Structure-Based Discovery of Novel Chemical Classes of Autotaxin Inhibitors

 ,
,  ,
, 
Abstract
1. Introduction
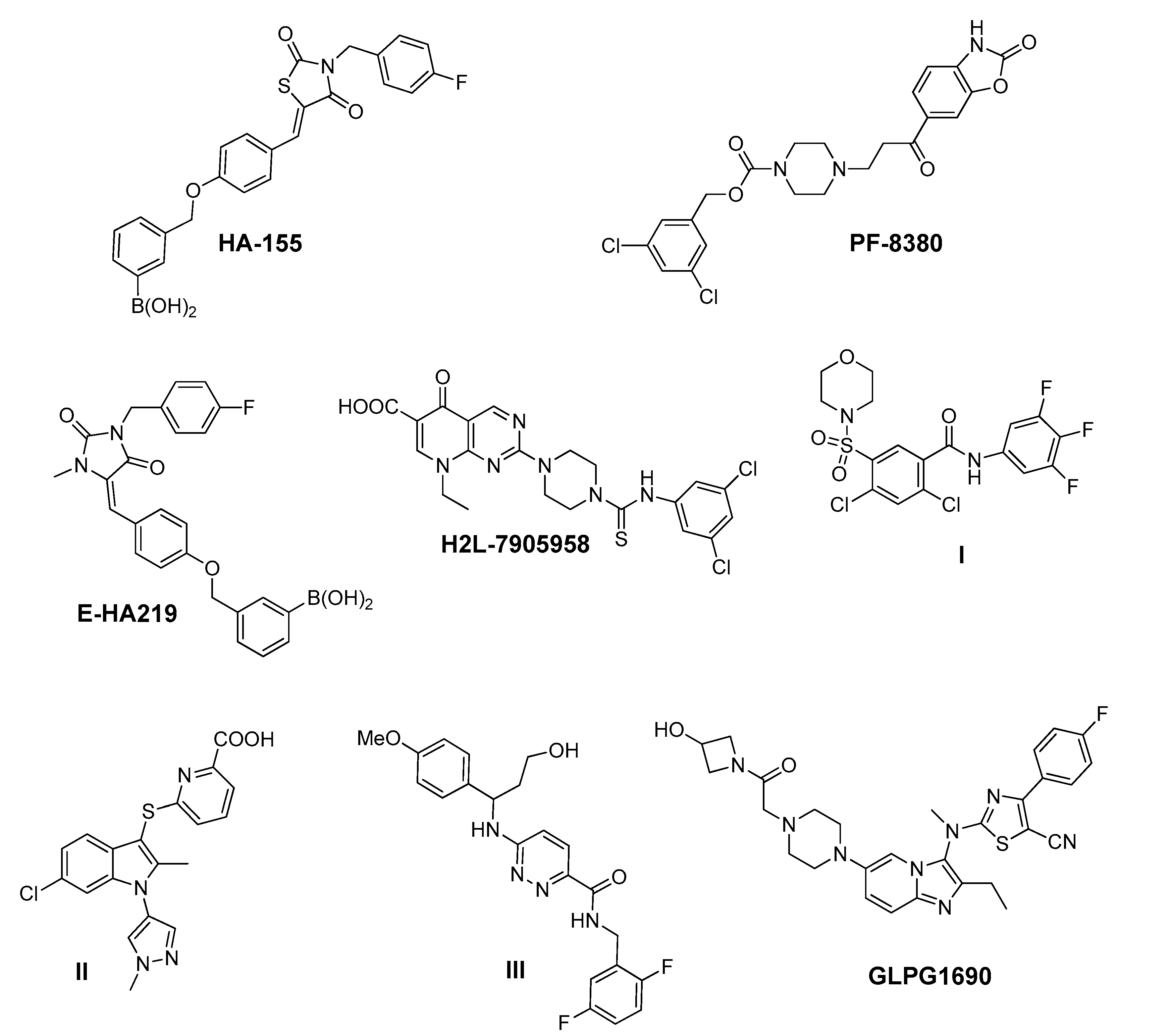
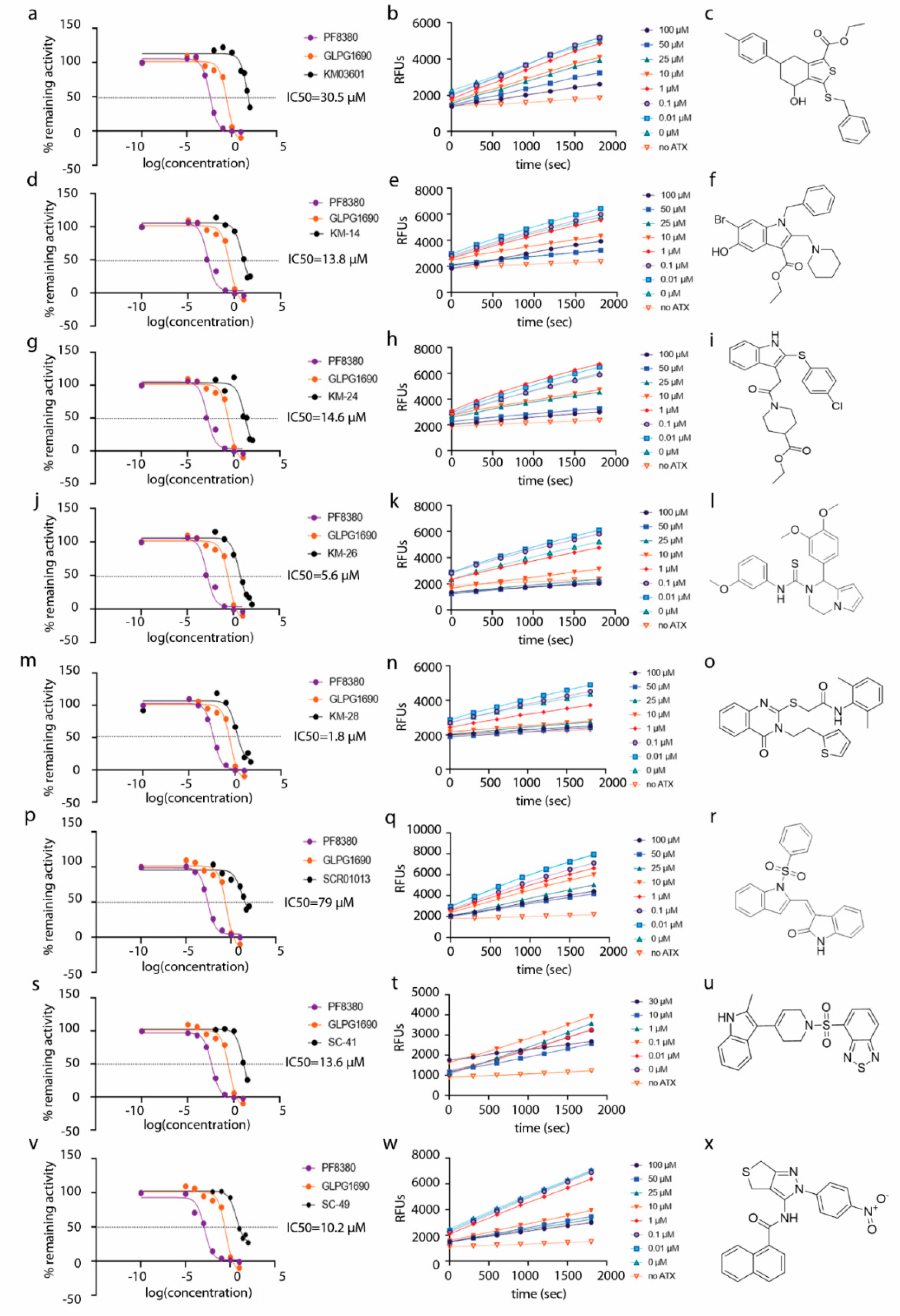
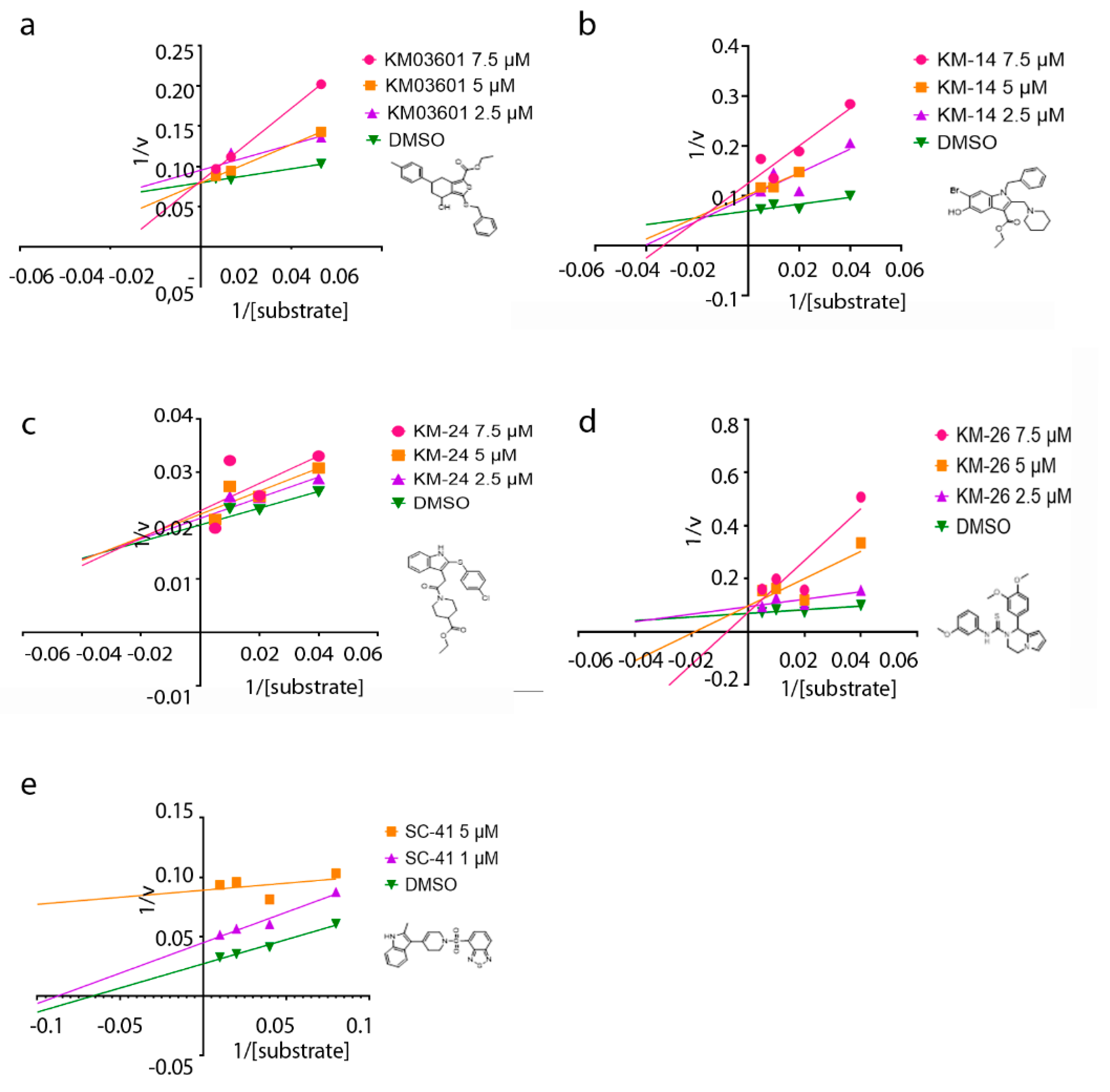
2. Results and Discussion
3. Materials and Methods
3.1. Database
3.2. Virtual Screening
3.3. Optimization of the Initial Hits
3.4. Pharmacological Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADMET | Absorption, distribution, metabolism, excretion and toxicity |
| ATX | Autotaxin |
| Asp | Aspartic acid |
| ENPP2 | Ectonucleotidepyrophosphatase/ phosphodiesterase |
| His | Histidine |
| LPC | Lysophosphatidylcholine |
| LPA | Lysophosphatidic acid |
| PK/PD | Pharmacokinetics/pharmacodynamics |
| Thr | Threonine |
References
- Magkrioti, C.; Galaris, A.; Kanellopoulou, P.; Stylianaki, E.A.; Kaffe, E.; Aidinis, V. Autotaxin and chronic inflammatory diseases. J. Autoimmun. 2019, 104, 102327. [Google Scholar] [CrossRef] [PubMed]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- Barbayianni, E.; Kaffe, E.; Aidinis, V.; Kokotos, G. Autotaxin, a secreted lysophospholipase D, as a promising therapeutic target in chronic inflammation and cancer. Prog. Lipid Res. 2015, 58, 76–96. [Google Scholar] [CrossRef]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The Bulk of Autotaxin Activity Is Dispensable for Adult Mouse Life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef]
- Sevastou, I.; Kaffe, E.; Mouratis, M.A.; Aidinis, V. Lysoglycerophospholipids in chronic inflammatory disorders: The PLA(2)/LPC and ATX/LPA axes. Biochim. Biophys. Acta 2013, 1831, 42–60. [Google Scholar] [CrossRef]
- Bourgoin, S.G.; Zhao, C. Autotaxin and lysophospholipids in rheumatoid arthritis. Curr. Opin. Investig. Drugs 2010, 11, 515–526. [Google Scholar]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary Autotaxin Expression Contributes to the Pathogenesis of Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef]
- Reeves, V.L.; Trybula, J.S.; Wills, R.C.; Goodpaster, B.H.; Dube, J.J.; Kienesberger, P.C.; Kershaw, E.E. Serum Autotaxin/ENPP2 correlates with insulin resistance in older humans with obesity. Obesity 2015, 23, 2371–2376. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.E.; Martens, J.J.; Kulik, W.; Rueff, F.; Kuiper, E.M.; van Buuren, H.R.; van Erpecum, K.J.; Kondrackiene, J.; Prieto, J.; Rust, C.; et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology 2010, 139, 1008–1018.e1. [Google Scholar] [CrossRef] [PubMed]
- Kremer, A.E.; van Dijk, R.; Leckie, P.; Schaap, F.G.; Kuiper, E.M.; Mettang, T.; Reiners, K.S.; Raap, U.; van Buuren, H.R.; van Erpecum, K.J.; et al. Serum autotaxin is increased in pruritus of cholestasis, but not of other origin, and responds to therapeutic interventions. Hepatology 2012, 56, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Schlatzer, D.M.; Sugalski, J.M.; Chen, Y.; Barnholtz-Sloan, J.; Davitkov, P.; Hazlett, F.E.; Funderburg, N.; Rodriguez, B.; Lederman, M.M.; Sieg, S.F.; et al. Plasma proteome analysis reveals overlapping, yet distinct mechanisms of immune activation in chronic HCV and HIV infections. J. Acquir. Immune Defic. Syndr. 2013, 63, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.B.; Wu, J.; Lu, D.; Maluccio, M.A. Is autotaxin (ENPP2) the link between hepatitis C and hepatocellular cancer? J. Gastrointest. Surg. 2007, 11, 1628–1634; discussion 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Ikeda, H.; Nakamura, K.; Ohkawa, R.; Kume, Y.; Aoki, J.; Hama, K.; Okudaira, S.; Tanaka, M.; Tomiya, T.; et al. Both plasma lysophosphatidic acid and serum autotaxin levels are increased in chronic hepatitis C. J. Clin. Gastroenterol. 2007, 41, 616–623. [Google Scholar] [CrossRef]
- Magkrioti, C.; Aidinis, V. Autotaxin and lysophosphatidic acid signalling in lung pathophysiology. World J. Respirol. 2013, 3, 77–103. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.K.; Brindley, D.N. Role of the autotaxin-lysophosphatidate axis in the development of resistance to cancer therapy. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2020, 1865, 158716. [Google Scholar] [CrossRef]
- Brandon, J.A.; Kraemer, M.; Vandra, J.; Halder, S.; Ubele, M.; Morris, A.J.; Smyth, S.S. Adipose-derived autotaxin regulates inflammation and steatosis associated with diet-induced obesity. PLoS ONE 2019, 14, e0208099. [Google Scholar] [CrossRef]
- Tang, X.; Wuest, M.; Benesch, M.G.K.; Dufour, J.; Zhao, Y.; Curtis, J.M.; Monjardet, A.; Heckmann, B.; Murray, D.; Wuest, F.; et al. Inhibition of Autotaxin with GLPG1690 Increases the Efficacy of Radiotherapy and Chemotherapy in a Mouse Model of Breast Cancer. Mol. Cancer Ther. 2020, 19, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Desroy, N.; Housseman, C.; Bock, X.; Joncour, A.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Rondet, E.; Peixoto, C.; Grassot, J.M.; et al. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methyli midazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a First-in-Class Autotaxin Inhibitor Undergoing Clinical Evaluation for the Treatment of Idiopathic Pulmonary Fibrosis. J. Med. Chem. 2017, 60, 3580–3590. [Google Scholar] [CrossRef]
- Hausmann, J.; Christodoulou, E.; Kasiem, M.; De Marco, V.; van Meeteren, L.A.; Moolenaar, W.H.; Axford, D.; Owen, R.L.; Evans, G.; Perrakis, A. Mammalian cell expression, purification, crystallization and microcrystal data collection of autotaxin/ENPP2, a secreted mammalian glycoprotein. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar]
- Perrakis, A.; Moolenaar, W.H. Autotaxin: Structure-function and signaling. J. Lipid Res. 2014, 55, 1010–1018. [Google Scholar] [CrossRef]
- Matralis, A.N.; Afantitis, A.; Aidinis, V. Development and therapeutic potential of autotaxin small molecule inhibitors: From bench to advanced clinical trials. Med. Res. Rev. 2019, 39, 976–1013. [Google Scholar] [CrossRef]
- Salgado-Polo, F.; Perrakis, A. The Structural Binding Mode of the Four Autotaxin Inhibitor Types that Differentially Affect Catalytic and Non-Catalytic Functions. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef]
- Parrill, A.L.; Baker, D.L. Autotaxin inhibitors: A perspective on initial medicinal chemistry efforts. Expert Opin. Ther. Pat. 2010, 20, 1619–1625. [Google Scholar] [CrossRef][Green Version]
- Banerjee, S.; Norman, D.D.; Lee, S.C.; Parrill, A.L.; Pham, T.C.; Baker, D.L.; Tigyi, G.J.; Miller, D.D. Highly Potent Non-Carboxylic Acid Autotaxin Inhibitors Reduce Melanoma Metastasis and Chemotherapeutic Resistance of Breast Cancer Stem Cells. J. Med. Chem. 2017, 60, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Fells, J.I.; Lee, S.C.; Norman, D.D.; Tsukahara, R.; Kirby, J.R.; Nelson, S.; Seibel, W.; Papoian, R.; Patil, R.; Miller, D.D.; et al. Targeting the hydrophobic pocket of autotaxin with virtual screening of inhibitors identifies a common aromatic sulfonamide structural motif. FEBS J. 2014, 281, 1017–1028. [Google Scholar] [CrossRef] [PubMed]
- Cuozzo, J.W.; Clark, M.A.; Keefe, A.D.; Kohlmann, A.; Mulvihill, M.; Ni, H.; Renzetti, L.M.; Resnicow, D.I.; Ruebsam, F.; Sigel, E.A.; et al. Novel Autotaxin Inhibitor for the Treatment of Idiopathic Pulmonary Fibrosis: A Clinical Candidate Discovered Using DNA-Encoded Chemistry. J. Med. Chem. 2020, 63, 7840–7856. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Guo, M.; Li, X.; Jia, F.; Li, C.; Yang, Y.; Cao, M.; Jiang, N.; Ma, E.; Zhai, X. Discovery of Novel Indole-Based Allosteric Highly Potent ATX Inhibitors with Great In Vivo Efficacy in a Mouse Lung Fibrosis Model. J. Med. Chem. 2020, 63, 7326–7346. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Okabe, T.; Okudaira, S.; Hama, K.; Kano, K.; Nishimasu, H.; Nakagawa, H.; Ishitani, R.; Kojima, H.; Nureki, O.; et al. Identification of Potent In Vivo Autotaxin Inhibitors that Bind to Both Hydrophobic Pockets and Channels in the Catalytic Domain. J. Med. Chem. 2020, 63, 3188–3204. [Google Scholar] [CrossRef]
- Nikolaou, A.; Kokotou, M.G.; Limnios, D.; Psarra, A.; Kokotos, G. Autotaxin inhibitors: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 815–829. [Google Scholar] [CrossRef]
- Barbayianni, E.; Magrioti, V.; Moutevelis-Minakakis, P.; Kokotos, G. Autotaxin inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 1123–1132. [Google Scholar] [CrossRef]
- Nikolaou, A.; Ninou, I.; Kokotou, M.G.; Kaffe, E.; Afantitis, A.; Aidinis, V.; Kokotos, G. Hydroxamic Acids Constitute a Novel Class of Autotaxin Inhibitors that Exhibit In Vivo Efficacy in a Pulmonary Fibrosis Model. J. Med. Chem. 2018, 61, 3697–3711. [Google Scholar] [CrossRef]
- Gerokonstantis, D.T.; Nikolaou, A.; Magkrioti, C.; Afantitis, A.; Aidinis, V.; Kokotos, G.; Moutevelis-Minakakis, P. Synthesis of novel 2-pyrrolidinone and pyrrolidine derivatives and study of their inhibitory activity against autotaxin enzyme. Bioorg. Med. Chem. 2020, 28, 115216. [Google Scholar] [CrossRef]
- Miller, L.M.; Keune, W.J.; Castagna, D.; Young, L.C.; Duffy, E.L.; Potjewyd, F.; Salgado-Polo, F.; Engel Garcia, P.; Semaan, D.; Pritchard, J.M.; et al. Structure-Activity Relationships of Small Molecule Autotaxin Inhibitors with a Discrete Binding Mode. J. Med. Chem. 2017, 60, 722–748. [Google Scholar] [CrossRef]
- Keune, W.J.; Potjewyd, F.; Heidebrecht, T.; Salgado-Polo, F.; Macdonald, S.J.; Chelvarajan, L.; Abdel Latif, A.; Soman, S.; Morris, A.J.; Watson, A.J.; et al. Rational Design of Autotaxin Inhibitors by Structural Evolution of Endogenous Modulators. J. Med. Chem. 2017, 60, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Clair, T.; Koh, E.; Ptaszynska, M.; Bandle, R.W.; Liotta, L.A.; Schiffmann, E.; Stracke, M.L. L-histidine inhibits production of lysophosphatidic acid by the tumor-associated cytokine, autotaxin. Lipids Health Dis. 2005, 4, 5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Durgam, G.G.; Virag, T.; Walker, M.D.; Tsukahara, R.; Yasuda, S.; Liliom, K.; van Meeteren, L.A.; Moolenaar, W.H.; Wilke, N.; Siess, W.; et al. Synthesis, structure-activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARgamma, and inhibitors of autotaxin. J. Med. Chem. 2005, 48, 4919–4930. [Google Scholar] [CrossRef] [PubMed]
- Durgam, G.G.; Tsukahara, R.; Makarova, N.; Walker, M.D.; Fujiwara, Y.; Pigg, K.R.; Baker, D.L.; Sardar, V.M.; Parrill, A.L.; Tigyi, G.; et al. Synthesis and pharmacological evaluation of second-generation phosphatidic acid derivatives as lysophosphatidic acid receptor ligands. Bioorg. Med. Chem. Lett. 2006, 16, 633–640. [Google Scholar] [CrossRef]
- Gududuru, V.; Zeng, K.; Tsukahara, R.; Makarova, N.; Fujiwara, Y.; Pigg, K.R.; Baker, D.L.; Tigyi, G.; Miller, D.D. Identification of Darmstoff analogs as selective agonists and antagonists of lysophosphatidic acid receptors. Bioorg. Med. Chem. Lett. 2006, 16, 451–456. [Google Scholar] [CrossRef]
- Scott, L.J. Fingolimod: A review of its use in the management of relapsing-remitting multiple sclerosis. Cns Drugs 2011, 25, 673–698. [Google Scholar] [CrossRef]
- Cui, P.; McCalmont, W.F.; Tomsig, J.L.; Lynch, K.R.; Macdonald, T.L. alpha- and beta-substituted phosphonate analogs of LPA as autotaxin inhibitors. Bioorg. Med. Chem. 2008, 16, 2212–2225. [Google Scholar] [CrossRef]
- Cui, P.; Tomsig, J.L.; McCalmont, W.F.; Lee, S.; Becker, C.J.; Lynch, K.R.; Macdonald, T.L. Synthesis and biological evaluation of phosphonate derivatives as autotaxin (ATX) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1634–1640. [Google Scholar] [CrossRef]
- Ferry, G.; Moulharat, N.; Pradere, J.P.; Desos, P.; Try, A.; Genton, A.; Giganti, A.; Beucher-Gaudin, M.; Lonchampt, M.; Bertrand, M.; et al. S32826, a nanomolar inhibitor of autotaxin: Discovery, synthesis and applications as a pharmacological tool. J. Pharmacol. Exp. Ther. 2008, 327, 809–819. [Google Scholar] [CrossRef]
- Albers, H.M.; Dong, A.; van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; van Tilburg, E.W.; Schuurman, K.; van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-Pierce, E.; Cortes-Burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S.; et al. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010, 334, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Saga, H.; Ohhata, A.; Hayashi, A.; Katoh, M.; Maeda, T.; Mizuno, H.; Takada, Y.; Komichi, Y.; Ota, H.; Matsumura, N.; et al. A novel highly potent autotaxin/ENPP2 inhibitor produces prolonged decreases in plasma lysophosphatidic acid formation in vivo and regulates urethral tension. PLoS ONE 2014, 9, e93230. [Google Scholar] [CrossRef]
- Iwaki, Y.; Ohhata, A.; Nakatani, S.; Hisaichi, K.; Okabe, Y.; Hiramatsu, A.; Watanabe, T.; Yamamoto, S.; Nishiyama, T.; Kobayashi, J.; et al. ONO-8430506: A Novel Autotaxin Inhibitor That Enhances the Antitumor Effect of Paclitaxel in a Breast Cancer Model. ACS Med. Chem. Lett. 2020, 11, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Parrill, A.L.; Echols, U.; Nguyen, T.; Pham, T.C.; Hoeglund, A.; Baker, D.L. Virtual screening approaches for the identification of non-lipid autotaxin inhibitors. Bioorg. Med. Chem. 2008, 16, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Hoeglund, A.B.; Howard, A.L.; Wanjala, I.W.; Pham, T.C.; Parrill, A.L.; Baker, D.L. Characterization of non-lipid autotaxin inhibitors. Bioorg. Med. Chem. 2010, 18, 769–776. [Google Scholar] [CrossRef]
- Ueda, K.; Yoshihara, M.; Nakao, M.; Tanaka, T.; Sano, S.; Fukuzawa, K.; Tokumura, A. Evaluation of inhibitory actions of flavonols and related substances on lysophospholipase d activity of serum autotaxin by a convenient assay using a chromogenic substrate. J. Agric. Food Chem. 2010, 58, 6053–6063. [Google Scholar] [CrossRef]
- Fells, J.I.; Lee, S.C.; Fujiwara, Y.; Norman, D.D.; Lim, K.G.; Tsukahara, R.; Liu, J.; Patil, R.; Miller, D.D.; Kirby, R.J.; et al. Hits of a high-throughput screen identify the hydrophobic pocket of autotaxin/lysophospholipase D as an inhibitory surface. Mol. Pharmacol. 2013, 84, 415–424. [Google Scholar] [CrossRef]
- Gibson, K.R.; Owen, D.R. Pyridazine Derivatives Useful in Therapy. U.S. Patent 20140275100A1, 1 March 2016. [Google Scholar]
- Long, S.A.; Thorarensen, A.; Schnute, M.E. Pyrimidine and Pyridine Derivatives Useful in Therapy. International Application No. PCT/IB2012/002073, 15 October 2012. [Google Scholar]
- Schultz, M.; Schiemann, K.; Staehle, W. Autotaxin Inhibitors. WO2010112124A1, 7 October 2010. [Google Scholar]
- Schultz, M.; Schiemann, K.; Botton, G.; Blaukat, A.; Kober, I. Imidazole Derivatives. CA2701568C, 6 June 2017. [Google Scholar]
- Staehle, W.; Kober, I.; Schiemann, K.; Schultz, M.; Wienke, D. Benzonaphtyridine Compounds Used as Inhibitors of Autotaxin. International Application No. PCT/EP2009/007930, 5 November 2009. [Google Scholar]
- Staehle, W.; Schultz, M.; Schiemann, K. Benzonaphthyridinamines as Autotaxin Inhibitors. International Application No. PCT/EP2011/000964, 28 February 2011. [Google Scholar]
- Maher, T.M.; Kreuter, M.; Lederer, D.J.; Brown, K.K.; Wuyts, W.; Verbruggen, N.; Stutvoet, S.; Fieuw, A.; Ford, P.; Abi-Saab, W.; et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir. Res. 2019, 6, e000422. [Google Scholar] [CrossRef]
- Maher, T.M.; van der Aar, E.M.; Van de Steen, O.; Allamassey, L.; Desrivot, J.; Dupont, S.; Fagard, L.; Ford, P.; Fieuw, A.; Wuyts, W. Safety, tolerability, pharmacokinetics, and pharmacodynamics of GLPG1690, a novel autotaxin inhibitor, to treat idiopathic pulmonary fibrosis (FLORA): A phase 2a randomised placebo-controlled trial. Lancet Respir. Med. 2018, 6, 627–635. [Google Scholar] [CrossRef]
- Taneja, A.; Desrivot, J.; Diderichsen, P.M.; Blanque, R.; Allamasey, L.; Fagard, L.; Fieuw, A.; Van der Aar, E.; Namour, F. Population Pharmacokinetic and Pharmacodynamic Analysis of GLPG1690, an Autotaxin Inhibitor, in Healthy Volunteers and Patients with Idiopathic Pulmonary Fibrosis. Clin. Pharmacokinet. 2019, 58, 1175–1191. [Google Scholar] [CrossRef]
- Albers, H.M.; Hendrickx, L.J.; van Tol, R.J.; Hausmann, J.; Perrakis, A.; Ovaa, H. Structure-based design of novel boronic acid-based inhibitors of autotaxin. J. Med. Chem. 2011, 54, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Afantitis, A.; Tsoumanis, A.; Melagraki, G. Enalos Suite of tools: Enhance Cheminformatics and Nanoinformatics through KNIME. Curr. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nikitopoulou, I.; Kaffe, E.; Sevastou, I.; Sirioti, I.; Samiotaki, M.; Madan, D.; Prestwich, G.D.; Aidinis, V. A metabolically-stabilized phosphonate analog of lysophosphatidic acid attenuates collagen-induced arthritis. PLoS ONE 2013, 8, e70941. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Polo, F.; Fish, A.; Matsoukas, M.T.; Heidebrecht, T.; Keune, W.J.; Perrakis, A. Lysophosphatidic acid produced by autotaxin acts as an allosteric modulator of its catalytic efficiency. J. Biol. Chem. 2018, 293, 14312–14327. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Pubchem CID | Name | Structure | IC50 (μΜ) [hATX/mATX] | Inhibition of 2nd/3rd Reaction | IC50 (μΜ) PDE Assay | Mode of Inhibition |
|---|---|---|---|---|---|---|---|
| KM03601 | 2820206 | ethyl 3-benzylsulfanyl-4-hydroxy-6-(4-methylphenyl)-4,5,6,7-tetrahydro-2-benzothiophene-1-carboxylate |  | 30.5/14.9 | no | n.d. | competitive |
| KM-14 | 1201991 | ethyl 1-benzyl-6-bromo-5-hydroxy-2-(piperidin-1-ylmethyl)-1H-indole-3-carboxylate |  | 13.8/5.9 | no | n.d. | mixed |
| KM-24 | 46379015 | ethyl 1-[2-[2-(4-chlorophenyl)sulfanyl-1H-indol-3-yl]acetyl]piperidine-4-carboxylate |  | 14.6/6.5 | no | n.d. | mixed |
| KM-26 | 4662990 | 1-(3,4-dimethoxyphenyl)-N-(3-methoxyphenyl)-3,4-dihydro-1H-pyrrolo[1,2-a]pyrazine-2-carbothioamide |  | 5.6/7.4 | yes | 2.8 | competitive |
| KM-28 | 46925664 | N-(2,6-dimethylphenyl)-2-[4-oxo-3-(2-thiophen-2-ylethyl)quinazolin-2-yl]sulfanylacetamide |  | 1.8/3.0 | no | 0.9 | n.d. |
| SCR01013 | 5702991 | (3Z)-3-[[1-(Benzenesulfonyl)indol-2-yl]methylidene]-1H-indol-2-one |  | 79.0/67.0 | no | n.d. | n.d. |
| SC-41 | 20867222 | 4-{[4-(2-Methyl-1H-indol-3-yl)-3,6-dihydropyridin-1(2H)-yl]sulfonyl}-2,1,3-benzothiadiazole |  | 13.6/4.0 | no | n.d. | uncompetitive |
| SC-49 | 3540718 | N-[2-(4-nitrophenyl)-4,6-dihydrothieno[3,4-c]pyrazol-3-yl]naphthalene-1-carboxamide |  | 10.2/9.7 | no | 73.0 | n.d. |
| HA155 | 46856189 | [4-[[4-[(Z)-[3-[(4-fluorophenyl)methyl]-2,4-dioxo-1,3-thiazolidin-5-ylidene]methyl]phenoxy]methyl]phenyl]boronic acid |  | 0.0025 (reported: 0.0057 [67])/n.d. | n.d. | n.d. | competitive |
| PF-8380 | 25265312 | 3,5-Dichlorobenzyl 4-(3-oxo-3-(2-oxo-2,3-dihydrobenzo[d]oxazol-6-yl)propyl)piperazine-1-carboxylate |  | 0.0025 (reported: 0.0017 [51])/n.d. | no | 0.0011 | competitive |
| GLPG1690 | 90420193 | 2-[[2-ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]-methylamino]-4-(4-fluorophenyl)-1,3-thiazole-5-carbonitrile |  | 0.229 (reported: 0.131 [23])/n.d. | n.d. | n.d. | n.d. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magkrioti, C.; Kaffe, E.; Stylianaki, E.-A.; Sidahmet, C.; Melagraki, G.; Afantitis, A.; Matralis, A.N.; Aidinis, V. Structure-Based Discovery of Novel Chemical Classes of Autotaxin Inhibitors. Int. J. Mol. Sci. 2020, 21, 7002. https://doi.org/10.3390/ijms21197002
Magkrioti C, Kaffe E, Stylianaki E-A, Sidahmet C, Melagraki G, Afantitis A, Matralis AN, Aidinis V. Structure-Based Discovery of Novel Chemical Classes of Autotaxin Inhibitors. International Journal of Molecular Sciences. 2020; 21(19):7002. https://doi.org/10.3390/ijms21197002
Chicago/Turabian StyleMagkrioti, Christiana, Eleanna Kaffe, Elli-Anna Stylianaki, Camelia Sidahmet, Georgia Melagraki, Antreas Afantitis, Alexios N. Matralis, and Vassilis Aidinis. 2020. "Structure-Based Discovery of Novel Chemical Classes of Autotaxin Inhibitors" International Journal of Molecular Sciences 21, no. 19: 7002. https://doi.org/10.3390/ijms21197002
APA StyleMagkrioti, C., Kaffe, E., Stylianaki, E.-A., Sidahmet, C., Melagraki, G., Afantitis, A., Matralis, A. N., & Aidinis, V. (2020). Structure-Based Discovery of Novel Chemical Classes of Autotaxin Inhibitors. International Journal of Molecular Sciences, 21(19), 7002. https://doi.org/10.3390/ijms21197002