A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43


Abstract
1. Introduction
2. Adverse Drug Reactions of CLZ
2.1. Seizure
2.2. Cardiotoxicity
2.2.1. Myocarditis
2.2.2. Cardiomyopathy
2.3. Pneumonia
2.4. Discontinuation Relapse Psychosis
3. Estimated Pathomechanisms of CLZ
3.1. Candidate Mechanisms of Clinical Efficicacy of CLZ on Treatment-Resistant Schizophrenia
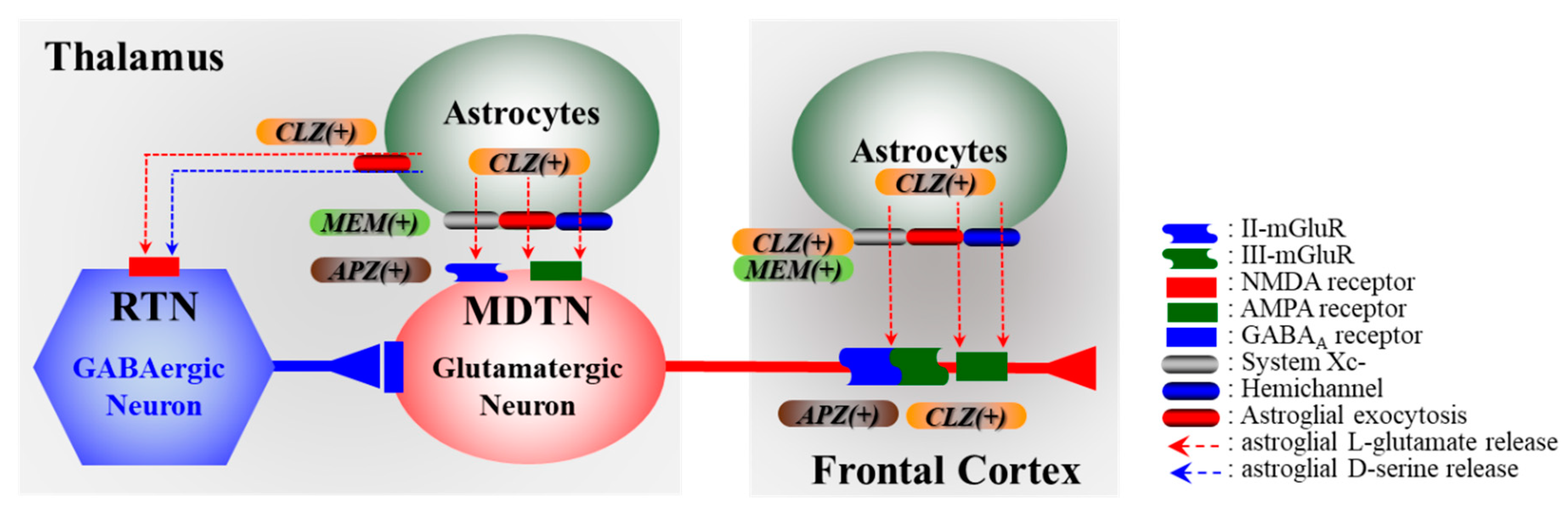
3.2. Impact of Tripartite Synaptic Transmission in Pathomechanisms of Neuropsychiatric Defeciency
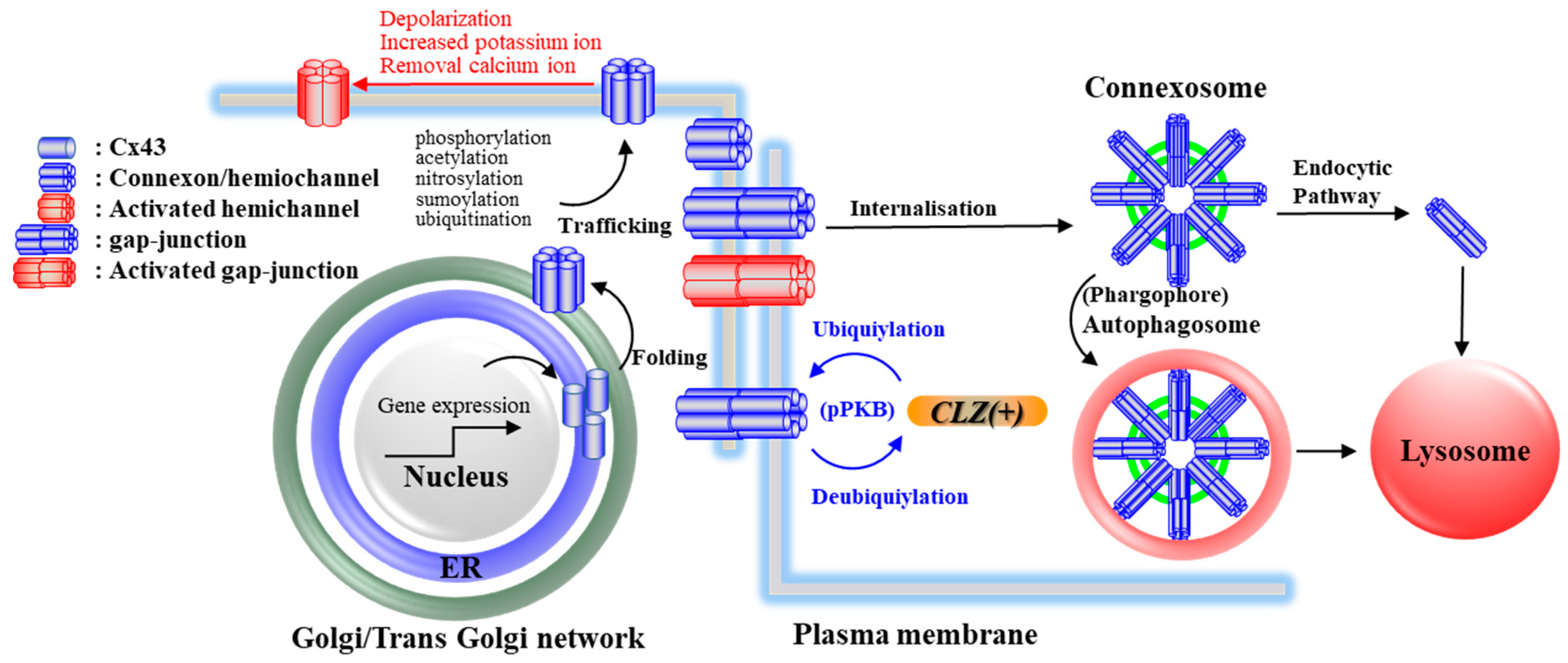
4. Effects of CLZ on Cx43 and Its Associated Signal Transduction System
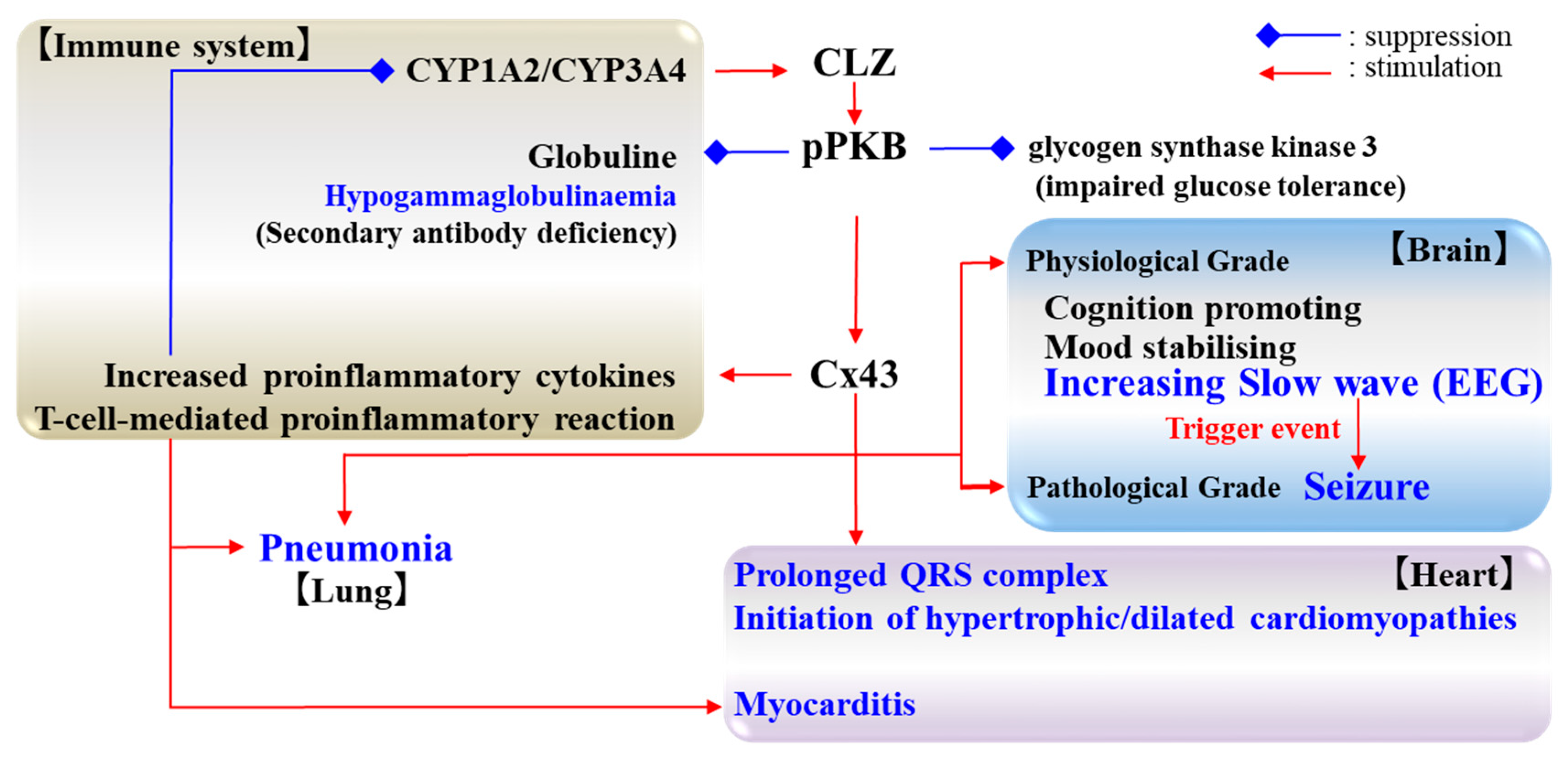
5. Candidate Pathomechanisms of CLZ Associated with Cx43
5.1. Cognition and Cx43
5.2. Seizure and Cx43
5.3. Cardiotoxicity and Cx43
5.4. Pneumonia and Cx43
6. Remaining Challenges and Conclusions
- (1)
- Cognitive promoting and mood stabilization.
- (2)
- Hyperactivation of excitatory tripartite synaptic transmission.
- (3)
- Dysfunction of the immune-responses in the heart and lungs.
- (4)
- Dysfunction of the propagation of the electrical impulses in the heart.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-HT | serotonin |
| 5-HT1AT | serotonin receptor type 1A |
| 5-HT2AR | serotonin receptor type 2A |
| cAMP | cyclic adenosine monophosphate |
| CLZ | clozapine |
| Cx | connexin |
| Cx43 | connexin43 |
| CYP | cytochromes P450 |
| D2R | dopamine D2 receptor |
| ECG | electrocardiogram |
| EEG | electroencephalograph |
| ER | endoplasmic reticulum |
| Erk | extracellular Signal-regulated Kinase |
| FDA | the Food and Drug Administration |
| FIN11 | 11-year follow-up of mortality in patients with schizophrenia: a population-based cohort |
| GABA | γ-aminobutyrate |
| HDAC | histone deacetylase |
| IFN | interferon |
| IL | interleukin |
| MAPK | mitogen-activated protein kinase |
| mPFC | medial prefrontal cortex |
| NAc | nucleus accumbens |
| NMDAR | N-methyl-D-aspartate/glutamate receptor |
| PI3K | phosphoinositide 3-kinase |
| PKB | protein kinase B |
| pPKB | phosphorylated protein kinase B |
| Sp1 | activator protein 1 complex |
| SSRI | selective serotonin reuptake inhibitor |
| TNF | tumour necrosis factor |
| VigiBase | the World Health Organization global database |
| VPA | valproate |
| VTA | ventral tegmental area |
| WHO | the World Health Organization |
| Wnt | wingless |
References
- Lally, J.; MacCabe, J.H. Antipsychotic medication in schizophrenia: A review. Br. Med. Bull. 2015, 114, 169–179. [Google Scholar] [CrossRef]
- Tandon, R. Schizophrenia and Other Psychotic Disorders in Diagnostic and Statistical Manual of Mental Disorders (DSM)-5: Clinical Implications of Revisions from DSM-IV. Indian J. Psychol. Med. 2014, 36, 223–225. [Google Scholar] [CrossRef]
- Tan, N.; Van Os, J. Schizofreniespectrum en andere psychotische stoornissen in de DSM-5. Tijdschr. Psychiatr. 2014, 56, 167–172. [Google Scholar]
- Kane, J.M.; Leucht, S.; Carpenter, D.; Docherty, J.P. The expert consensus guideline series. Optimizing pharmacologic treatment of psychotic disorders. Introduction: Methods, commentary, and summary. J. Clin. Psychiatry 2003, 64, 5–19. [Google Scholar]
- Kreyenbuhl, J.; Buchanan, R.; Dickerson, F.; Dixon, L. The Schizophrenia Patient Outcomes Research Team (PORT): Updated treatment recommendations 2009. Schizophr. Bull. 2010, 36, 94–103. [Google Scholar] [CrossRef]
- Falkai, P.; Wobrock, T.; Lieberman, J.; Glenthoj, B.; Gattaz, W.F.; Moller, H.J.; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: Long-term treatment of schizophrenia. World J. Biol. Psychiatry 2006, 7, 5–40. [Google Scholar] [CrossRef]
- Tiihonen, J.; Mittendorfer-Rutz, E.; Majak, M.; Mehtala, J.; Hoti, F.; Jedenius, E.; Enkusson, D.; Leval, A.; Sermon, J.; Tanskanen, A.; et al. Real-World Effectiveness of Antipsychotic Treatments in a Nationwide Cohort of 29823 Patients with Schizophrenia. JAMA Psychiatry 2017, 74, 686–693. [Google Scholar] [CrossRef]
- Hu, R.J.; Malhotra, A.K.; Pickar, D. Predicting response to clozapine. CNS Drugs 1999, 11, 317–326. [Google Scholar] [CrossRef]
- Kane, J.; Honigfeld, G.; Singer, J.; Meltzer, H. Clozapine for the treatment-resistant schizophrenic: A double-blind comparison with chlorpromazine. Arch. Gen. Psychiatry 1988, 45, 789–796. [Google Scholar] [CrossRef]
- Meltzer, H.Y.; Bobo, W.V.; Roy, A.; Jayathilake, K.; Chen, Y.; Ertugrul, A.; Yağcıoğlu, A.; Small, J.G. A randomized, double-blind comparison of clozapine and high-dose olanzapine in treatment-resistant patients with schizophrenia. J. Clin. Psychiatry 2008, 69, 274–285. [Google Scholar] [CrossRef]
- Masuda, T.; Misawa, F.; Takase, M.; Kane, J.M.; Correll, C.U. Association with Hospitalization and All-Cause Discontinuation among Patients with Schizophrenia on Clozapine vs Other Oral Second-Generation Antipsychotics: A Systematic Review and Meta-analysis of Cohort Studies. JAMA Psychiatry 2019, 76, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Farooq, S.; Taylor, M. Clozapine: Dangerous orphan or neglected friend? Br. J. Psychiatry 2011, 198, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; Young, C.; Paton, C. Prior antipsychotic prescribing in patients currently receiving clozapine: A case note review. J. Clin. Psychiatry 2003, 64, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Doyle, R.; Behan, C.; O’Keeffe, D.; Masterson, S.; Kinsella, A.; Kelly, A.; Sheridan, A.; Keating, D.; Hynes, C.; Madigan, K.; et al. Clozapine Use in a Cohort of First-Episode Psychosis. J. Clin. Psychopharmacol. 2017, 37, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Howes, O.D.; Vergunst, F.; Gee, S.; McGuire, P.; Kapur, S.; Taylor, D. Adherence to treatment guidelines in clinical practice: Study of antipsychotic treatment prior to clozapine initiation. Br. J. Psychiatry 2012, 201, 481–485. [Google Scholar] [CrossRef]
- Gee, S.; Vergunst, F.; Howes, O.; Taylor, D. Practitioner attitudes to clozapine initiation. Acta Psychiatr. Scand. 2014, 130, 16–24. [Google Scholar] [CrossRef]
- Verdoux, H.; Quiles, C.; Bachmann, C.J.; Siskind, D. Prescriber and institutional barriers and facilitators of clozapine use: A systematic review. Schizophr. Res. 2018, 201, 10–19. [Google Scholar] [CrossRef]
- Okhuijsen-Pfeifer, C.; Cohen, D.; Bogers, J.; de Vos, C.M.H.; Huijsman, E.A.H.; Kahn, R.S.; Luykx, J.J. Differences between physicians’ and nurse practitioners’ viewpoints on reasons for clozapine underprescription. Brain Behav. 2019, 9, e01318. [Google Scholar] [CrossRef]
- Aringhieri, S.; Kolachalam, S.; Gerace, C.; Carli, M.; Verdesca, V.; Brunacci, M.G.; Rossi, C.; Ippolito, C.; Solini, A.; Corsini, G.U.; et al. Clozapine as the most efficacious antipsychotic for activating ERK 1/2 kinases: Role of 5-HT2A receptor agonism. Eur. Neuropsychopharmacol. 2017, 27, 383–398. [Google Scholar] [CrossRef]
- Huhn, M.; Nikolakopoulou, A.; Schneider-Thoma, J.; Krause, M.; Samara, M.; Peter, N.; Arndt, T.; Backers, L.; Rothe, P.; Cipriani, A.; et al. Comparative efficacy and tolerability of 32 oral antipsychotics for the acute treatment of adults with multi-episode schizophrenia: A systematic review and network meta-analysis. Lancet 2019, 394, 939–951. [Google Scholar] [CrossRef]
- Tiihonen, J.; Lönnqvist, J.; Wahlbeck, K.; Klaukka, T.; Niskanen, L.; Tanskanen, A.; Haukka, J. 11-year follow-up of mortality in patients with schizophrenia: A population-based cohort study (FIN11 study). Lancet 2009, 374, 620–627. [Google Scholar] [CrossRef]
- Haidary, H.A.; Padhy, R.K. Clozapine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020; pp. 1–10. Available online: http://www.ncbi.nlm.nih.gov/books/NBK535399/ (accessed on 24 March 2020).
- De Leon, J.; Sanz, E.J.; De Las Cuevas, C. Data from the World Health Organization’s Pharmacovigilance Database Supports the Prominent Role of Pneumonia in Mortality Associated with Clozapine Adverse Drug Reactions. Schizophr. Bull. 2020, 46, 1–3. [Google Scholar] [CrossRef]
- Nielsen, J.; Young, C.; Ifteni, P.; Kishimoto, T.; Xiang, Y.T.; Schulte, P.F.; Correll, C.U.; Taylor, D. Worldwide Differences in Regulations of Clozapine Use. CNS Drugs 2016, 30, 149–161. [Google Scholar] [CrossRef]
- Naheed, M.; Green, B. Focus on clozapine. Curr Med. Res. Opin. 2001, 17, 223–229. [Google Scholar] [CrossRef]
- Aronson, J.K.; Ferner, R.E. Clarification of terminology in drug safety. Drug Saf. 2005, 28, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.M.; Park, S.H. Seizure associated with clozapine: Incidence, etiology, and management. CNS Drugs 2015, 29, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, Y.S.; Sato, W.; Ataka, K.; Yagisawa, K.; Omori, Y.; Kanbayashi, T.; Shimizu, T. Clozapine-induced seizures, electroencephalography abnormalities, and clinical responses in Japanese patients with schizophrenia. Neuropsychiatry Dis. Treat. 2014, 10, 1973–1978. [Google Scholar] [CrossRef]
- Grover, S.; Hazari, N.; Chakrabarti, S.; Avasthi, A. Association of Clozapine with Seizures: A Brief Report Involving 222 Patients Prescribed Clozapine. East Asian Arch. Psychiatry 2015, 25, 73–78. [Google Scholar]
- Varma, S.; Bishara, D.; Besag, F.M.; Taylor, D. Clozapine-related EEG changes and seizures: Dose and plasma-level relationships. Ther. Adv. Psychopharmacol. 2011, 1, 47–66. [Google Scholar] [CrossRef]
- Neufeld, M.Y.; Rabey, J.M.; Orlov, E.; Korczyn, A.D. Electroencephalographic findings with low-dose clozapine treatment in psychotic Parkinsonian patients. Clin. Neuropharmacol. 1996, 19, 81–86. [Google Scholar] [CrossRef]
- Freudenreich, O.; Weiner, R.D.; McEvoy, J.P. Clozapine-induced electroencephalogram changes as a function of clozapine serum levels. Biol. Psychiatry 1997, 42, 132–137. [Google Scholar] [PubMed]
- Risby, E.D.; Epstein, C.M.; Jewart, R.D.; Nguyen, B.V.; Morgan, W.N.; Risch, S.C.; Thrivikraman, K.V.; Lewine, R.L. Clozapine-induced EEG abnormalities and clinical response to clozapine. J. Neuropsychiatry Clin. Neurosci. 1995, 7, 466–470. [Google Scholar] [PubMed]
- Haring, C.; Neudorfer, C.; Schwitzer, J.; Hummer, M.; Saria, A.; Hinterhuber, H.; Fleischhacker, W.W. EEG alterations in patients treated with clozapine in relation to plasma levels. Psychopharmacology 1994, 114, 97–100. [Google Scholar] [PubMed]
- Chung, S.J.; Jeong, S.H.; Ahn, Y.M.; Kang, U.G.; Koo, Y.J.; Ha, J.H.; Lee, S.G.; Kim, Y.S. A retrospective study of clozapine and electroencephalographic abnormalities in schizophrenic patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2002, 26, 139–144. [Google Scholar]
- Schuld, A.; Kuhn, M.; Haack, M.; Kraus, T.; Hinze-Selch, D.; Lechner, C.; Pollmacher, T. A comparison of the effects of clozapine and olanzapine on the EEG in patients with schizophrenia. Pharmacopsychiatry 2000, 33, 109–111. [Google Scholar]
- Treves, I.A.; Neufeld, M.Y. EEG abnormalities in clozapine-treated schizophrenic patients. Eur. Neuropsychopharmacol. 1996, 6, 93–94. [Google Scholar] [CrossRef]
- Wu, C.S.; Wang, S.C.; Yeh, I.J.; Liu, S.K. Comparative risk of seizure with use of first- and second-generation antipsychotics in patients with schizophrenia and mood disorders. J. Clin. Psychiatry 2016, 77, e573–e579. [Google Scholar] [CrossRef]
- Ronaldson, K.J. Cardiovascular Disease in Clozapine-Treated Patients: Evidence, Mechanisms and Management. CNS Drugs 2017, 31, 777–795. [Google Scholar]
- De Berardis, D.; Serroni, N.; Campanella, D.; Olivieri, L.; Ferri, F.; Carano, A.; Cavuto, M.; Martinotti, G.; Cicconetti, A.; Piersanti, M.; et al. Update on the adverse effects of clozapine: Focus on myocarditis. Curr. Drug Saf. 2012, 7, 55–62. [Google Scholar] [CrossRef]
- Rohde, C.; Polcwiartek, C.; Kragholm, K.; Ebdrup, B.H.; Siskind, D.; Nielsen, J. Adverse cardiac events in out-patients initiating clozapine treatment: A nationwide register-based study. Acta Psychiatr. Scand. 2018, 137, 47–53. [Google Scholar]
- Ronaldson, K.J.; Fitzgerald, P.B.; McNeil, J.J. Clozapine-induced myocarditis, a widely overlooked adverse reaction. Acta Psychiatr. Scand. 2015, 132, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Siskind, D.; Sidhu, A.; Cross, J.; Chua, Y.T.; Myles, N.; Cohen, D.; Kisely, S. Systematic review and meta-analysis of rates of clozapine-associated myocarditis and cardiomyopathy. Aust. N. Z. J. Psychiatry 2020, 54, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Moore, A.M.; Piper, S.; Sweeney, M.; Whiskey, E.; Cole, G.; Shergill, S.S.; Plymen, C.M. Clozapine and cardiotoxicity—A guide for psychiatrists written by cardiologists. Psychiatry Res. 2019, 282, 112491. [Google Scholar] [CrossRef] [PubMed]
- Layland, J.J.; Liew, D.; Prior, D.L. Clozapine-induced cardiotoxicity: A clinical update. Med. J. Aust. 2009, 190, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Razminia, M.; Salem, Y.; Devaki, S.; Shah, N.; Khosla, S. Clozapine induced myopericarditis: Early recognition improves clinical outcome. Am. J. Ther. 2006, 13, 274–276. [Google Scholar] [CrossRef]
- Ronaldson, K.; Fitzgerald, P.; Taylor, A.; Topliss, D.; McNeil, J. A new monitoring protocol for clozapine-induced myocarditis based on an analysis of 75 cases and 94 controls. Aust. N. Z. J. Psychiatry 2011, 45, 458. [Google Scholar] [CrossRef]
- Ronaldson, K.; Taylor, A.; Fitzgerald, P.; Topliss, D.; Elsik, M.; McNeil, J. Diagnostic characteristics of clozapine-induced myocarditis identified by an analysis of 38 cases and 47 controls. J. Clin. Psychiatry 2010, 71, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.L.; Narayanan, P.; Gill, N. Incidence and risk factors for clozapine-induced myocarditis and cardiomyopathy at a regional mental health service in Australia. Australas. Psychiatry 2016, 24, 176–180. [Google Scholar] [CrossRef]
- Ronaldson, K.J.; Fitzgerald, P.B.; Taylor, A.J.; Topliss, D.J.; Wolfe, R.; McNeil, J.J. Rapid clozapine dose titration and concomitant sodium valproate increase the risk of myocarditis with clozapine: A case–control study. Schizophr. Res. 2012, 141, 173–178. [Google Scholar] [CrossRef]
- Cooper, L.T., Jr. Myocarditis. N. Engl. J. Med. 2009, 360, 1526–1538. [Google Scholar] [CrossRef]
- Alawami, M.; Wasywich, C.; Cicovic, A.; Kenedi, C. A systematic review of clozapine induced cardiomyopathy. Inter. J. Cardiol. 2014, 176, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.A.; Dent, S.; Brezden-Masley, C.; Clarke, B.; Davis, M.K.; Jassal, D.S.; Johnson, C.; Lemieux, J.; Paterson, I.; Sebag, I.A.; et al. Canadian Cardiovascular Society Guidelines for Evaluation and Management of Cardiovascular Complications of Cancer Therapy. Can. J. Cardiol. 2016, 32, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Katoh, C.; Osanai, T.; Tomita, H.; Okumura, K. Brain natriuretic peptide is released from human astrocytoma cell line U373MG under hypoxia: A possible role in anti-apoptosis. J. Endocrinol. 2011, 208, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Waschek, J.A. Developmental actions of natriuretic peptides in the brain and skeleton. Cell Mol. Life Sci. 2004, 61, 2332–2342. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Yang, S.Y.; Liao, Y.T.; Chen, W.J.; Lee, W.C.; Shau, W.Y.; Chang, Y.T.; Tsai, S.Y.; Chen, C.C. Second-generation antipsychotic medications and risk of pneumonia in schizophrenia. Schizophr. Bull. 2013, 39, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; Siskind, D.; de Leon, J.; Nielsen, J. Antipsychotic medication exposure, clozapine, and pneumonia: Results from a self-controlled study. Acta Psychiatry Scand. 2020, 142, 78–86. [Google Scholar] [CrossRef]
- Mustafa, F.A.; Burke, J.G.; Abukmeil, S.S.; Scanlon, J.J.; Cox, M. “Schizophrenia past clozapine”: Reasons for clozapine discontinuation, mortality, and alternative antipsychotic prescribing. Pharmacopsychiatry 2015, 48, 11–14. [Google Scholar] [CrossRef]
- Ruan, C.J.; de Leon, J. Thirty Years of Both Ignorance and Clinical Experience Suggest That Clozapine Intoxication during Co-Occurring Infections and Inflammation May Have Higher Morbidity and Mortality than Is Currently Believed. Psychosomatics 2019, 60, 221–222. [Google Scholar] [CrossRef]
- de Leon, J.; Sanz, E.J.; Noren, G.N.; De Las Cuevas, C. Pneumonia may be more frequent and have more fatal outcomes with clozapine than with other second-generation antipsychotics. World Psychiatry 2020, 19, 120–121. [Google Scholar] [CrossRef]
- Leung, J.G.; Nelson, S.; Takala, C.R.; Gören, J.L. Infection and inflammation leading to clozapine toxicity and intensive care: A case series. Ann. Pharmacother. 2014, 48, 801–805. [Google Scholar] [CrossRef]
- Haack, M.-J.; Bak, M.; Beurskens, R.; Maes, M.; Stolk, L.; Delespaul, P.A. Toxic rise of clozapine plasma concentrations in relation to inflammation. Eur. Neuropsychopharmacol. 2003, 13, 381–385. [Google Scholar] [PubMed]
- de Leon, J.; Diaz, F.J. Serious respiratory infections can increase clozapine levels and contribute to side effects: A case report. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 1059–1063. [Google Scholar]
- Albitar, O.; Harun, S.N.; Zainal, H.; Ibrahim, B.; Sheikh Ghadzi, S.M. Population Pharmacokinetics of Clozapine: A Systematic Review. BioMed Res. Int. 2020, 2020, 9872936. [Google Scholar] [PubMed]
- Shah, R.R.; Smith, R.L. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: Hypothesis with implications for personalized medicine. Drug Metab. Dispos. 2015, 43, 400–410. [Google Scholar] [PubMed]
- de Leon, J.; Ruan, C.J.; Verdoux, H.; Wang, C. Clozapine is strongly associated with the risk of pneumonia and inflammation. Gen. Psychiatr. 2020, 33, e100183. [Google Scholar]
- de Leon, J.; Ruan, C.J.; Schoretsanitis, G.; De Las Cuevas, C. A Rational Use of Clozapine Based on Adverse Drug Reactions, Pharmacokinetics, and Clinical Pharmacopsychology. Psychother. Psychosom. 2020, 89, 200–214. [Google Scholar]
- Citrome, L. Introduction. In Handbook of Treatment-Resistant Schizophrenia; Springer: London, UK, 2013; pp. 1–11. [Google Scholar]
- Kuha, S. The consequences of sudden stopping of clozapine in Finland. In Proceedings of the Sixth World Congress of Psychiatry, Honolulu, HI, USA, 28 August–3 September 1977. Abstract No. 813. [Google Scholar]
- Moncrieff, J. Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse. Acta Psychiatr. Scand. 2006, 114, 3–13. [Google Scholar]
- Gilbert, P.L.; Harris, M.J.; McAdams, L.A.; Jeste, D.V. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch. Gen. Psychiatr. 1995, 52, 173–188. [Google Scholar]
- Meltzer, H.Y.; Li, Z.; Kaneda, Y.; Ichikawa, J. Serotonin receptors: Their key role in drugs to treat schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 1159–1172. [Google Scholar]
- Meltzer, H.Y.; Massey, B.W. The role of serotonin receptors in the action of atypical antipsychotic drugs. Curr. Opin. Pharmacol. 2011, 11, 59–67. [Google Scholar]
- Su, T.P.; Malhotra, A.K.; Hadd, K.; Breier, A.; Pickar, D. D2 dopamine receptor occupancy: A crossover comparison of risperidone with clozapine therapy in schizophrenic patients. Arch. Gen. Psychiatry 1997, 54, 972–973. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.Y. The mechanism of action of novel antipsychotic drugs. Schizophr. Bull. 1991, 17, 263–287. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Ueda, Y. Lurasidone inhibits NMDA receptor antagonist-induced functional abnormality of thalamocortical glutamatergic transmission via 5-HT7 receptor blockade. Br. J. Pharmacol. 2019, 176, 4002–4018. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Okubo, R.; Shiroyama, T.; Ueda, Y. Lurasidone Sub-Chronically Activates Serotonergic Transmission via Desensitization of 5-HT1A and 5-HT7 Receptors in Dorsal Raphe Nucleus. Pharmaceuticals (Basel) 2019, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Dopamine D2 and serotonin 5-HT1A receptors mediate the actions of aripiprazole in mesocortical and mesoaccumbens transmission. Neuropharmacology 2012, 62, 765–774. [Google Scholar] [CrossRef]
- Ohoyama, K.; Yamamura, S.; Hamaguchi, T.; Nakagawa, M.; Motomura, E.; Shiroyama, T.; Tanii, H.; Okada, M. Effect of novel atypical antipsychotic, blonanserin, on extracellular neurotransmitter level in rat prefrontal cortex. Eur. J. Pharmacol. 2011, 653, 47–57. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Nakagawa, M.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Tanii, H.; Shiroyama, T.; Okada, M. Effects of zotepine on extracellular levels of monoamine, GABA and glutamate in rat prefrontal cortex. Br. J. Pharmacol. 2009, 157, 656–665. [Google Scholar] [CrossRef]
- Yamamura, S.; Ohoyama, K.; Hamaguchi, T.; Kashimoto, K.; Nakagawa, M.; Kanehara, S.; Suzuki, D.; Matsumoto, T.; Motomura, E.; Shiroyama, T.; et al. Effects of quetiapine on monoamine, GABA, and glutamate release in rat prefrontal cortex. Psychopharmacology 2009, 206, 243–258. [Google Scholar] [CrossRef]
- Huang, M.; Panos, J.J.; Kwon, S.; Oyamada, Y.; Rajagopal, L.; Meltzer, H.Y. Comparative effect of lurasidone and blonanserin on cortical glutamate, dopamine, and acetylcholine efflux: Role of relative serotonin (5-HT)2A and DA D2 antagonism and 5-HT1A partial agonism. J. Neurochem. 2014, 128, 938–949. [Google Scholar] [CrossRef]
- Zocchi, A.; Fabbri, D.; Heidbreder, C.A. Aripiprazole increases dopamine but not noradrenaline and serotonin levels in the mouse prefrontal cortex. Neurosci. Lett. 2005, 387, 157–161. [Google Scholar] [CrossRef]
- Fukuyama, K.; Kato, R.; Murata, M.; Shiroyama, T.; Okada, M. Clozapine Normalizes a Glutamatergic Transmission Abnormality Induced by an Impaired NMDA Receptor in the Thalamocortical Pathway via the Activation of a Group III Metabotropic Glutamate Receptor. Biomolecules 2019, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Hasegawa, T.; Okada, M. Cystine/Glutamate Antiporter and Aripiprazole Compensate NMDA Antagonist-Induced Dysfunction of Thalamocortical L-Glutamatergic Transmission. Int. J. Mol. Sci. 2018, 19, 3645. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, S.; Yamamura, S.; Nakagawa, M.; Motomura, E.; Okada, M. Clozapine, but not haloperidol, enhances glial D-serine and L-glutamate release in rat frontal cortex and primary cultured astrocytes. Br. J. Pharmacol. 2012, 165, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W.T.; O’Shea, S.D. Clozapine and GABA transmission in schizophrenia disease models: Establishing principles to guide treatments. Pharmacol. Ther. 2015, 150, 47–80. [Google Scholar] [CrossRef]
- Ruderfer, D.M.; Charney, A.W.; Readhead, B.; Kidd, B.A.; Kahler, A.K.; Kenny, P.J.; Keiser, M.J.; Moran, J.L.; Hultman, C.M.; Scott, S.A.; et al. Polygenic overlap between schizophrenia risk and antipsychotic response: A genomic medicine approach. Lancet Psychiatry 2016, 3, 350–357. [Google Scholar] [CrossRef]
- Hons, J.; Vasatova, M.; Cermakova, E.; Doubek, P.; Libiger, J. Different serine and glycine metabolism in patients with schizophrenia receiving clozapine. J. Psychiatr. Res. 2012, 46, 811–818. [Google Scholar] [CrossRef]
- Yamamori, H.; Hashimoto, R.; Fujita, Y.; Numata, S.; Yasuda, Y.; Fujimoto, M.; Ohi, K.; Umeda-Yano, S.; Ito, A.; Ohmori, T.; et al. Changes in plasma D-serine, L-serine, and glycine levels in treatment-resistant schizophrenia before and after clozapine treatment. Neurosci. Lett. 2014, 582, 93–98. [Google Scholar] [CrossRef]
- Schwieler, L.; Engberg, G.; Erhardt, S. Clozapine modulates midbrain dopamine neuron firing via interaction with the NMDA receptor complex. Synapse 2004, 52, 114–122. [Google Scholar] [CrossRef]
- Potkin, S.G.; Kane, J.M.; Correll, C.U.; Lindenmayer, J.P.; Agid, O.; Marder, S.R.; Olfson, M.; Howes, O.D. The neurobiology of treatment-resistant schizophrenia: Paths to antipsychotic resistance and a roadmap for future research. NPJ Schizophr. 2020, 6, 1. [Google Scholar] [CrossRef]
- Ninan, I.; Jardemark, K.E.; Wang, R.Y. Olanzapine and clozapine but not haloperidol reverse subchronic phencyclidine-induced functional hyperactivity of N-methyl-D-aspartate receptors in pyramidal cells of the rat medial prefrontal cortex. Neuropharmacology 2003, 44, 462–472. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Nakano, T.; Ueda, Y. Pharmacological Discrimination of Effects of MK801 on Thalamocortical, Mesothalamic, and Mesocortical Transmissions. Biomolecules 2019, 9, 746. [Google Scholar]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Ueda, Y. Memantine protects thalamocortical hyper-glutamatergic transmission induced by NMDA receptor antagonism via activation of system xc−. Pharmacol. Res. Perspect. 2019, 7, e00457. [Google Scholar] [PubMed]
- Fukuyama, K.; Okubo, R.; Murata, M.; Shiroyama, T.; Okada, M. Activation of Astroglial Connexin is Involved in Concentration-Dependent Double-Edged Sword Clinical Action of Clozapine. Cells 2020, 9, 414. [Google Scholar]
- Izquierdo, A. Functional Heterogeneity within Rat Orbitofrontal Cortex in Reward Learning and Decision Making. J. Neurosci. 2017, 37, 10529–10540. [Google Scholar]
- Izquierdo, A.; Murray, E.A. Selective bilateral amygdala lesions in rhesus monkeys fail to disrupt object reversal learning. J. Neurosci. 2007, 27, 1054–1062. [Google Scholar]
- McCormick, D.A.; Wang, Z. Serotonin and noradrenaline excite GABAergic neurones of the guinea-pig and cat nucleus reticularis thalami. J. Physiol. 1991, 442, 235–255. [Google Scholar]
- Porrino, L.J.; Crane, A.M.; Goldman-Rakic, P.S. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J. Comp. Neurol. 1981, 198, 121–136. [Google Scholar]
- Russchen, F.T.; Amaral, D.G.; Price, J.L. The afferent input to the magnocellular division of the mediodorsal thalamic nucleus in the monkey, Macaca fascicularis. J. Comp. Neurol. 1987, 256, 175–210. [Google Scholar]
- Okada, M.; Fukuyama, K. Interaction between Mesocortical and Mesothalamic Catecholaminergic Transmissions Associated with NMDA Receptor in the Locus Coeruleus. Biomolecules 2020, 10, 990. [Google Scholar]
- Fukuyama, K.; Ueda, Y.; Okada, M. Effects of Carbamazepine, Lacosamide and Zonisamide on Gliotransmitter Release Associated with Activated Astroglial Hemichannels. Pharmaceuticals (Basel) 2020, 13, 117. [Google Scholar] [CrossRef]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathomechanism of nocturnal paroxysmal dystonia in autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Biomed. Pharmacother. 2020, 126, 110070. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Fukuzawa, M.; Okubo, R.; Okada, M. Upregulated Connexin 43 Induced by Loss-of-Functional S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor Contributes to Pathomechanisms of Autosomal Dominant Sleep-Related Hypermotor Epilepsy. Pharmaceuticals (Basel) 2020, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Fukuzawa, M.; Okada, M. Upregulated and Hyperactivated Thalamic Connexin 43 Plays Important Roles in Pathomechanisms of Cognitive Impairment and Seizure of Autosomal Dominant Sleep-Related Hypermotor Epilepsy with S284L-Mutant alpha4 Subunit of Nicotinic ACh Receptor. Pharmaceuticals (Basel) 2020, 13, 99. [Google Scholar] [CrossRef]
- Okada, M.; Fukuyama, K.; Kawano, Y.; Shiroyama, T.; Suzuki, D.; Ueda, Y. Effects of acute and sub-chronic administrations of guanfacine on catecholaminergic transmissions in the orbitofrontal cortex. Neuropharmacology 2019, 156, 107547. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, K.; Fukuzawa, M.; Shiroyama, T.; Okada, M. Pathogenesis and pathophysiology of autosomal dominant sleep-related hypermotor epilepsy with S284L-mutant alpha4 subunit of nicotinic ACh receptor. Br. J. Pharmacol. 2020, 177, 2143–2162. [Google Scholar] [CrossRef]
- Nakano, T.; Hasegawa, T.; Suzuki, D.; Motomura, E.; Okada, M. Amantadine Combines Astroglial System Xc(-) Activation with Glutamate/NMDA Receptor Inhibition. Biomolecules 2019, 9, 191. [Google Scholar] [CrossRef]
- Lapato, A.S.; Tiwari-Woodruff, S.K. Connexins and pannexins: At the junction of neuro-glial homeostasis & disease. J. Neurosci. Res. 2018, 96, 31–44. [Google Scholar]
- Li, Q.; Li, Q.Q.; Jia, J.N.; Liu, Z.Q.; Zhou, H.H.; Mao, X.Y. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed. Pharmacother. 2019, 109, 57–65. [Google Scholar] [CrossRef]
- Medina-Ceja, L.; Salazar-Sanchez, J.C.; Ortega-Ibarra, J.; Morales-Villagran, A. Connexins-Based Hemichannels/Channels and Their Relationship with Inflammation, Seizures and Epilepsy. Int. J. Mol. Sci. 2019, 20, 5976. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girao, H. Role of connexin 43 in different forms of intercellular communication–gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef]
- Dallerac, G.; Rouach, N. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.B.; Chibane, F.L.; Wang, Z.; Chuang, D.M. Potential roles of HDAC inhibitors in mitigating ischemia-induced brain damage and facilitating endogenous regeneration and recovery. Curr. Pharm. Des. 2013, 19, 5105–5120. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.; Shao, Q.; Yang, X.J.; Luh, S.P.; Kandouz, M.; Batist, G.; Laird, D.W.; Alaoui-Jamali, M.A. A histone deacetylation-dependent mechanism for transcriptional repression of the gap junction gene cx43 in prostate cancer cells. Prostate 2006, 66, 1151–1161. [Google Scholar] [CrossRef]
- Ogawa, T.; Hayashi, T.; Tokunou, M.; Nakachi, K.; Trosko, J.E.; Chang, C.C.; Yorioka, N. Suberoylanilide hydroxamic acid enhances gap junctional intercellular communication via acetylation of histone containing connexin 43 gene locus. Cancer Res. 2005, 65, 9771–9778. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Akhtar, M.; Asklund, T.; Juliusson, B.; Almqvist, P.M.; Ekstrom, T.J. HDAC inhibition amplifies gap junction communication in neural progenitors: Potential for cell-mediated enzyme prodrug therapy. Exp. Cell Res. 2007, 313, 2958–2967. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Specific Cx43 phosphorylation events regulate gap junction turnover in vivo. FEBS Lett. 2014, 588, 1423–1429. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Calloe, K.; Holstein-Rathlou, N.H.; Nielsen, M.S. Managing the complexity of communication: Regulation of gap junctions by post-translational modification. Front. Pharmacol. 2013, 4, 130. [Google Scholar] [CrossRef]
- Su, V.; Nakagawa, R.; Koval, M.; Lau, A.F. Ubiquitin-independent proteasomal degradation of endoplasmic reticulum-localized connexin43 mediated by CIP75. J. Biol. Chem. 2010, 285, 40979–40990. [Google Scholar] [CrossRef]
- Dunn, C.A.; Su, V.; Lau, A.F.; Lampe, P.D. Activation of Akt, not connexin 43 protein ubiquitination, regulates gap junction stability. J. Biol. Chem. 2012, 287, 2600–2607. [Google Scholar] [CrossRef]
- Einoch, R.; Weinreb, O.; Mandiuk, N.; Youdim, M.B.H.; Bilker, W.; Silver, H. The involvement of BDNF-CREB signaling pathways in the pharmacological mechanism of combined SSRI-antipsychotic treatment in schizophrenia. Eur. Neuropsychopharmacol. 2017, 27, 470–483. [Google Scholar] [CrossRef]
- Smith, G.C.; McEwen, H.; Steinberg, J.D.; Shepherd, P.R. The activation of the Akt/PKB signalling pathway in the brains of clozapine-exposed rats is linked to hyperinsulinemia and not a direct drug effect. Psychopharmacology 2014, 231, 4553–4560. [Google Scholar] [PubMed]
- Liu, X.; Wu, Z.; Lian, J.; Hu, C.H.; Huang, X.F.; Deng, C. Time-dependent changes and potential mechanisms of glucose-lipid metabolic disorders associated with chronic clozapine or olanzapine treatment in rats. Sci. Rep. 2017, 7, 2762. [Google Scholar] [PubMed]
- Zeng, Z.; Wang, X.; Bhardwaj, S.K.; Zhou, X.; Little, P.J.; Quirion, R.; Srivastava, L.K.; Zheng, W. The Atypical Antipsychotic Agent, Clozapine, Protects Against Corticosterone-Induced Death of PC12 Cells by Regulating the Akt/FoxO3a Signaling Pathway. Mol. Neurobiol. 2017, 54, 3395–3406. [Google Scholar] [PubMed]
- Smith, G.C.; Chaussade, C.; Vickers, M.; Jensen, J.; Shepherd, P.R. Atypical antipsychotic drugs induce derangements in glucose homeostasis by acutely increasing glucagon secretion and hepatic glucose output in the rat. Diabetologia 2008, 51, 2309–2317. [Google Scholar] [PubMed]
- Zhao, M.; Hou, S.; Feng, L.; Shen, P.; Nan, D.; Zhang, Y.; Wang, F.; Ma, D.; Feng, J. Vinpocetine Protects Against Cerebral Ischemia-Reperfusion Injury by Targeting Astrocytic Connexin43 via the PI3K/AKT Signaling Pathway. Front. Neurosci. 2020, 14, 223. [Google Scholar] [PubMed]
- Ock, S.; Lee, W.S.; Kim, H.M.; Park, K.S.; Kim, Y.K.; Kook, H.; Park, W.J.; Lee, T.J.; Abel, E.D.; Kim, J. Connexin43 and zonula occludens-1 are targets of Akt in cardiomyocytes that correlate with cardiac contractile dysfunction in Akt deficient hearts. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1183–1191. [Google Scholar] [CrossRef]
- Mitterauer, B. Loss of function of glial gap junctions may cause severe cognitive impairments in schizophrenia. Med. Hypotheses 2009, 73, 393–397. [Google Scholar] [CrossRef]
- Gawlik, M.; Wagner, M.; Pfuhlmann, B.; Stober, G. The role of Pannexin gene variants in schizophrenia: Systematic analysis of phenotypes. Eur. Arch. Psychiatry Clin. Neurosci. 2016, 266, 433–437. [Google Scholar]
- Hagger, C.; Buckley, P.; Kenny, J.T.; Friedman, L.; Ubogy, D.; Meltzer, H.Y. Improvement in cognitive functions and psychiatric symptoms in treatment-refractory schizophrenic patients receiving clozapine. Biol. Psychiatry 1993, 34, 702–712. [Google Scholar]
- Meltzer, H.Y. Clozapine: Balancing safety with superior antipsychotic efficacy. Clin. Schizophr. Relat. Psychoses 2012, 6, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.; Velosa, J.; Zhang, J.; Dursun, S.M.; Kapczinski, F.; de Azevedo Cardoso, T. Clozapine in bipolar disorder: A systematic review and meta-analysis. J. Psychiatr. Res. 2020, 125, 21–27. [Google Scholar] [CrossRef] [PubMed]
- López-Villarreal, A.; Sánchez-Morla, E.M.; Jiménez-López, E.; Martínez-Vizcaíno, V.; Aparicio, A.I.; Mateo-Sotos, J.; Rodriguez-Jimenez, R.; Vieta, E.; Santos, J.L. Progression of the functional deficit in a group of patients with bipolar disorder: A cluster analysis based on longitudinal data. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 1–11. [Google Scholar] [CrossRef]
- Van Rheenen, T.E.; Lewandowski, K.E.; Bauer, I.E.; Kapczinski, F.; Miskowiak, K.; Burdick, K.E.; Balanzá-Martínez, V. Current understandings of the trajectory and emerging correlates of cognitive impairment in bipolar disorder: An overview of evidence. Bipolar. Disorders 2020, 22, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Mitterauer, B. Imbalance of glial-neuronal interaction in synapses: A possible mechanism of the pathophysiology of bipolar disorder. Neuroscientist 2004, 10, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Mitterauer, B.J. Downregulation and upregulation of glial connexins may cause synaptic imbalances responsible for the pathophysiology of bipolar disorder. CNS Neurosci. Ther. 2011, 17, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Flores, C.E.; Nannapaneni, S.; Davidson, K.G.; Yasumura, T.; Bennett, M.V.; Rash, J.E.; Pereda, A.E. Trafficking of gap junction channels at a vertebrate electrical synapse in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E573–E582. [Google Scholar] [CrossRef]
- Garbelli, R.; Frassoni, C.; Condorelli, D.; Salinaro, A.T.; Musso, N.; Medici, V.; Tassi, L.; Bentivoglio, M.; Spreafico, R. Expression of connexin 43 in the human epileptic and drug-resistant cerebral cortex. Neurology 2011, 76, 895–902. [Google Scholar] [CrossRef]
- Das, A.; Wallace IV, G.C.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef]
- Fonseca, C.G.; Green, C.R.; Nicholson, L.F. Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 2002, 929, 105–116. [Google Scholar] [CrossRef]
- Kosaka, T.; Deans, M.; Paul, D.; Kosaka, K. Neuronal gap junctions in the mouse main olfactory bulb: Morphological analyses on transgenic mice. Neuroscience 2005, 134, 757–769. [Google Scholar] [CrossRef] [PubMed]
- Collignon, F.; Wetjen, N.M.; Cohen-Gadol, A.A.; Cascino, G.D.; Parisi, J.; Meyer, F.B.; Marsh, W.R.; Roche, P.; Weigand, S.D. Altered expression of connexin subtypes in mesial temporal lobe epilepsy in humans. J. Neurosurg. 2006, 105, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Fukuyama, K.; Shiroyama, T.; Ueda, Y. Carbamazepine Attenuates Astroglial L-Glutamate Release Induced by Pro-Inflammatory Cytokines via Chronically Activation of Adenosine A2A Receptor. Int. J. Mol. Sci. 2019, 20, 3727. [Google Scholar] [CrossRef]
- Zheng, W.; Xiang, Y.T.; Yang, X.H.; Xiang, Y.Q.; de Leon, J. Clozapine Augmentation with Antiepileptic Drugs for Treatment-Resistant Schizophrenia: A Meta-Analysis of Randomized Controlled Trials. J. Clin. Psychiatry 2017, 78, e498–e505. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.A.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef]
- Lampe, P.D.; Lau, A.F. The effects of connexin phosphorylation on gap junctional communication. Int. J. Biochem. Cell Biol. 2004, 36, 1171–1186. [Google Scholar] [CrossRef]
- Nambara, C.; Kawasaki, Y.; Yamasaki, H. Role of the cytoplasmic loop domain of Cx43 in its intracellular localization and function: Possible interaction with cadherin. J. Membr. Biol. 2007, 217, 63–69. [Google Scholar] [CrossRef]
- Iwasaki, Y.K.; Nishida, K.; Kato, T.; Nattel, S. Atrial fibrillation pathophysiology: Implications for management. Circulation 2011, 124, 2264–2274. [Google Scholar] [CrossRef]
- van Rijen, H.V.; Eckardt, D.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; de Bakker, J.M. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef]
- Michela, P.; Velia, V.; Aldo, P.; Ada, P. Role of connexin 43 in cardiovascular diseases. Eur. J. Pharmacol. 2015, 768, 71–76. [Google Scholar] [CrossRef]
- Zhong, C.; Chang, H.; Wu, Y.; Zhou, L.; Wang, Y.; Wang, M.; Wu, P.; Qi, Z.; Zou, J. Up-regulated Cx43 phosphorylation at Ser368 prolongs QRS duration in myocarditis. J. Cell Mol. Med. 2018, 22, 3537–3547. [Google Scholar] [CrossRef]
- Gao, J.; Zhao, Y.; Wang, Y.; Xin, J.; Cui, J.; Ma, S.; Lu, F.; Qin, L.; Yu, X. Anti-arrhythmic effect of acupuncture pretreatment in the rats subjected to simulative global ischemia and reperfusion--involvement of intracellular Ca2+ and connexin 43. BMC Complementary Altern. Med. 2015, 15, 5. [Google Scholar] [CrossRef]
- Fontes, M.S.; van Veen, T.A.; de Bakker, J.M.; van Rijen, H.V. Functional consequences of abnormal Cx43 expression in the heart. Biochim. Biophys. Acta 2012, 1818, 2020–2029. [Google Scholar] [CrossRef]
- Formigli, L.; Ibba-Manneschi, L.; Perna, A.M.; Pacini, A.; Polidori, L.; Nediani, C.; Modesti, P.A.; Nosi, D.; Tani, A.; Celli, A.; et al. Altered Cx43 expression during myocardial adaptation to acute and chronic volume overloading. Histol. Histopathol. 2003, 18, 359–369. [Google Scholar] [PubMed]
- Itoh, M.; Takeishi, Y.; Nakada, S.; Miyamoto, T.; Tsunoda, Y.; Takahashi, H.; Kubota, I.; Tomoike, H. Long-term treatment with angiotensin II type 1 receptor antagonist, CV-11974, restores beta-catenin mRNA expression in volume-overloaded rabbit hearts. Heart Vessels 2002, 17, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Emdad, L.; Uzzaman, M.; Takagishi, Y.; Honjo, H.; Uchida, T.; Severs, N.J.; Kodama, I.; Murata, Y. Gap junction remodeling in hypertrophied left ventricles of aortic-banded rats: Prevention by angiotensin II type 1 receptor blockade. J. Mol. Cell. Cardiol. 2001, 33, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Uzzaman, M.; Honjo, H.; Takagishi, Y.; Emdad, L.; Magee, A.I.; Severs, N.J.; Kodama, I. Remodeling of gap junctional coupling in hypertrophied right ventricles of rats with monocrotaline-induced pulmonary hypertension. Circ. Res. 2000, 86, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Behera, D. Text Book of Pulmonary Medicine. Indian J. Chest Dis. Allied Sci. 2010, 52, 173. [Google Scholar]
- Zhang, H.C.; Zhang, Z.S.; Zhang, L.; Wang, A.; Zhu, H.; Li, L.; Si, J.Q.; Li, X.Z.; Ma, K.T. Connexin 43 in splenic lymphocytes is involved in the regulation of CD4+CD25+ T lymphocyte proliferation and cytokine production in hypertensive inflammation. Int. J. Mol. Med. 2018, 41, 13–24. [Google Scholar] [CrossRef]
- Ni, X.; Wang, A.; Zhang, L.; Shan, L.Y.; Zhang, H.C.; Li, L.; Si, J.Q.; Luo, J.; Li, X.Z.; Ma, K.T. Up-regulation of gap junction in peripheral blood T lymphocytes contributes to the inflammatory response in essential hypertension. PLoS ONE 2017, 12, e0184773. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Li, X.Z.; Fan, Z.R.; Wang, A.; Zhang, H.C.; Zhang, L.; Li, L.; Si, J.Q.; Ma, K.T. Increased expression and functionality of the gap junction in peripheral blood lymphocytes is associated with hypertension-mediated inflammation in spontaneously hypertensive rats. Cell. Mol. Biol. Lett. 2018, 23, 40. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Zhang, L.; Peng, M.; Shen, T.W.; Yu, X.S.; Shan, L.Y.; Li, L.; Si, J.Q.; Li, X.Z.; Ma, K.T. Hydrogen Sulfide Attenuates Hypertensive Inflammation via Regulating Connexin Expression in Spontaneously Hypertensive Rats. Med. Sci. Monit. 2018, 24, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Zhang, L.; Ma, X.; Shan, L.Y.; Li, L.; Si, J.Q.; Li, X.Z.; Zhang, Y.Y.; Ma, K.T. Betaestradiol alleviates hypertension and concanavalin Amediated inflammatory responses via modulation of connexins in peripheral blood lymphocytes. Mol. Med. Rep. 2019, 19, 3743–3755. [Google Scholar]
- Ram, A.; Singh, S.K.; Singh, V.P.; Kumar, S.; Ghosh, B. Inhaled carbenoxolone prevents allergic airway inflammation and airway hyperreactivity in a mouse model of asthma. Int. Arch. Allergy Immunol. 2009, 149, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Endong, L.; Shijie, J.; Sonobe, Y.; Di, M.; Hua, L.; Kawanokuchi, J.; Mizuno, T.; Suzumura, A. The gap-junction inhibitor carbenoxolone suppresses the differentiation of Th17 cells through inhibition of IL-23 expression in antigen presenting cells. J. Neuroimmunol. 2011, 240–241, 58–64. [Google Scholar] [CrossRef]
- Zhang, L.; Fan, Z.R.; Wang, L.; Liu, L.Q.; Li, X.Z.; Li, L.; Si, J.Q.; Ma, K.T. Carbenoxolone decreases monocrotalineinduced pulmonary inflammation and pulmonary arteriolar remodeling in rats by decreasing the expression of connexins in T lymphocytes. Int. J. Mol. Med. 2020, 45, 81–92. [Google Scholar]
- Ponsford, M.J.; Pecoraro, A.; Jolles, S. Clozapine-associated secondary antibody deficiency. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 553–562. [Google Scholar]
- Hung, G.C.; Liu, H.C.; Yang, S.Y.; Pan, C.H.; Liao, Y.T.; Chen, C.C.; Kuo, C.J. Antipsychotic reexposure and recurrent pneumonia in schizophrenia: A nested case-control study. J. Clin. Psychiatry 2016, 77, 60–66. [Google Scholar] [CrossRef]
- Leung, J.G.; Hasassri, M.E.; Barreto, J.N.; Nelson, S.; Morgan, R.J., III. Characterization of Admission Types in Medically Hospitalized Patients Prescribed Clozapine. Psychosomatics 2017, 58, 164–172. [Google Scholar] [CrossRef]
- Ponsford, M.; Castle, D.; Tahir, T.; Robinson, R.; Wade, W.; Steven, R.; Bramhall, K.; Moody, M.; Carne, E.; Ford, C.; et al. Clozapine is associated with secondary antibody deficiency. Br. J. Psychiatry 2018, 214, 83–89. [Google Scholar]
- Lucas, C.L.; Kuehn, H.S.; Zhao, F.; Niemela, J.E.; Deenick, E.K.; Palendira, U.; Avery, D.T.; Moens, L.; Cannons, J.L.; Biancalana, M.; et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat. Immunol. 2014, 15, 88–97. [Google Scholar] [PubMed]
- Deau, M.C.; Heurtier, L.; Frange, P.; Suarez, F.; Bole-Feysot, C.; Nitschke, P.; Cavazzana, M.; Picard, C.; Durandy, A.; Fischer, A.; et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J. Clin. Investig. 2014, 124, 3923–3928. [Google Scholar] [PubMed]
- Angulo, I.; Vadas, O.; Garcon, F.; Banham-Hall, E.; Plagnol, V.; Leahy, T.R.; Baxendale, H.; Coulter, T.; Curtis, J.; Wu, C.; et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 2013, 342, 866–871. [Google Scholar] [PubMed]
- Hojlund, K. Metabolism and insulin signaling in common metabolic disorders and inherited insulin resistance. Dan. Med. J. 2014, 61, B4890. [Google Scholar]
- Zhao, Z.; Ksiezak-Reding, H.; Riggio, S.; Haroutunian, V.; Pasinetti, G.M. Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr. Res. 2006, 84, 1–14. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Adverse Reaction | Cases | Fatal Outcomes | Relative Lethality (%) |
|---|---|---|---|
| Agranulocytosis | 34,931 | 550 | 1.6 |
| Pneumonia | 6983 | 2077 | 29.7 |
| Arrhythmia | 6927 | 319 | 4.6 |
| Myocarditis | 4586 | 539 | 11.8 |
| Sudden death/Cardiac arrests | 1614 | 1449 | 89.8 |
| Cardiomyopathy | 1132 | 131 | 11.6 |
| Seizure | 6231 | 308 | 4.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, M.; Fukuyama, K.; Shiroyama, T.; Murata, M. A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43. Int. J. Mol. Sci. 2020, 21, 7019. https://doi.org/10.3390/ijms21197019
Okada M, Fukuyama K, Shiroyama T, Murata M. A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43. International Journal of Molecular Sciences. 2020; 21(19):7019. https://doi.org/10.3390/ijms21197019
Chicago/Turabian StyleOkada, Motohiro, Kouji Fukuyama, Takashi Shiroyama, and Masahiko Murata. 2020. "A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43" International Journal of Molecular Sciences 21, no. 19: 7019. https://doi.org/10.3390/ijms21197019
APA StyleOkada, M., Fukuyama, K., Shiroyama, T., & Murata, M. (2020). A Working Hypothesis Regarding Identical Pathomechanisms between Clinical Efficacy and Adverse Reaction of Clozapine via the Activation of Connexin43. International Journal of Molecular Sciences, 21(19), 7019. https://doi.org/10.3390/ijms21197019