Dynamical Behavior of the Human Ferroportin Homologue from Bdellovibrio bacteriovorus: Insight into the Ligand Recognition Mechanism


 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
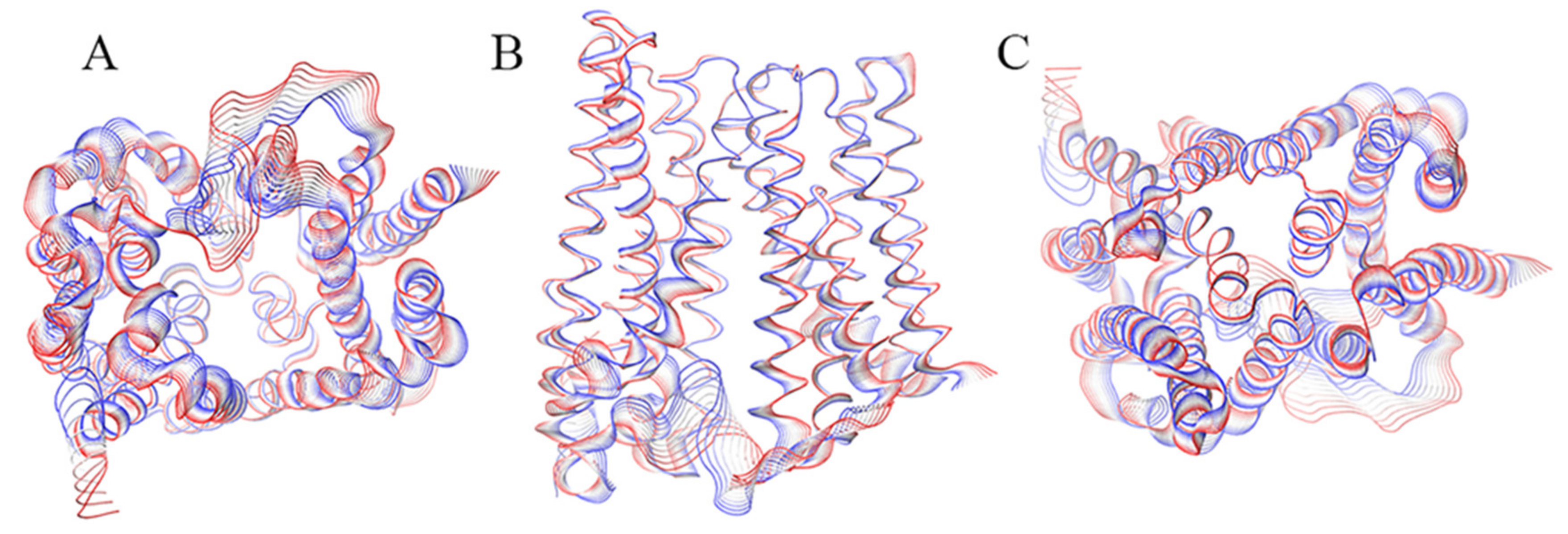
2. Results
2.1. Inward_Apo
- D162-R17—the interaction between D162 (TM 5) and R17 (TM 1) is present in 93.6% of the simulation time for the three replicas;
- D229-R371—the salt-bridge between the D229 (loop TM6-TM7) and R371 (TM 11) can be observed in 97.1% of the simulation time for the three replicas;
- D28-R284—the salt-bridge between D28 and R284, present for about 95.8% of the simulation time for the three replicas, is located in the upper TM region and stabilizes the interaction between the N (TM1) and C (TM8) domains;
- E10-K150—this salt bridge, present in 52.5% of the simulation time for the three replicas, connects the C-terminal end of TM1 with the N-terminal end of TM5;
- E277-H180—this interaction (present in about 62.8% of the simulation time for the three replicas) appears to be essential for establishing the interaction of L-TM5-TM6 with TM8, characterizing the inward-facing conformation.
2.2. Outward_Apo
2.3. Inward_Fe
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MSF | Major Facilitator Superfamily |
| MD | Molecular Dynamics |
| hFpn | Human Ferroportin |
| BdFpn | Bdellovibrio bacteriovorus Ferroportin |
References
- Pao, S.S.; Paulsen, I.T.; Saier, M.H. Major Facilitator Superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Quistgaard, E.M.; Löw, C.; Guettou, F.; Nordlund, P.; Löw, C. Understanding transport by the major facilitator superfamily (MFS): Structures pave the way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Marger, M.D.; Saier, M.H. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem. Sci. 1993, 18, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sands, Z.A.; Biggin, P. A Numbering System for MFS Transporter Proteins. Front. Mol. Biosci. 2016, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Yan, N. Structural advances for the major facilitator superfamily (MFS) transporters. Trends Biochem. Sci. 2013, 38, 151–159. [Google Scholar] [CrossRef]
- Law, C.J.; Maloney, P.C.; Wang, D.-N. Ins and Outs of Major Facilitator Superfamily Antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef]
- Dang, S.; Sun, L.; Huang, Y.; Lu, F.; Liu, Y.; Gong, H.; Wang, J.; Yan, N. Structure of a fucose transporter in an outward-open conformation. Nature 2010, 467, 734–738. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-dependent multidrug efflux systems. Microbiol. Mol. Biol. Rev. 1996, 60, 575–608. [Google Scholar] [CrossRef]
- Heng, J.; Zhao, Y.; Liu, M.; Liu, Y.; Fan, J.; Wang, X.; Zhao, Y.; Zhang, X.C. Substrate-bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res. 2015, 25, 1060–1073. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, Y.; Wang, X.; Fan, J.; Heng, J.; Liu, X.-H.; Feng, W.; Kang, X.; Huang, B.; Liu, J.; et al. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc. Natl. Acad. Sci. USA 2013, 110, 14664–14669. [Google Scholar] [CrossRef]
- Zhang, X.C.; Zhao, Y.; Heng, J.; Jiang, D. Energy coupling mechanisms of MFS transporters. Protein Sci. 2015, 24, 1560–1579. [Google Scholar] [CrossRef] [PubMed]
- Pasrija, R.; Banerjee, D.; Prasad, R. Structure and Function Analysis of CaMdr1p, a Major Facilitator Superfamily Antifungal Efflux Transporter Protein of Candida albicans: Identification of Amino Acid Residues Critical for Drug/H+ Transport. Eukaryot. Cell 2007, 6, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Adler, J.; Bibi, E. Promiscuity in the Geometry of Electrostatic Interactions between theEscherichia coliMultidrug Resistance Transporter MdfA and Cationic Substrates. J. Biol. Chem. 2004, 280, 2721–2729. [Google Scholar] [CrossRef] [PubMed]
- Sigal, N.; Fluman, N.; Siemion, S.; Bibi, E. The Secondary Multidrug/Proton Antiporter MdfA Tolerates Displacements of an Essential Negatively Charged Side Chain. J. Biol. Chem. 2009, 284, 6966–6971. [Google Scholar] [CrossRef] [PubMed]
- Fluman, N.; Ryan, C.M.; Whitelegge, J.P.; Bibi, E. Dissection of Mechanistic Principles of a Secondary Multidrug Efflux Protein. Mol. Cell 2012, 47, 777–787. [Google Scholar] [CrossRef]
- Edgar, R.; Bibi, E. A single membrane-embedded negative charge is critical for recognizing positively charged drugs by the Escherichia coli multidrug resistance protein MdfA. EMBO J. 1999, 18, 822–832. [Google Scholar] [CrossRef]
- Jardetzky, O. Simple Allosteric Model for Membrane Pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef]
- Huang, Y.; Lemieux, M.J.; Song, J.; Auer, M.; Wang, D.N. Structure and Mechanism of the Glycerol-3-Phosphate Transporter from Escherichia coli. Science 2003, 301, 616–620. [Google Scholar] [CrossRef]
- Di Patti, M.C.B.; Polticelli, F.; Cece, G.; Cutone, A.; Felici, F.; Persichini, T.; Musci, G. A structural model of human ferroportin and of its iron binding site. FEBS J. 2014, 281, 2851–2860. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1426–1433. [Google Scholar]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Billesbølle, C.B.; Azumaya, C.M.; Kretsch, R.C.; Powers, A.S.; Gonen, S.; Schneider, S.; Arvedson, T.; Dror, R.O.; Cheng, Y.; Manglik, A. Structure of human ferroportin bound to hepcidin reveals mechanisms of iron homeostasis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Di Patti, M.C.B.; Polticelli, F.; Tortosa, V.; Furbetta, P.A.; Musci, G. A bacterial homologue of the human iron exporter ferroportin. FEBS Lett. 2015, 589, 3829–3835. [Google Scholar] [CrossRef]
- Taniguchi, R.; Kato, H.E.; Font, J.; Deshpande, C.N.; Wada, M.; Ito, K.; Ishitani, R.; Jormakka, M.; Nureki, O. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Tortosa, V.; Di Patti, M.C.B.; Brandi, V.; Musci, G.; Polticelli, F. An improved structural model of the human iron exporter ferroportin. Insight into the role of pathogenic mutations in hereditary hemochromatosis type 4. Bio-Algorithms Med-Syst. 2017, 13, 215–222. [Google Scholar] [CrossRef]
- Guellec, J.; Elbahnsi, A.; Le Tertre, M.; Uguen, K.; Gourlaouen, I.; Férec, C.; Ka, C.; Callebaut, I.; Le Gac, G. Molecular model of the ferroportin intracellular gate and implications for the human iron transport cycle and hemochromatosis type 4A. FASEB J. 2019, 33, 14625–14635. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [PubMed]
- Case, D.A.; Iii, T.E.C.; Darden, T.; Gohlke, H.; Luo, R., Jr.; Luo, R.; Kenneth, M.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; et al. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C.L. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, J.K.M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Brünger, A.; Brooks, C.L.; Karplus, M. Stochastic boundary conditions for molecular dynamics simulations of ST2 water. Chem. Phys. Lett. 1984, 105, 495–500. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, U.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Abraham, M.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 19–25. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, I.T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-S. Molecular Dynamics Simulations of the Human Glucose Transporter GLUT1. PLoS ONE 2015, 10, 1–18. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Duration (ns) | Conformational State | Iron | Number of Simulations |
|---|---|---|---|---|
| Inward_Apo | 200 | Inward-facing | - | 3 |
| Outward_Apo | 200 | Outward-facing | - | 3 |
| Inward_Fe | 200 | Inward-facing | + | 3 |
| Residue | Occupancy |
|---|---|
| D24 | 18.2% |
| D134 | 33.3% |
| S130 | 18.3% |
| E203 | 11.1% |
| D229 | 13.8% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tortosa, V.; Bonaccorsi di Patti, M.C.; Iacovelli, F.; Pasquadibisceglie, A.; Falconi, M.; Musci, G.; Polticelli, F. Dynamical Behavior of the Human Ferroportin Homologue from Bdellovibrio bacteriovorus: Insight into the Ligand Recognition Mechanism. Int. J. Mol. Sci. 2020, 21, 6785. https://doi.org/10.3390/ijms21186785
Tortosa V, Bonaccorsi di Patti MC, Iacovelli F, Pasquadibisceglie A, Falconi M, Musci G, Polticelli F. Dynamical Behavior of the Human Ferroportin Homologue from Bdellovibrio bacteriovorus: Insight into the Ligand Recognition Mechanism. International Journal of Molecular Sciences. 2020; 21(18):6785. https://doi.org/10.3390/ijms21186785
Chicago/Turabian StyleTortosa, Valentina, Maria Carmela Bonaccorsi di Patti, Federico Iacovelli, Andrea Pasquadibisceglie, Mattia Falconi, Giovanni Musci, and Fabio Polticelli. 2020. "Dynamical Behavior of the Human Ferroportin Homologue from Bdellovibrio bacteriovorus: Insight into the Ligand Recognition Mechanism" International Journal of Molecular Sciences 21, no. 18: 6785. https://doi.org/10.3390/ijms21186785
APA StyleTortosa, V., Bonaccorsi di Patti, M. C., Iacovelli, F., Pasquadibisceglie, A., Falconi, M., Musci, G., & Polticelli, F. (2020). Dynamical Behavior of the Human Ferroportin Homologue from Bdellovibrio bacteriovorus: Insight into the Ligand Recognition Mechanism. International Journal of Molecular Sciences, 21(18), 6785. https://doi.org/10.3390/ijms21186785