Inorganic Phosphate (Pi) Signaling in Endothelial Cells: A Molecular Basis for Generation of Endothelial Microvesicles in Uraemic Cardiovascular Disease

 ,
,  ,
,
Abstract
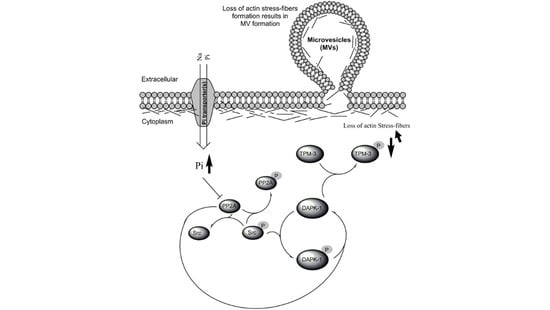
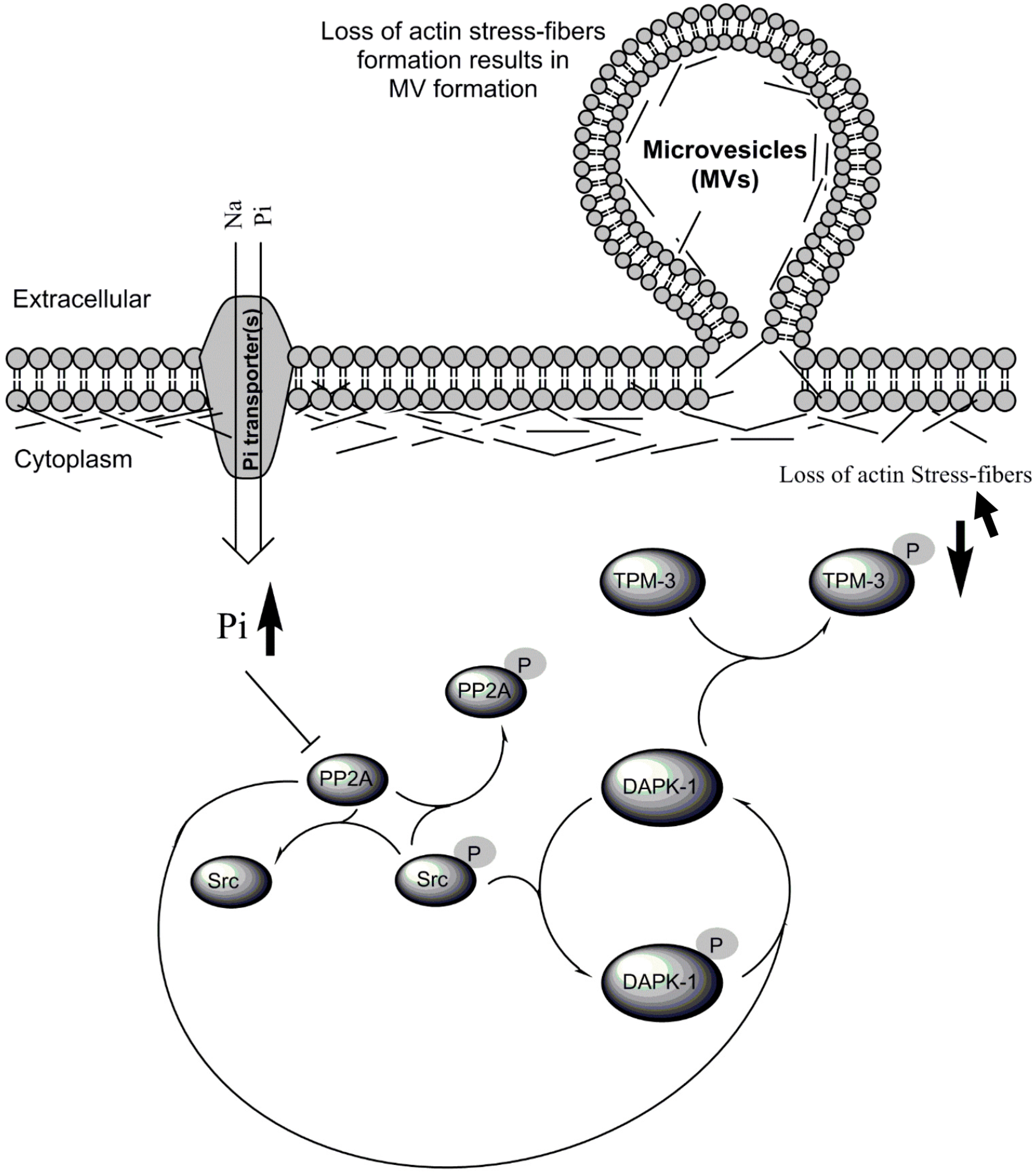
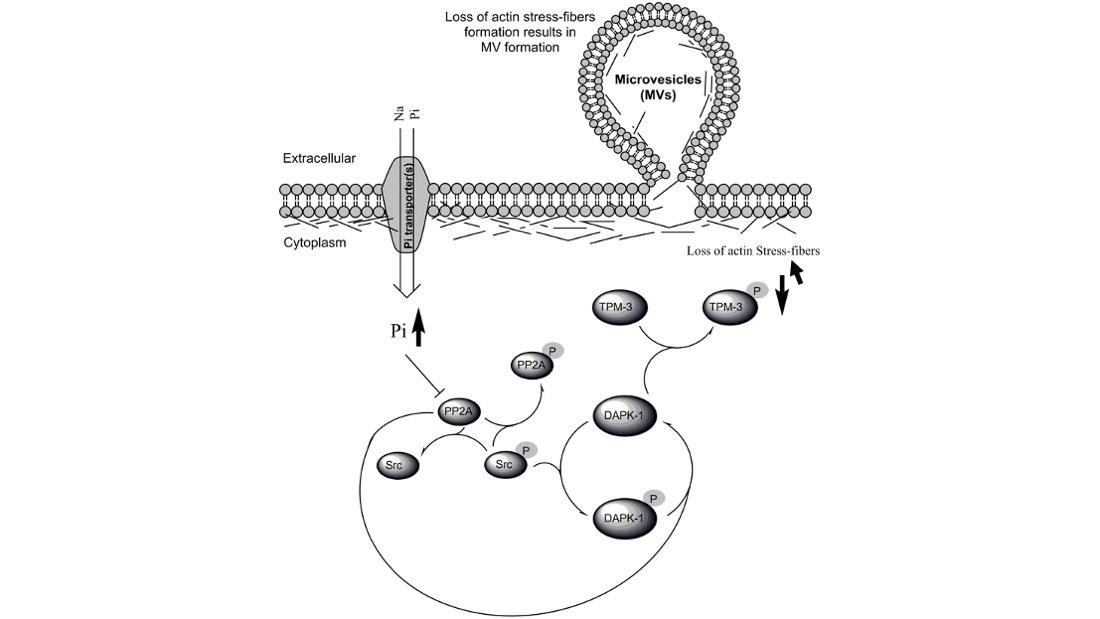{kind=link}
{kind=link}
{kind=link}
{kind=link}
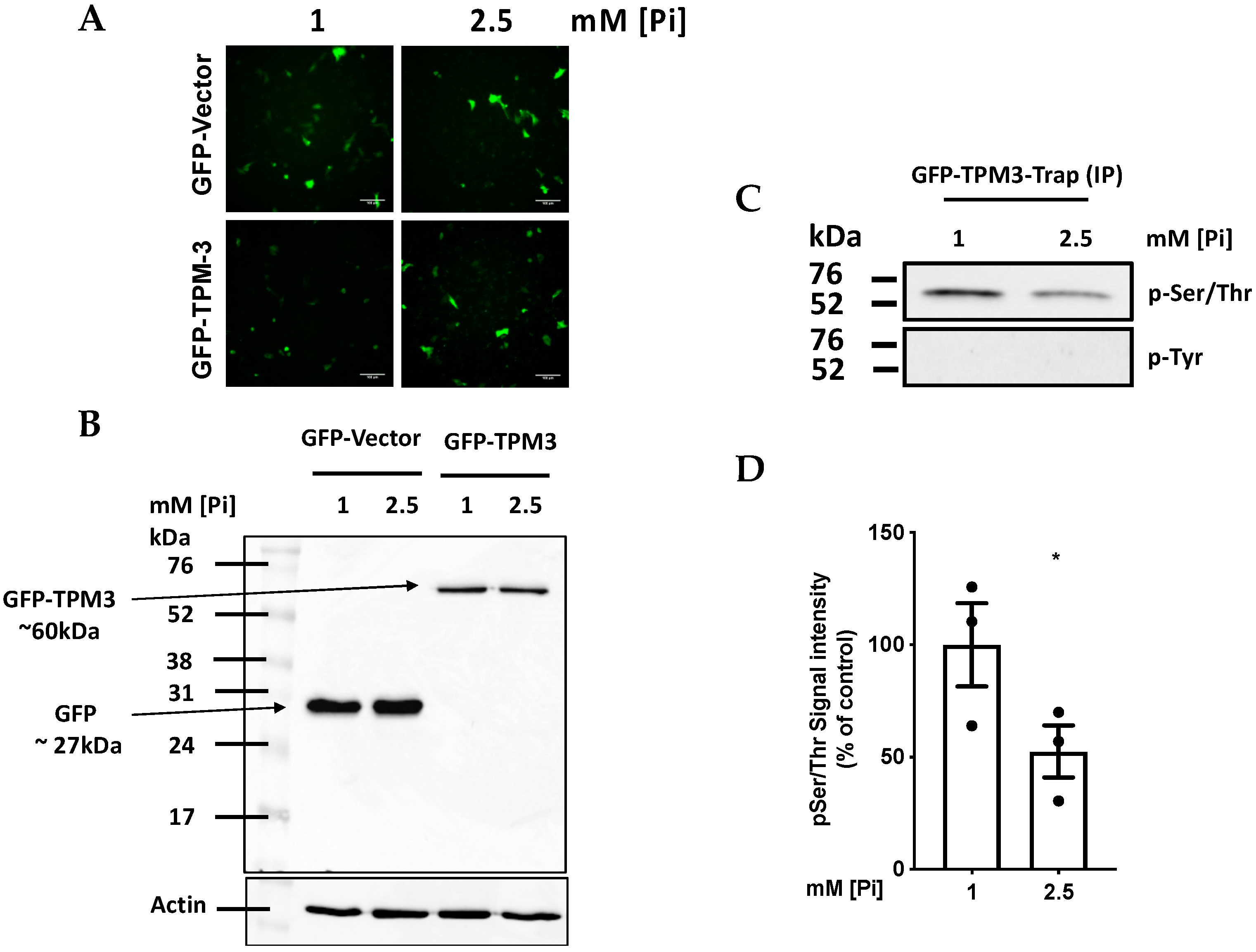{kind=link}
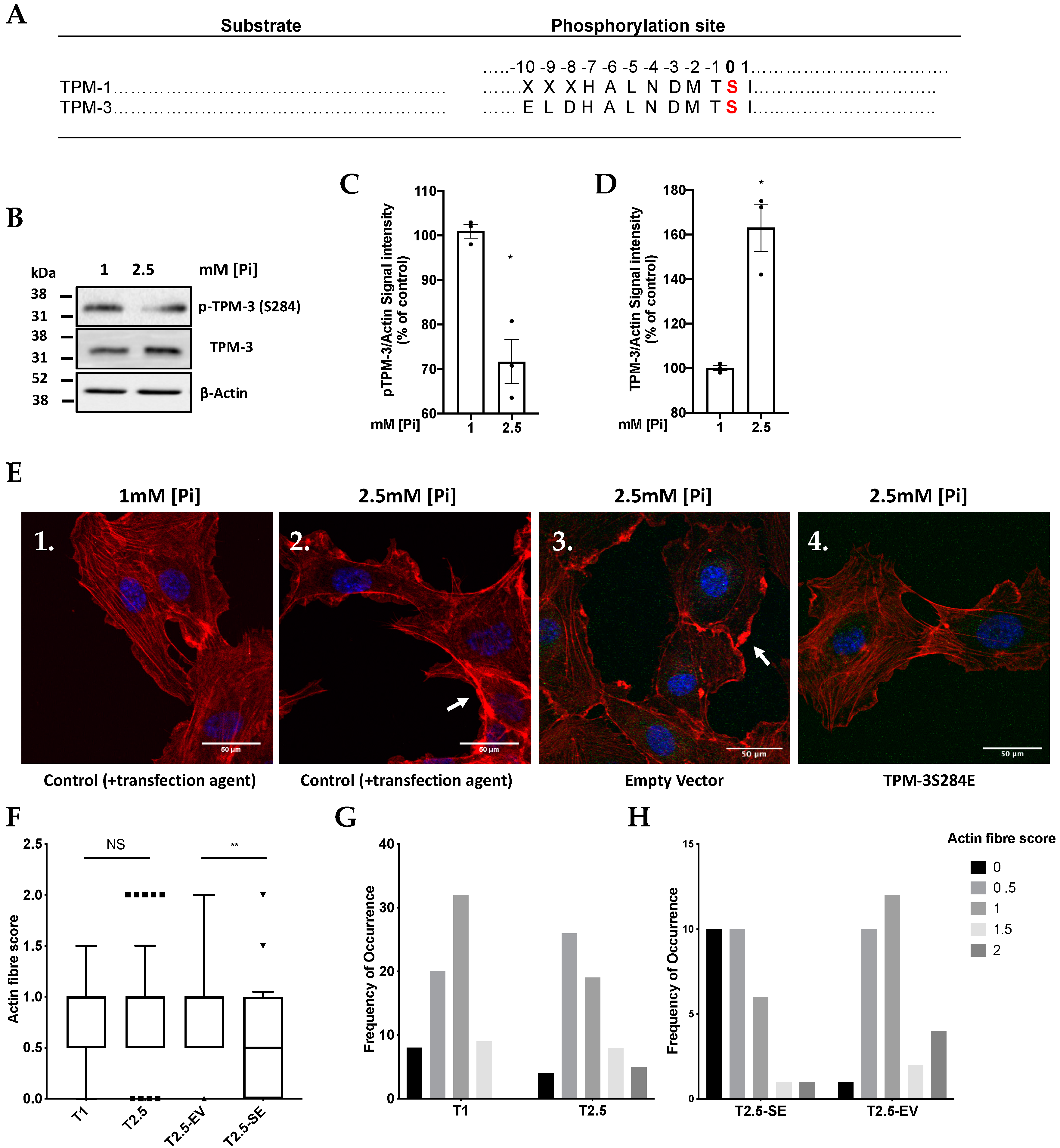{kind=link}
1. Introduction
2. Results
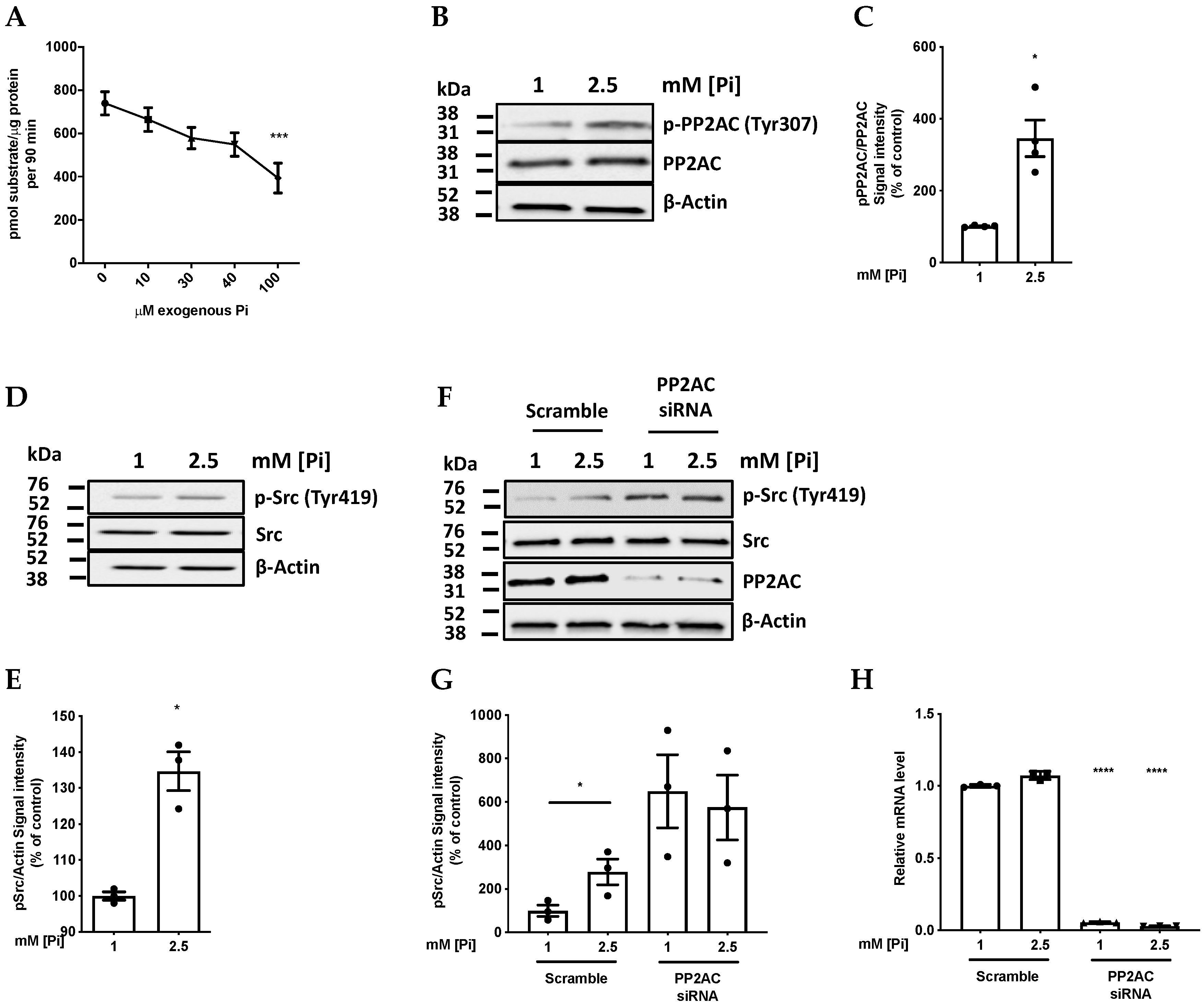
2.1. Pi Directly Inhibits PP2A Catalytic Activity
2.2. High Pi Activates Src and PP2A-C Partial Silencing Preserves Src Phosphorylation
2.3. High Pi Inactivates DAPK-1 by Inducing Phosphorylation at Ser 308
2.4. High Pi induces Hypophosphorylation of TPM-3 on Ser284
2.5. Pi and TPM-3 Phosphorylation Alters Actin Stress-Fibre Organisation within ECs
3. Discussion
3.1. Pi Sensing through Signaling from PP2A to Src
3.2. The Need for Intracellular Amplification in the Sensing of Extracellular Pi by Cells
3.3. Pi Sensing by Proteins Other Than PP2A
3.4. Biological Significance of Pi Signaling through PP2A/Src to DAPK-1
4. Materials and Methods
4.1. Cell Culture and Incubations
4.2. Nanoparticle Tracking Analysis (NTA)
4.3. Plasmids and siRNA
4.4. RNA Extraction and qRT-PCR
4.5. Immunoblotting
4.6. Immunoprecipitation
4.7. GFP-Trap (IP) TPM-3 Coupled Proteomics
4.8. Determination of the Inhibitory Effect of Pi on PP2A Activity
4.9. Confocal Microscopy
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Razzaque, M. Phosphate toxicity: New insights into an old problem. Clin. Sci. 2011, 120, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G. Cholesterol And Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Sullivan, L.M.; Fox, C.S.; Wang, T.J.; D’Agostino, R.B.; Gaziano, J.M.; Vasan, R.S. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch. Intern. Med. 2007, 167, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Ellam, T.J.; Chico, T.J. Phosphate: The new cholesterol? The role of the phosphate axis in non-uremic vascular disease. Atherosclerosis 2012, 220, 310–318. [Google Scholar] [CrossRef]
- Tan, X.; Xu, X.; Zeisberg, E.M.; Zeisberg, M. High inorganic phosphate causes DNMT1 phosphorylation and subsequent fibrotic fibroblast activation. Biochem. Biophys. Res. Commun. 2016, 472, 459–464. [Google Scholar] [CrossRef]
- Lin, Y.; McKinnon, K.E.; Ha, S.W.; Beck, G.R. Inorganic phosphate induces cancer cell mediated angiogenesis dependent on forkhead box protein C2 (FOXC2) regulated osteopontin expression. Mol. Carcinog. 2015, 54, 926–934. [Google Scholar] [CrossRef]
- Wulaningsih, W.; Michaelsson, K.; Garmo, H.; Hammar, N.; Jungner, I.; Walldius, G.; Holmberg, L.; Van Hemelrijck, M. Inorganic phosphate and the risk of cancer in the Swedish AMORIS study. BMC Cancer 2013, 13, 257. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.; Hong, S.; Lee, A.; Park, E.; Seo, H.W.; Chae, C.; Doble, P.; Bishop, D.; Cho, M. High inorganic phosphate intake promotes tumorigenesis at early stages in a mouse model of lung cancer. PLoS ONE 2015, 10, e0135582. [Google Scholar] [CrossRef]
- Tan, X.; Xu, X.; Zeisberg, M.; Zeisberg, E.M. DNMT1 and HDAC2 cooperate to facilitate aberrant promoter methylation in inorganic phosphate-induced endothelial-mesenchymal transition. PLoS ONE 2016, 11, e0147816. [Google Scholar] [CrossRef]
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, phosphoprotein phosphatases, and microparticle release in vascular endothelial cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef]
- Bergwitz, C.; Jüppner, H. Phosphate sensing. Adv. Chronic Kidney Dis. 2011, 18, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Santos-Beneit, F. The pho regulon: A huge regulatory network in bacteria. Front. Microbiol. 2015, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Ticconi, C.A.; Abel, S. Short on phosphate: Plant surveillance and countermeasures. Trends Plant Sci. 2004, 9, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Bon, N.; Couasnay, G.; Bourgine, A.; Sourice, S.; Beck-Cormier, S.; Guicheux, J.; Beck, L. Phosphate (Pi)-regulated heterodimerization of the high-affinity sodium-dependent pi transporters PiT1/Slc20a1 and PiT2/Slc20a2 underlies extracellular pi sensing independently of pi uptake. J. Biol. Chem. 2018, 293, 2102–2114. [Google Scholar] [CrossRef] [PubMed]
- Kemp, G.J.; Bevington, A.; Khodja, D.; Graham, R.; Russell, G. Net fluxes of orthophosphate across the plasma membrane in human red cells following alteration of pH and extracellular Pi concentration. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1988, 969, 148–157. [Google Scholar] [CrossRef]
- Kemp, G.J.; Khouja, H.I.; Ahmado, A.; Graham, R.; Russell, G.; Bevington, A. Regulation of the phosphate (pi) concentration in UMR 106 osteoblast-like cells: Effect of Pi, Na+ and K+. Cell Biochem. Funct. 1993, 11, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Buzalaf, M.A.; Taga, E.M.; Granjeiro, J.M.; Ferreira, C.; Lourencao, V.A.; Ortega, M.M.; Poletto, D.W.; Aoyama, H. Kinetic characterization of bovine lung low-molecular-weight protein tyrosine phosphatase. Exp. Lung Res. 1998, 24, 269–272. [Google Scholar] [CrossRef]
- Szajerka, G.; Kwiatkowska, J. The effect of cortisol on rabbit red cell acid phosphatase isoenzymes. Mol. Cell. Biochem. 1984, 59, 183–186. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; VanEtten, R.L. Pre-steady-state and steady-state kinetic analysis of the low molecular weight phosphotyrosyl protein phosphatase from bovine heart. J. Biol. Chem. 1991, 266, 1516–1525. [Google Scholar] [PubMed]
- Nematullah, M.; Hoda, M.; Khan, F. Protein phosphatase 2A: A double-faced phosphatase of cellular system and its role in neurodegenerative disorders. Mol. Neurobiol. 2017, 55, 1750–1761. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Alrehani, N.; Patel, V.; Khatlani, T.; Vijayan, K.V. Cross-talk between serine/threonine protein phosphatase 2A and protein tyrosine phosphatase 1B regulates src activation and adhesion of integrin alphaIIbbeta3 to fibrinogen. J. Biol. Chem. 2010, 285, 29059–29068. [Google Scholar] [CrossRef] [PubMed]
- Guenebeaud, C.; Goldschneider, D.; Castets, M.; Guix, C.; Chazot, G.; Delloye-Bourgeois, C.; Eisenberg-Lerner, A.; Shohat, G.; Zhang, M.; Laudet, V. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol. Cell 2010, 40, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Widau, R.C.; Jin, Y.; Dixon, S.A.; Wadzinski, B.E.; Gallagher, P.J. Protein phosphatase 2A (PP2A) holoenzymes regulate death-associated protein kinase (DAPK) in ceramide-induced anoikis. J. Biol. Chem. 2010, 285, 13827–13838. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Src protein–tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004, 324, 1155–1164. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Chen, M.; Du, T.; Duan, C.; Gao, G.; Yang, H. The novel mechanism of rotenone-induced α-synuclein phosphorylation via reduced protein phosphatase 2A activity. Int. J. Biochem. Cell Biol. 2016, 75, 34–44. [Google Scholar] [CrossRef]
- Xu, J.; Xu, Z.; Zhou, J.Y.; Zhuang, Z.; Wang, E.; Boerner, J.; Wu, G.S. Regulation of the Src-PP2A interaction in tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J. Biol. Chem. 2013, 288, 33263–33271. [Google Scholar] [CrossRef]
- Bjorge, J.D.; Jakymiw, A.; Fujita, D.J. Selected glimpses into the activation and function of Src kinase. Oncogene 2000, 19, 5620. [Google Scholar] [CrossRef]
- Wang, W.; Kuo, J.; Ku, W.; Lee, Y.; Lin, F.; Chang, Y.; Lin, Y.; Chen, C.; Huang, Y.; Chiang, M. The tumor suppressor DAPK is reciprocally regulated by tyrosine kinase src and phosphatase LAR. Mol. Cell 2007, 27, 701–716. [Google Scholar] [CrossRef]
- Barisic, S.; Schmidt, C.; Walczak, H.; Kulms, D. Tyrosine phosphatase inhibition triggers sustained canonical serine-dependent NFκB activation via src-dependent blockade of PP2A. Biochem. Pharmacol. 2010, 80, 439–447. [Google Scholar] [CrossRef][Green Version]
- Jin, Y.; Blue, E.K.; Gallagher, P.J. Control of death-associated protein kinase (DAPK) activity by phosphorylation and proteasomal degradation. J. Biol. Chem. 2006, 281, 39033–39040. [Google Scholar] [CrossRef]
- Houle, F.; Poirier, A.; Dumaresq, J.; Huot, J. DAP kinase mediates the phosphorylation of tropomyosin-1 downstream of the ERK pathway, which regulates the formation of stress fibers in response to oxidative stress. J. Cell Sci. 2007, 120, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstadt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal. Physiol. 2008, 294, F1381–F1387. [Google Scholar] [CrossRef] [PubMed]
- Peng, A.; Wu, T.; Zeng, C.; Rakheja, D.; Zhu, J.; Ye, T.; Hutcheson, J.; Vaziri, N.D.; Liu, Z.; Mohan, C.; et al. Adverse effects of simulated hyper-and hypo-phosphatemia on endothelial cell function and viability. PLoS ONE 2011, 6, e23268. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Kohler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2012, 83, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.O.; Hamali, H.A.; Singh, R.; Abbasian, N.; Parsons, R.; Patel, A.K.; Goodall, A.H.; Brunskill, N.J. Elevated levels of procoagulant plasma microvesicles in dialysis patients. PLoS ONE 2013, 8, e72663. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Janssens, V.; Van Hoof, C.; Merlevede, W.; Goris, J. PTPA regulating PP2A as a dual specificity phosphatase. Protein Phosphatase Protoc. 1998, 93, 103–115. [Google Scholar]
- Bevington, A.; Mundy, K.; Yates, A.; Kanis, J.; Russell, R. A study of intracellular orthophosphate concentration in human muscle and erythrocytes by 31P nuclear magnetic resonance spectroscopy and selective chemical assay. Clin. Sci. 1986, 71, 729–735. [Google Scholar] [CrossRef]
- Chobanian, M.C.; Anderson, M.E.; Brazy, P.C. An NMR study of cellular phosphates and membrane transport in renal proximal tubules. Am. J. Physiol. 1995, 268, F375–F384. [Google Scholar] [CrossRef]
- Huyer, G.; Liu, S.; Kelly, J.; Moffat, J.; Payette, P.; Kennedy, B.; Tsaprailis, G.; Gresser, M.J.; Ramachandran, C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997, 272, 843–851. [Google Scholar] [CrossRef]
- Luo, Y.; Nie, Y.; Shi, H.; Ni, Z.; Wang, Q.; Wang, J.; Liu, G. PTPA activates protein phosphatase-2A through reducing its phosphorylation at tyrosine-307 with upregulation of protein tyrosine phosphatase 1B. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 1235–1243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shimizu, S.; Ugi, S.; Maegawa, H.; Egawa, K.; Nishio, Y.; Yoshizaki, T.; Shi, K.; Nagai, Y.; Morino, K.; Nemoto, K. Protein-tyrosine phosphatase 1B as new activator for hepatic lipogenesis via sterol regulatory element-binding protein-1 gene expression. J. Biol. Chem. 2003, 278, 43095–43101. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, X.; Yin, Y.; Liu, B.; Luo, D.; Liu, D.; Chen, N.; Ni, Z.; Wang, X.; Wang, Q. Glycogen synthase kinase-3β regulates Tyr307 phosphorylation of protein phosphatase-2A via protein tyrosine phosphatase 1B but not src. Biochem. J. 2011, 437, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Ravanan, P.; Talwar, P. Death associated protein kinase 1 (DAPK1): A regulator of apoptosis and autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef]
- Rath, D.P.; Bailey, M.; Zhang, H.; Jiang, Z.; Abduljalil, A.M.; Weisbrode, S.; Hamlin, R.L.; Robitaille, P.M. 31P-nuclear magnetic resonance studies of chronic myocardial ischemia in the yucatan micropig. J. Clin. Investig. 1995, 95, 151–157. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbasian, N.; Bevington, A.; Burton, J.O.; Herbert, K.E.; Goodall, A.H.; Brunskill, N.J. Inorganic Phosphate (Pi) Signaling in Endothelial Cells: A Molecular Basis for Generation of Endothelial Microvesicles in Uraemic Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 6993. https://doi.org/10.3390/ijms21196993
Abbasian N, Bevington A, Burton JO, Herbert KE, Goodall AH, Brunskill NJ. Inorganic Phosphate (Pi) Signaling in Endothelial Cells: A Molecular Basis for Generation of Endothelial Microvesicles in Uraemic Cardiovascular Disease. International Journal of Molecular Sciences. 2020; 21(19):6993. https://doi.org/10.3390/ijms21196993
Chicago/Turabian StyleAbbasian, Nima, Alan Bevington, James O. Burton, Karl E. Herbert, Alison H. Goodall, and Nigel J. Brunskill. 2020. "Inorganic Phosphate (Pi) Signaling in Endothelial Cells: A Molecular Basis for Generation of Endothelial Microvesicles in Uraemic Cardiovascular Disease" International Journal of Molecular Sciences 21, no. 19: 6993. https://doi.org/10.3390/ijms21196993
APA StyleAbbasian, N., Bevington, A., Burton, J. O., Herbert, K. E., Goodall, A. H., & Brunskill, N. J. (2020). Inorganic Phosphate (Pi) Signaling in Endothelial Cells: A Molecular Basis for Generation of Endothelial Microvesicles in Uraemic Cardiovascular Disease. International Journal of Molecular Sciences, 21(19), 6993. https://doi.org/10.3390/ijms21196993