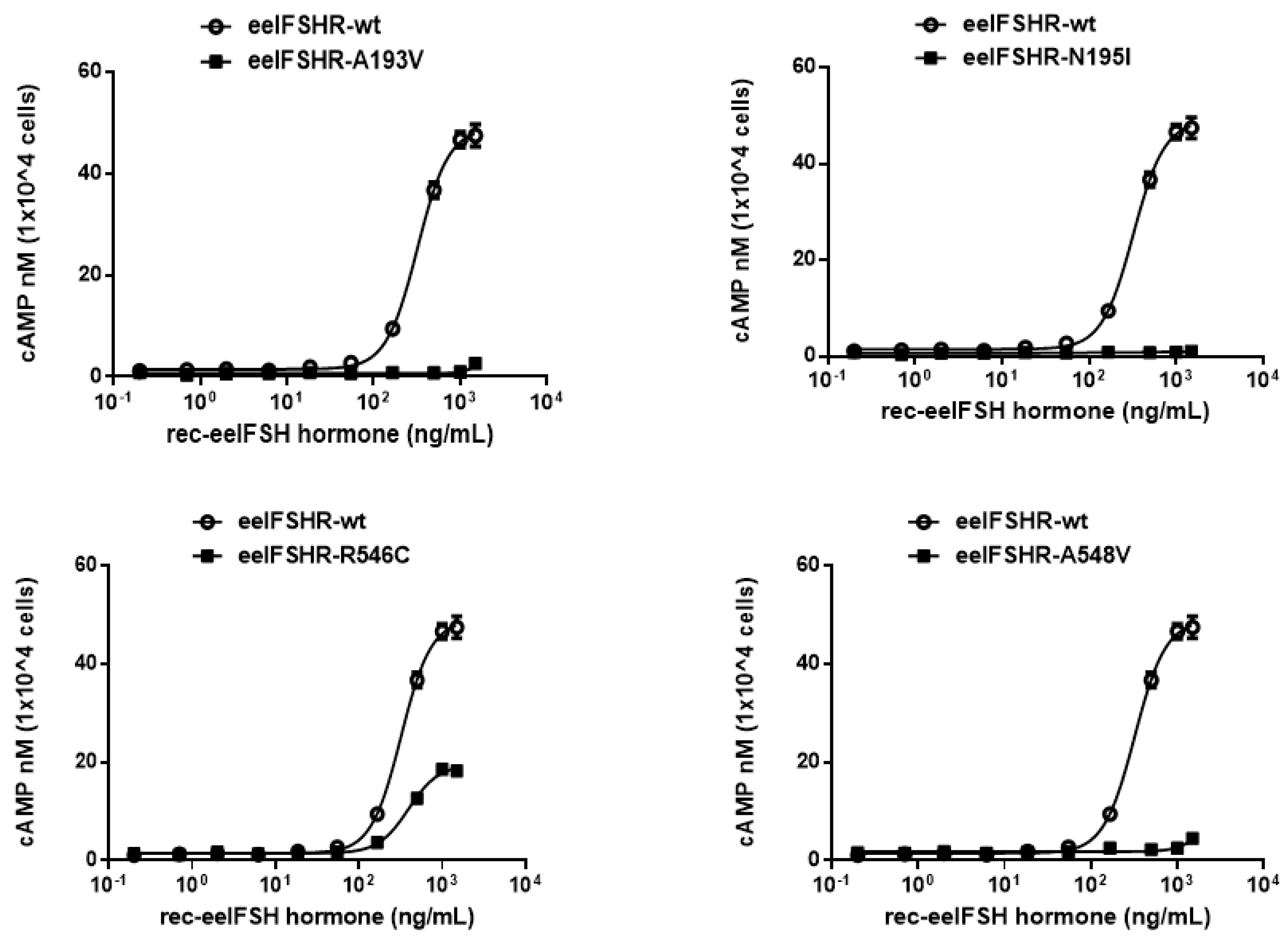
2.2. cAMP Responsiveness Induced by Agonist in Activating and Inactivating Mutants
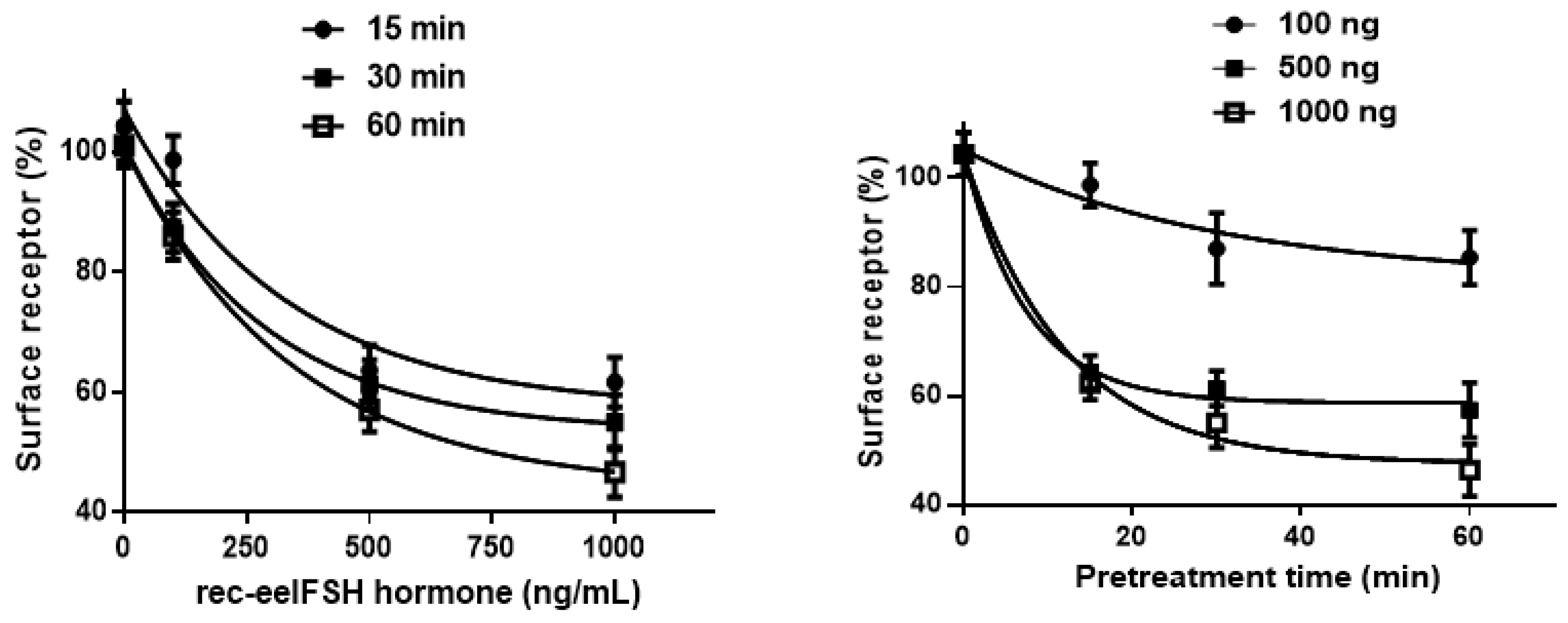
2.3. Cell Surface Receptor Loss Induced by Treatment with the eelFSH Agonist
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Site-Directed Mutagenesis and Vector Construction
4.3. Transient Transfection
4.4. Production and ELISA Analysis of Recombinant eelFSH Protein (rec-eelFSH)
4.5. cAMP Analysis by Homogeneous Time-Resolved Fluorescence (HTRF)
4.6. Agonist-Induced Cell Surface Loss
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kim, J.M.; Munkhuu, O.; Byambragchaa, M.; Lee, B.I.; Kim, S.K.; Kang, M.H.; Kim, D.J.; Min, K.S. Site-specific roles of N-linked oligosaccharides in recombinant eel follicle-stimulating hormone for secretion and signal transduction. Gen. Comp. Endocrinol. 2019, 276, 37–44. [Google Scholar] [CrossRef]
- Simoni, M.; Gromoll, J. Monitoring the transfection efficiency of the human follicle-stimulating hormone receptor cDNA in COS-7 cells: Evaluation of the growth hormone transient gene expression assay system. J. Endocrinol. Investig. 1996, 19, 359–364. [Google Scholar] [CrossRef]
- Foster, S.R.; Brauner-Osborne, H. Investigating internalization and intracellular trafficking of GPCRs: New techniques and real-time experimental approaches. Handb. Exp. Pharmacol. 2018, 245, 41–61. [Google Scholar]
- Gromoll, J.; Pekel, E.; Nieschlag, E. The structure and organization of the human follicle-stimulating hormone receptor (FSHR) gene. Genomics 1996, 35, 308–311. [Google Scholar] [CrossRef]
- Layman, L.C.; McDonough, P.G. Mutations of follicle stimulating hormone-β and its receptor in human and mouse: Genotype/phenotype. Mol. Cell Endocrinol. 2000, 161, 9–17. [Google Scholar] [CrossRef]
- Simoni, M.; Gromoll, J.; Nieschlag, E. The follicle-stimulating hormone receptor: Biochemistry, molecular biology, physiology, and pathophysiology. Endoc. Rev. 1997, 18, 739–773. [Google Scholar] [CrossRef]
- Bhaskaran, R.S.; Ascoli, M. The post-endocytotic fate of the gonadotropin receptors is an important determinant of the desensitization of gonadotropin responses. J. Mol. Endocrinol. 2005, 34, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Dhanwada, K.R.; Vijapurkar, U.; Ascoli, M. Two mutations of the lutropin/choriogonadotropin receptor that impair signal transduction also interfere with receptor-mediated endocytosis. Mol. Endocrinol. 1996, 10, 544–554. [Google Scholar] [PubMed]
- Min, K.S.; Liu, X.; Fabritz, J.; Jaquette, J.; Abell, A.N.; Ascoli, M. Mutations that induce constitutive activations and mutations that impair signal transduction modulate the basal and/or agonist-stimulated internalization of the lutropin/choriogonadotropin receptor. J. Biol. Chem. 1998, 273, 34911–34919. [Google Scholar] [CrossRef]
- Wang, Z.; Hipkin, R.W.; Ascoli, M. Progressive cytoplasmic tail truncations of the lutropin-choriogonadotropin receptor prevent agonist- or phorbol ester-induced phosphorylation, impair agonist- or phorbol ester-induced desensitization, and enhance agonist-induced receptor down-regulation. Mol. Endocrinol. 1996, 10, 748–759. [Google Scholar]
- Wang, Z.; Liu, X.; Ascoli, M. Phosphorylation of the lutropin/choriogonadotropin receptor facilities uncoupling of the receptor from adenylyl cyclase and endocytosis of the bound hormone. Mol. Endocrinol. 1997, 11, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Tapanainen, J.S.; Vaskivuo, T.; Aittomaki, K.; Huhtaniemi, I.T. Inactivating FSH receptor mutations and gonadal dysfunction. Mol. Cell. Endocrinol. 1998, 145, 129–135. [Google Scholar] [CrossRef]
- Dierich, A.; Sairam, M.R.; Monaco, L.; Fimia, G.M.; Gansmuller, A.; LeMeur, M.; Sassone-Corsi, P. Impairing follicle-stimulating hormone (FSH) signaling in vivo: Targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc. Natl. Acad. Sci. USA 1998, 95, 13612–13617. [Google Scholar] [CrossRef]
- Huhtaniemi, I.T.; Aittomaki, K. Mutations of follicle-stimulating hormone and its receptor: Effects on gonadal function. Eur. J. Endocrinol. 1998, 138, 473–481. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gromoll, J.; Simoni, M.; Nieschlag, E. An activating mutation of the follicle stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J. Clin. Endocrinol. Metab. 1996, 84, 1367–1370. [Google Scholar]
- Gromoll, J.; Simoni, M.; Nordhoff, V.; Behre, H.M.; De Geyter, C.; Nieschlag, E. Functional and clinical consequences of mutations in the FSH receptor. Mol. Cell. Endocrinol. 1996, 125, 177–182. [Google Scholar] [CrossRef]
- Laue, L.; Chan, W.Y.; Hsueh, A.J.W.; Kudo, M.; Hsu, S.Y.; Wu, S.M.; Blomberg, L.; Cutler, G.B.J. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc. Natl. Acad. Sci. USA 1995, 92, 1906–1910. [Google Scholar] [CrossRef]
- Kudo, M.; Osuga, Y.; Kobilka, B.K.; Hsueh, A.J. Transmembrane region V and VI of the human luteinizing hormone receptor are required for constitutive activation by a mutation in the third intracellular loop. J. Biol. Chem. 1996, 271, 22470–22478. [Google Scholar] [CrossRef]
- De Leener, A.; Montanelli, L.; Van Durme, J.; Chae, H.; Smits, G.; Costagliola, S. Presence and absence of follicle-stimulating hormone receptor mutation provide some insights into spontaneous ovarian hyperstimulation syndrome physiopatology. J. Clin. Endocrinol. Metab. 2006, 91, 555–562. [Google Scholar] [CrossRef]
- Aittomaki, K.; Dieguez-Lucena, J.L.; Pakarinen, P.; Sistonen, P.; Tapanainen, J.; Gromoll, J.; Kaskikari, R.; Sankila, E.M.; Lehvaslaiho, H.; Engel, A.R.; et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 1995, 82, 956–968. [Google Scholar] [CrossRef]
- Banerjee, A.; Achrekar, S.K.; Joseph, S.; Pathak, B.R.; Mahale, S.D. Functional characterization of two naturally occurring mutation V221G and T449N in the follicle stimulating hormone receptor. Mol. Cell. Endocrinol. 2017, 440, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Bramble, M.S.; Goldstein, E.H.; Lipson, A.; Ngun, T.; Eskin, A.; Gosschalk, J.E.; Roach, L.; Vashist, N.; Barseghyan, H.; Lee, E.; et al. A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: A fertility application of whole exome sequencing. Hum. Reprod. 2016, 31, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Ghezelayagh, Z.; Totonchi, M.; Zarei-Moradi, S.; Asadpour, O.; Maroufizadeh, S.; Eftekhari-Yazdi, P.; Gourabi, H.; Mohseni-Meybodi, A. The impact of genetic variation and gene expression level of the follicle-stimulating hormone receptor on ovarian reserve. Cell J. 2018, 19, 620–626. [Google Scholar] [PubMed]
- Katari, S.; Wood-Trageser, M.A.; Jiang, H.; Kalynchuk, E.; Muzumdar, R.; Yatsenko, S.A.; Rajkovic, A. Novel inactivating mutation of the FSH receptor in two siblings of Indian origin with premature ovarian failure. J. Clin. Endocrinol. Metab. 2015, 100, 2154–2157. [Google Scholar] [CrossRef]
- Liu, H.; Xu, X.; Han, T.; Yan, L.; Cheng, L.; Qin, Y.; Liu, W.; Zhao, S.; Chen, Z.J. A novel homozygous mutation in the FSHR gene is causative for primary ovarian insufficiency. Fertil. Steril. 2017, 108, 1050–1055. [Google Scholar]
- Bas, F.; Pescovitz, O.H.; Steinmetz, R. No activating mutations of FSH receptor in four children with ovarian juvenile granulosa cell tumors and the association of these tumors with central precocious puberty. J. Pediatric Adolesc. Gynecol. 2009, 22, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.S.; Achrekar, S.K.; Sahasrabuddhe, K.A.; Meharji, P.K.; Desai, S.K.; Mangoli, V.S.; Mahale, S.D. Functional characterization of two naturally occurring mutations (Val514Ala and Ala575Val) in follicle-stimulating hormone receptor. J. Clin. Endocrinol. Metab. 2015, 100, E635–E645. [Google Scholar]
- Hugon-Rodin, J.; Sonigo, C.; Gompel, A.; Dode, C.; Grynberg, M.; Binart, N.; Beau, I. First mutation in the FSHR cytoplasmic tail identified in a non-pregnant woman with spontaneous ovarian hyperstimulation syndrome. BMC Med. Genet. 2017, 18, 44. [Google Scholar]
- Ji, I.; Ji, T.H. Differential roles of exoloop 1 of the human follicle-stimulating hormone receptor in hormone binding and receptor activation. J. Biol. Chem. 1995, 270, 15970–15973. [Google Scholar]
- Touraine, P.; Beau, I.; Gougeon, A.; Meduri, G.; Desroches, A.; Pichard, C.; Detoeuf, M.; Paniel, B.; Prieur, M.; Zom, J.R.; et al. New natural inactivating mutations of the follicle-stimulating hormone receptor: Correlations between receptor function and phenotype. Mol. Endocrinol. 1999, 13, 1844–1854. [Google Scholar] [CrossRef]
- Banerjee, A.A.; Mahale, S.D. Role of the extracellular and intracellular loops of follicle-stimulating hormone receptor in its function. Front. Endocrinol. 2015, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Beau, I.; Touraine, P.; Meduri, G.; Gougeon, A.; Desroches, A.; Matuchansky, C.; Milgrom, E.; Kuttenn, F.; Misrahi, M. A novel phenotype related to partial loss of function mutations of the follicle stimulating hormone receptor. J. Clin. Investig. 1998, 102, 1352–1359. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.; Ammendrup-Johnsen, I.; Jansen, A.M.; Gether, U.; Madsen, K.L.; Brauner-Osborne, H. The GPRC6A receptor displays constitutive internalization and sorting to the slow recycling pathway. J. Biol. Chem. 2017, 292, 6910–6926. [Google Scholar] [CrossRef] [PubMed]
- Mos, I.; Jacobsen, S.E.; Foster, S.R.; Brauner-Osborne, H. Calcium-sensing receptor internalization is β-arrestin-dependent and modulated by allosteric ligands. Mol. Pharmacol. 2019, 96, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Norskov-Lauritsen, L.; Jorgensen, S.; Brauner-Osborne, H. N-glycosylation and disulfide bonding affects GPRC6A receptor expression, function, and dimerization. FEBS Lett. 2015, 589, 588–597. [Google Scholar] [CrossRef]
- Spiess, K.; Bagger, S.O.; Torz, L.J.; Jensen, K.H.R.; Walser, A.L.; Kvam, J.M.; Mogelmose, A.S.K.; Daugvilaite, V.; Junnila, R.K.; Hjorto, G.M.; et al. Arrestin-independent constitutive endocytosis of GPR125/ADGRA3. Ann. N. Y. Acad. Sci. 2019, 1456, 186–199. [Google Scholar] [CrossRef]
- Krishnamurthy, H.; Kishi, H.; Shi, M.; Galct, C.; Bhaskaran, R.S.; Hirakawa, T.; Ascoli, M. Post-endocytotic trafficking of the FSH/FSH receptor complex. Mol. Endocrinol. 2003, 17, 2162–2176. [Google Scholar] [CrossRef]
- Kara, E.; Crepieux, P.; Gauthier, C.; Martinat, N.; Piketty, V.; Guillou, F.; Reiter, E. A phosphorylation cluster of five serine and threonine residues in the C-terminus of the follicle-stimulating hormone receptor is important for desensitization but not for beta-arrestin-mediated ERK activation. Mol. Endocrinol. 2006, 20, 3014–3026. [Google Scholar] [CrossRef]
- Thomas, R.M.; Mechamen, C.A.; Mazurkiewicz, J.E.; Muda, M.; Palmer, S.; Dias, J.A. Follicle-stimulating hormone receptor forms oligomers and shows evidence of carboxyl-terminal proteolytic processing. Endocrinology 2007, 148, 987–995. [Google Scholar] [CrossRef][Green Version]
- Nakamura, K.; Hipkin, R.W.; Ascoli, M. The agonist-induced phosphorylation of the rat follitropin receptor maps to the first and third intracellular loops. Mol. Endocrinol. 1998, 12, 580–590. [Google Scholar] [CrossRef]
- Kim, D.J.; Park, C.W.; Byambaragchaa, M.; Kim, S.K.; Lee, B.I.; Hwang, H.K.; Myeong, J.I.; Hong, S.M.; Kang, M.H.; Min, K.S. Data on the characterization of follicle-stimulating hormone monoclonal antibodies and localization in Japanese eel pituitary. Data Brief 2016, 8, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Park, C.W.; Kim, D.W.; Park, H.K.; Byambaragchaa, M.; Lee, N.S.; Hong, S.M.; Seo, M.Y.; Kang, M.H.; Min, K.S. Production and characterization of monoclonal antibodies against recombinant tethered follicle-stimulating homrone from Japanese eel anguilla japonica. Gen. Comp. Endocrinol. 2016, 233, 8–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Byambaragchaa, M.; Kim, D.J.; Kang, M.H.; Min, K.S. Site specificity of eel luteinizing hormone N-linked oligosaccharides in signal transduction. Gen. Comp. Endocrinol. 2018, 268, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Smits, G.; Olatunbosun, O.; Delbaere, A.; Pierson, R.; Vassart, G.; Costagliola, S. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. N. Engl. J. Med. 2003, 349, 760–766. [Google Scholar] [CrossRef]
- Moore, C.A.; Milano, S.K.; Benovic, J.L. Regulation of receptor trafficking by GRKs and arrestins. Annu. Rev. Physiol. 2007, 69, 451–482. [Google Scholar] [CrossRef]
- von Zastrow, M.; Kobilka, B.K. Ligand-regulated internalization and recycling of human beta 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J. Biol. Chem. 1992, 267, 3530–3538. [Google Scholar] [PubMed]
- Jean-Alphonse, F.; Bowersox, S.; Chen, S.; Beard, G.; Puthenveedu, M.A.; Hanyaloglu, A.C. Spatially restricted G protein-coupled receptor activity via divergent endocytic compartments. J. Biol. Chem. 2014, 289, 3960–3977. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Zwier, J.M.; Jaracz-Ros, A.; Klipfel, L.; Cottet, M.; Maurel, D.; Bdioui, S.; Balabanian, K.; Prezeau, L.; Trinquet, E.; et al. A broad G protein-coupled receptor internalization assay that combines SNAP-tag labeling, diffusion-enhanced resonance energy transfer, and a highly emissive terbium cryptate. Front. Endocrinol. 2015, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Mundell, S.J.; Matharu, A.L.; Nisar, S.; Palmer, T.M.; Benovic, J.L.; Kelly, E. Deletion of the distal COOH-terminus of the A2B adenosine receptor switches internalization to an arrestin- and clathrin-independent pathway and inhibits recycling. Br. J. Pharmacol. 2010, 159, 518–533. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| eelFSH Receptors | cAMP Responses | ||
|---|---|---|---|
| Basal a (nM/104 Cells) | EC50 (ng/mL) | Rmax b (nM/104 Cells) | |
| eelFSHR-WT | 0.6 ± 0.1 (1-fold) | 523.8 (476.5 to 575.7)c | 37.5 ± 2.6 (100%) |
| eelFSHR-D540G | 13.9 ± 0.9 (23.2-fold) | 358.8 (207.3 to 620) | 24.8 ± 2.3 (66%) |
| eelFSHR-D540N | 8.7 ± 0.7 (14.5-fold) | 544.3 (399.2 to 742.3) | 33.9 ± 3.7 (90%) |
| eelFSHR-D540F | - | -d | - |
| eelFSH Receptors | cAMP Responses | ||
|---|---|---|---|
| Basal a (nM/104 Cells) | EC50 (ng/mL) | Rmax b (nM/104 Cells) | |
| eelFSHR-WT | 1.1 ± 0.2 | 314.1 (289.5 to 340.7)c | 49.7 ± 5.6 (100%) |
| eelFSHR-A193V | 0.6 ± 0.1 | - d | - |
| eelFSHR-N195I | 0.6 ± 0.1 | - | - |
| eelFSHR-R546C | 1.5 ± 0.3 | 385.4 (309.4 to 480.0) | 18.9 ± 1.6 (38%) |
| eelLFSR-A548V | 1.2 ± 0.1 | - | - |
| eelFSHR Cell Lines | t1/2 (min) | Plateau (% of Control) |
|---|---|---|
| eelFSHR-WT | 3.0 ± 0.8 | 49.9 ± 3.1 |
| eelFSHR-D540G | 1.3 ± 0.2 | 47.7 ± 3.8 |
| eelFSHR-A193V | - a | - |
| eelFSHR-N195I | 0.9 ± 0.6 | 31.7 ± 1.9 |
| eelFSHR-R546C | - | 82.8 ± 5.9 |
| eelFSHR-A548V | 5.7 ± 1.3 | 65.7 ± 4.7 |
| Primer Name | Primer Sequence | |
|---|---|---|
| 1 | eelFSHR-wt Forward | 5′-ATGAATTCATGTCCAATCTGCTCTTGTGGACGATG-3′ * EcoRI Site |
| 2 | eelFSHR-wt reverse | 5′-CCTCGAGTTATTTAGGACCTCTGTTGAGAAT-3′ * XhoI Site |
| 3 | A193V forward | 5′-GCTGAATCACGTCTTCAATGGCACC-3′ |
| 4 | A193V reverse | 5′-GGTGCCATTGAAGACGTGATTCAGC-3′ |
| 5 | N195I forward | 5′-GAATCACGCTTTCATTGGCACCAAAC-3′ |
| 6 | N195I reverse | 5′-GTTTTGGTGCCAATGAAAGCGTGATTC-3′ |
| 7 | D540G forward | 5′-GCCAATGCCGGTATGCGCATC-3′ |
| 8 | D540G reverse | 5′-GATGCGCATACCGGCATTGGC-3′ |
| 9 | D540N forward | 5′-GCCAATGCCAATATGCGCATC-3′ |
| 10 | D540N reverse | 5′-GATGCGCATATTGGCATTGGC-3′ |
| 11 | D540F forward | 5′-GCCAATGCCTTTATGCGCATC-3′ |
| 12 | D540F reverse | 5′-GATGCGCATAAAGGCATTGGC-3′ |
| 13 | R546C forward | 5′-CGCATCGCCAAGTGCATGGCCGTG-3′ |
| 14 | R546C reverse | 5′-CACGGCCATGCACTTGGCGATGCG-3′ |
| 15 | A548V forward | 5′-GCCAAGCGCATGGTCGTGCTCATC-3′ |
| 16 | A548V reverse | 5′-GATGAGCACGACCATGCGCTTGGC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byambaragchaa, M.; Kim, J.-S.; Park, H.-K.; Kim, D.-J.; Hong, S.-M.; Kang, M.-H.; Min, K.-S. Constitutive Activation and Inactivation of Mutations Inducing Cell Surface Loss of Receptor and Impairing of Signal Transduction of Agonist-Stimulated Eel Follicle-Stimulating Hormone Receptor. Int. J. Mol. Sci. 2020, 21, 7075. https://doi.org/10.3390/ijms21197075
Byambaragchaa M, Kim J-S, Park H-K, Kim D-J, Hong S-M, Kang M-H, Min K-S. Constitutive Activation and Inactivation of Mutations Inducing Cell Surface Loss of Receptor and Impairing of Signal Transduction of Agonist-Stimulated Eel Follicle-Stimulating Hormone Receptor. International Journal of Molecular Sciences. 2020; 21(19):7075. https://doi.org/10.3390/ijms21197075
Chicago/Turabian StyleByambaragchaa, Munkhzaya, Jeong-Soo Kim, Hong-Kyu Park, Dae-Jung Kim, Sun-Mee Hong, Myung-Hwa Kang, and Kwan-Sik Min. 2020. "Constitutive Activation and Inactivation of Mutations Inducing Cell Surface Loss of Receptor and Impairing of Signal Transduction of Agonist-Stimulated Eel Follicle-Stimulating Hormone Receptor" International Journal of Molecular Sciences 21, no. 19: 7075. https://doi.org/10.3390/ijms21197075
APA StyleByambaragchaa, M., Kim, J.-S., Park, H.-K., Kim, D.-J., Hong, S.-M., Kang, M.-H., & Min, K.-S. (2020). Constitutive Activation and Inactivation of Mutations Inducing Cell Surface Loss of Receptor and Impairing of Signal Transduction of Agonist-Stimulated Eel Follicle-Stimulating Hormone Receptor. International Journal of Molecular Sciences, 21(19), 7075. https://doi.org/10.3390/ijms21197075