The Epigenetic Overlap between Obesity and Mood Disorders: A Systematic Review

 , ,
, ,
Abstract
1. Introduction
2. Methods
2.1. Search Strategy
2.1.1. Step 1: Identification of Candidate Genes for Obesity
2.1.2. Step 2: Exploration of the Role of Differentiated Methylated Obesity Genes in Mood Disorders
2.1.3. Search Term
2.2. Screening
2.3. Inclusion Criteria
2.4. Exclusion Criteria
2.5. The Following Outcome Measures Were Considered
2.6. Types of Tissue Samples Included in the Review
2.7. Format of Data Input for Factors
3. Results
4. Discussion
5. Limitation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ArgBP2 | Arg/c-Abl kinase binding protein 2 |
| BMI | body mass index |
| DMR | differentiated methylated regions |
| ER | endoplasmic reticulum |
| EWAS | Epigenome wide association study |
| IRS1 | insulin receptor substrate 1 |
| MDD | Major Depressive Disorder |
| MHC | major histocompatibility complex |
| OVAT | omental visceral adipose tissue |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
| PROSPERO | Prospective Register of Systematic Reviews |
| SOHOs | orbin homology |
| TAP | transporter associated with antigen processing |
References
- Popkin, B.M.; Adair, L.S.; Ng, S.W. Global nutrition transition and the pandemic of obesity in developing countries. Nutr. Rev. 2012, 70, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.; Yang, W.; Chen, C.-S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; Aguilar-Gaxiola, S.; Alonso, J.; Chatterji, S.; Lee, S.; Ormel, J.; Üstün, T.B.; Wang, P.S. The global burden of mental disorders: An update from the WHO World Mental Health (WMH) surveys. Epidemiologia e Psichiatr. Soc. 2009, 18, 23–33. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Veisani, Y.; Mohamadian, F.; Delpisheh, A. Prevalence and comorbidity of common mental disorders and associations with suicidal ideation in the adult population. Epidemiol. Heal. 2017, 39, e2017031. [Google Scholar] [CrossRef] [PubMed]
- Palou, A.; Serra, F.; Bonet, M.; Picó, C. Obesity: Molecular bases of a multifactorial problem. Eur. J. Nutr. 2000, 39, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Akiskal, H.S. New insights into the nature and heterogeneity of mood disorders. J. Clin. Psychiatry 1989, 50, 50. [Google Scholar]
- Morris, W.N.; Reilly, N.P. Toward the self-regulation of mood: Theory and research. Motiv. Emot. 1987, 11, 215–249. [Google Scholar] [CrossRef]
- Afari, N.; Noonan, C.; Goldberg, J.; Roy-Byrne, P.; Schur, E.; Golnari, G.; Buchwald, D. Depression and obesity: Do shared genes explain the relationship? Depression Anxiety 2010, 27, 799–806. [Google Scholar] [CrossRef]
- Khan, A.; Schwartz, K.A.; Kolts, R.L.; Brown, W.A. BMI, sex, and antidepressant response. J. Affect. Disord. 2007, 99, 101–106. [Google Scholar] [CrossRef]
- Hinze-Selch, D.; Schuld, A.; Kraus, T.; Kühn, M.; Uhr, M.; Haack, M.; Pollmacher, T. Effects of Antidepressants on Weight and on the Plasma Levels of Leptin, TNF-α and Soluble TNF Receptors A Longitudinal Study in Patients Treated with Amitriptyline or Paroxetine. Neuropsychopharmacol 2000, 23, 13–19. [Google Scholar] [CrossRef]
- Gafoor, R.; Booth, H.P.; Gulliford, M.C. Antidepressant utilisation and incidence of weight gain during 10 years’ follow-up: Population based cohort study. BMJ 2018, 361, k1951. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; EpiSCOPE, M.O.; Molloy, P.L.; Varinli, H.; Morrison, J.L.; Muhlhausler, B.S. Epigenetics and human obesity. Int. J. Obes. 2014, 39, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.M.; Schroeder, F.A.; Perlis, R.H.; Haggarty, S.J. Epigenetic mechanisms in mood disorders: Targeting neuroplasticity. Neuroscience 2013, 264, 112–130. [Google Scholar] [CrossRef] [PubMed]
- Schones, D.E.; Leung, A.; Natarajan, R. Chromatin Modifications Associated with Diabetes and Obesity. Arter. Thromb. Vasc. Boil. 2015, 35, 1557–1561. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, K.; Molina-Márquez, A.M.; Saavedra, N.; Zambrano, T.; Salazar, L.A. Epigenetic Modifications of Major Depressive Disorder. Int. J. Mol. Sci. 2016, 17, 1279. [Google Scholar] [CrossRef]
- Alegría-Torres, J.A.; Baccarelli, A.A.; Bollati, V. Epigenetics and lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef]
- Attwood, J.; Yung, R.; Richardson, B. DNA methylation and the regulation of gene transcription. Cell. Mol. Life Sci. 2002, 59, 241–257. [Google Scholar] [CrossRef]
- Kouzmenko, A.; Ohtake, F.; Fujiki, R.; Kato, S. Hormonal gene regulation through DNA methylation and demethylation. Epigenomics 2010, 2, 765–774. [Google Scholar] [CrossRef]
- Sayols-Baixeras, S.; Subirana, I.; Fernández-Sanlés, A.; Sentí, M.; Lluís-Ganella, C.; Marrugat, J.; Elosua, R. DNA methylation and obesity traits: An epigenome-wide association study. The REGICOR study. Epigenetics 2017, 12, 909–916. [Google Scholar] [CrossRef]
- Sonne, S.B.; Yadav, R.; Yin, G.; Dalgaard, M.D.; Myrmel, L.S.; Gupta, R.; Wang, J.; Madsen, L.; Kajimura, S.; Kristiansen, K. Obesity is associated with depot-specific alterations in adipocyte DNA methylation and gene expression. Adipocyte 2017, 6, 124–133. [Google Scholar] [CrossRef]
- De Mello, V.D.; Pulkkinen, L.; Lalli, M.; Kolehmainen, M.; Pihlajamäki, J.; Uusitupa, M. DNA methylation in obesity and type 2 diabetes. Ann. Med. 2014, 46, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kader, F.; Ghai, M.; Maharaj, L. The effects of DNA methylation on human psychology. Behav. Brain Res. 2018, 346, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Meng, L.; Pei, F.; Zheng, Y.; Leng, J. A review of DNA methylation in depression. J. Clin. Neurosci. 2017, 43, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Downs, S.H.; Black, N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J. Epidemiol. Community Heal. 1998, 52, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Monteiro, C.; Matos, A.; You, J.; Fraga, A.; Pereira, C.; Catalán, V.; Rodríguez, A.; Gómez-Ambrosi, J.; Frühbeck, G.; et al. Epigenome-wide DNA methylation profiling of periprostatic adipose tissue in prostate cancer patients with excess adiposity—A pilot study. Clin. Epigenetics 2018, 10, 54. [Google Scholar] [CrossRef]
- Murphy, T.M.; Crawford, B.; Dempster, E.L.; Hannon, E.; Burrage, J.; Turecki, G.; Kaminsky, Z.; Mill, J. Methylomic profiling of cortex samples from completed suicide cases implicates a role for PSORS1C3 in major depression and suicide. Transl. Psychiatry 2017, 7, e989. [Google Scholar] [CrossRef]
- Martin, C.L.; Jima, D.; Sharp, G.C.; McCullough, L.E.; Park, S.S.; Gowdy, K.M.; Skaar, D.; Cowley, M.; Maguire, R.L.; Fuemmeler, B.; et al. Maternal pre-pregnancy obesity, offspring cord blood DNA methylation, and offspring cardiometabolic health in early childhood: An epigenome-wide association study. Epigenetics 2019, 14, 325–340. [Google Scholar] [CrossRef]
- Rhee, J.K.; Lee, J.H.; Yang, H.K.; Kim, T.M.; Yoon, K.H. DNA Methylation profiles of blood cells are distinct between early-onset obese and control individuals. Genom. Inform. 2017, 15, 28. [Google Scholar] [CrossRef]
- Zhu, Y.; Strachan, E.; Fowler, E.; Bacus, T.; Roy-Byrne, P.; Zhao, J. Genome-Wide profiling of DNA methylome and transcriptome in peripheral blood monocytes for major depression: A Monozygotic Discordant Twin Study. Transl. Psychiatry 2019, 9, 215. [Google Scholar] [CrossRef]
- Keller, S.; Sarchiapone, M.; Zarrilli, F.; Videtic, A.; Ferraro, A.; Carli, V.; Sacchetti, S.; Lembo, F.; Angiolillo, A.; Jovanović, N.; et al. Increased BDNF Promoter Methylation in the Wernicke Area of Suicide Subjects. Arch. Gen. Psychiatry 2010, 67, 258–267. [Google Scholar] [CrossRef]
- Perroud, N.; Salzmann, A.; Prada, P.; Nicastro, R.; Hoeppli, M.E.; Furrer, S.; Ardu, S.; Krejci, I.; Karege, F.; Malafosse, A. Response to psychotherapy in borderline personality disorder and methylation status of the BDNF gene. Transl. Psychiatry 2013, 3, e207. [Google Scholar] [CrossRef] [PubMed]
- Januar, V.; Ancelin, M.-L.; Ritchie, K.; Saffery, R.; Ryan, J. BDNF promoter methylation and genetic variation in late-life depression. Transl. Psychiatry 2015, 5, e619. [Google Scholar] [CrossRef] [PubMed]
- Voisin, S.; Almén, M.S.; Zheleznyakova, G.Y.; Lundberg, L.; Zarei, S.; Castillo, S.; Eriksson, F.E.; Nilsson, E.K.; Blüher, M.; Böttcher, Y.; et al. Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 2015, 7, 1–6. [Google Scholar]
- Gardner, K.; Sapienza, C.; Fisher, J.J.P. Genetic and epigenetic associations to obesity-related appetite phenotypes among A frican–A merican children. Pediatric Obesity 2015, 10, 476–482. [Google Scholar] [CrossRef]
- Chapman, D.P.; Perry, G.S. Peer reviewed: Depression as a major component of public health for older adults. Prev. Chron. Dis. 2008, 5, 1. [Google Scholar]
- Nigatu, Y.T.; Reijneveld, S.A.; De Jonge, P.; Van Rossum, E.; Bültmann, U. The Combined Effects of Obesity, Abdominal Obesity and Major Depression/Anxiety on Health-Related Quality of Life: The LifeLines Cohort Study. PLoS ONE 2016, 11, e0148871. [Google Scholar] [CrossRef]
- Chen, K.-W.; Chen, L. Epigenetic Regulation of BDNF Gene during Development and Diseases. Int. J. Mol. Sci. 2017, 18, 571. [Google Scholar] [CrossRef]
- Mayer, W.E.; Klein, J. Is tapasin a modified Mhc class I molecule? Immunogenetics 2001, 53, 719–723. [Google Scholar] [CrossRef]
- Herberg, J.A.; Sgouros, J.; Jones, T.; Copeman, J.; Humphray, S.J.; Sheer, D. Genomic analysis of the Tapasin gene, located close to the TAP loci in the MHC. Eur. J. Immunol. 1998, 28, 459–467. [Google Scholar] [CrossRef]
- Montfort, A.; Martin, P.G.; Levade, T.; Benoist, H.; Ségui, B. FAN (factor associated with neutral sphingomyelinase activation), a moonlighting protein in TNF-R1 signaling. J. Leukoc. Boil. 2010, 88, 897–903. [Google Scholar] [CrossRef]
- Cui, D.; Wang, J.; Zeng, Y.; Rao, L.; Chen, H.; Li, W.; Li, Y.; Li, H.; Cui, C.; Xiao, L. Generating hESCs with reduced immunogenicity by disrupting TAP1 or TAPBP. Biosci. Biotechnol. Biochem. 2016, 80, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Abarca-Heidemann, K.; Friederichs, S.; Klamp, T.; Boehm, U.; Guethlein, L.A.; Ortmann, B. Regulation of the expression of mouse TAP-associated glycoprotein (tapasin) by cytokines. Immunol. Lett. 2002, 83, 197–207. [Google Scholar] [CrossRef]
- Lukong, K.E.; Chang, K.-W.; Khandjian, E.W.; Richard, S. RNA-Binding proteins in human genetic disease. Trends Genet. 2008, 24, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Tanaka, S.; Ishii, A.; Watanabe, M.; Fujitani, N.; Sugeo, A. A brain-specific Grb2-associated regulator of extracellular signal-regulated kinase (Erk)/mitogen-activated protein kinase (MAPK)(GAREM) subtype, GAREM2, contributes to neurite outgrowth of neuroblastoma cells by regulating Erk signaling. J. Cell Sci. 2013, 288, 29934. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bei, Y.; Shen, S.; Huang, P.; Shi, J.; Zhang, J.; Sun, Q.; Chen, Y.; Yang, Y.; Xu, T.; et al. miR-21-3p controls sepsis-associated cardiac dysfunction via regulating SORBS2. J. Mol. Cell. Cardiol. 2016, 94, 43–53. [Google Scholar] [CrossRef]
- Zhang, Q.; Gao, X.; Li, C.; Feliciano, C.; Wang, N.; Zhou, D.; Mei, Y.; Monteiro, P.; Anand, M.; Itohara, S.; et al. Impaired Dendritic Development and Memory in Sorbs2 Knock-Out Mice. J. Neurosci. 2016, 36, 2247–2260. [Google Scholar] [CrossRef]
- Lu, P.; Qiao, J.; He, W.; Wang, J.; Jia, Y.; Sun, Y.; Tang, S.; Fu, L.; Qin, Y. Genome-Wide Gene Expression Profile Analyses Identify CTTN as a Potential Prognostic Marker in Esophageal Cancer. PLoS ONE 2014, 9, e88918. [Google Scholar] [CrossRef]
- Cheli, S.; Francois, S.; Bodega, B.; Ferrari, F.; Tenedini, E.; Roncaglia, E.; Ferrari, S.; Ginelli, E.; Meneveri, R. Expression Profiling of FSHD-1 and FSHD-2 Cells during Myogenic Differentiation Evidences Common and Distinctive Gene Dysregulation Patterns. PLoS ONE 2011, 6, e20966. [Google Scholar] [CrossRef]
- McCarthy, M.J.; Leckband, S.G.; Kelsoe, J.R. Pharmacogenetics of lithium response in bipolar disorder. Pharmacogenomics 2010, 11, 1439–1465. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Hopp, L.; Liu, X.; Wohland, T.; Rohde, K.; Cancello, R.; Klös, M.; Bacos, K.; Kern, M.; Eichelmann, F.; et al. Genome-wide DNA promoter methylation and transcriptome analysis in human adipose tissue unravels novel candidate genes for obesity. Mol. Metab. 2017, 6, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Lorente, R.; Bejar, M.T.; Badimon, L. Notch Signaling Pathway Activation in Normal and Hyperglycemic Rats Differs in the Stem Cells of Visceral and Subcutaneous Adipose Tissue. Stem Cells Dev. 2014, 23, 3034–3048. [Google Scholar] [CrossRef] [PubMed]
- Fung, E.; Tang, S.-M.T.; Canner, J.P.; Morishige, K.; Arboleda-Velasquez, J.F.; Cardoso, A.A.; Carlesso, N.; Aster, J.C.; Aikawa, E. Delta-Like 4 Induces Notch Signaling in Macrophages. Circulation 2007, 115, 2948–2956. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, B.; Deng, Y.; Shangguan, S.; Zhou, F.; Zhou, W.; Li, X.; Li, Y.; Chen, G. Notch signaling in cerebrovascular diseases (Review). Mol. Med. Rep. 2016, 14, 2883–2898. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, E.P.; Speer, M.C.; Chen, Y.; Steffens, D.C.; Cassidy, F.; Van Meter, S.; Provenzale, J.M.; Weisler, R.H.; Krishnan, K.R.R. Investigation of Notch3 as a candidate gene for bipolar disorder using brain hyperintensities as an endophenotype. Am. J. Med. Genet. 2002, 114, 652–658. [Google Scholar] [CrossRef]
- Aoyama, T.; Takeshita, K.; Kikuchi, R.; Yamamoto, K.; Cheng, X.W.; Liao, J.K.; Murohara, T. γ-Secretase inhibitor reduces diet-induced atherosclerosis in apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2009, 383, 216–221. [Google Scholar] [CrossRef]
- Ando, K.; Kanazawa, S.; Tetsuka, T.; Ohta, S.; Jiang, X.; Tada, T.; Kobayashi, M.; Matsui, N.; Okamoto, T. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene 2003, 22, 7796–7803. [Google Scholar] [CrossRef]
- Hu, X.; Chung, A.Y.; Wu, I.; Foldi, J.; Chen, J.; Ji, J.D.; Tateya, T.; Kang, Y.J.; Han, J.; Gessler, M.; et al. Integrated Regulation of Toll-like Receptor Responses by Notch and Interferon-γ Pathways. Immunity 2008, 29, 691–703. [Google Scholar] [CrossRef]
- Hanson, I.M.; Seawright, A.; Van Heyningen, V. The human BDNF gene maps between FSHB and HVBS1 at the boundary of 11p13–p14. Genomics 1992, 13, 1331–1333. [Google Scholar] [CrossRef]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef]
- Gray, J.; Yeo, G.S.H.; Cox, J.J.; Morton, J.; Adlam, A.; Keogh, J.M.; A Yanovski, J.; El Gharbawy, A.; Han, J.C.; Tung, Y.L.; et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 2006, 55, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorso, S.; Sodhi, M.; Li, J.; Bobo, W.V.; Chen, Y.; Tumuklu, M.; Theleritis, C.; Jayathilake, K.; Meltzer, H.Y. The brain-derived neurotrophic factor (BDNF) Val66Met polymorphism is associated with increased body mass index and insulin resistance measures in bipolar disorder and schizophrenia. Bipolar. Disord. 2015, 17, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.; Wynne, K.; McGowan, B.; Bloom, S.R. Hormonal Regulation of Food Intake. Physiol. Rev. 2005, 85, 1131–1158. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.T.; Weickert, C.S.; Wyatt, E.; Webster, M.J. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry Neurosci. 2011, 36, 195–203. [Google Scholar] [CrossRef]
- Martinowich, K.; Manji, H.; Lu, B. New insights into BDNF function in depression and anxiety. Nat. Neurosci. 2007, 10, 1089–1093. [Google Scholar] [CrossRef]
- Jin, Y.; Sun, L.H.; Yang, W.; Cui, R.J.; Xu, S.B. The Role of BDNF in the Neuroimmune Axis Regulation of Mood Disorders. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Polyakova, M.; Stuke, K.; Schuemberg, K.; Mueller, K.; Schoenknecht, P.; Schroeter, M.L. BDNF as a biomarker for successful treatment of mood disorders: A systematic & quantitative meta-analysis. J. Affect. Disord. 2015, 174, 432–440. [Google Scholar] [CrossRef]
- Fernandes, B.S.; Molendijk, M.L.; Köhler, C.A.; Soares, J.C.; Leite, C.M.G.S.; Machado-Vieira, R.; Ribeiro, T.L.; Silva, J.C.; Sales, P.M.G.; Quevedo, J.; et al. Peripheral brain-derived neurotrophic factor (BDNF) as a biomarker in bipolar disorder: A meta-analysis of 52 studies. BMC Med. 2015, 13, 289. [Google Scholar] [CrossRef]
- Sanada, K.; Zorrilla, I.; Iwata, Y.; Bermúdez-Ampudia, C.; Graff-Guerrero, A.; Martínez-Cengotitabengoa, M. The efficacy of non-pharmacological interventions on brain-derived neurotrophic factor in schizophrenia: A systematic review and meta-analysis. Int. J. Mol. Sci. 2016, 17, 1766. [Google Scholar] [CrossRef]
- Green, M.J.; Matheson, S.L.; Shepherd, A.; Weickert, C.S.; Carr, V.J. Brain-Derived neurotrophic factor levels in schizophrenia: A systematic review with meta-analysis. Mol. Psychiatry 2010, 16, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Jin, Y.; Wang, J.; Weng, X.; Li, C. Serum brain-derived neurotrophic factor (BDNF) levels in schizophrenia: A systematic review. Shanghai Arch. Psychiatry 2012, 24, 250–261. [Google Scholar] [PubMed]
- Fernandes, B.S.; Steiner, J.; Berk, M.; Molendijk, M.L.; Gonzàlez-Pinto, A.; Turck, C.W.; Nardin, P.; Gonçalves, C.-A. Peripheral brain-derived neurotrophic factor in schizophrenia and the role of antipsychotics: Meta-Analysis and implications. Mol. Psychiatry 2014, 20, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Papathanassoglou, E.D.; Miltiadous, P.; Karanikola, M.N. May BDNF Be Implicated in the Exercise-Mediated Regulation of Inflammation? Critical Review and Synthesis of Evidence. Boil. Res. Nurs. 2014, 17, 521–539. [Google Scholar] [CrossRef]
- Zhang, J.-C.; Yao, W.; Hashimoto, K. Brain-Derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr. Neuropharmacol. 2016, 14, 721–731. [Google Scholar] [CrossRef]
- Chaldakov, G.N.; Fiore, M.; Stankulov, I.S.; Manni, L.; Hristova, M.G.; Antonelli, A.; Ghenev, P.I.; Aloe, L. Neurotrophin presence in human coronary atherosclerosis and metabolic syndrome: A role for NGF and BDNF in cardiovascular disease? In Progress in Brain Research; Elsevier BV: Amsterdam, The Netherlands, 2004; Volume 146, pp. 279–289. [Google Scholar]
- Sandrini, L.; Di Minno, G.; Amadio, P.; Ieraci, A.; Tremoli, E.; Barbieri, S.S. Association between Obesity and Circulating Brain-Derived Neurotrophic Factor (BDNF) Levels: Systematic Review of Literature and Meta-Analysis. Int. J. Mol. Sci. 2018, 19, 2281. [Google Scholar] [CrossRef]
- Silviera, M.L.; Smith, B.P.; Powell, J.; Sapienza, C. Epigenetic differences in normal colon mucosa of cancer patients suggest altered dietary metabolic pathways. Cancer Prev. Res. 2012, 5, 374–384. [Google Scholar] [CrossRef]
- Marosi, K.; Mattson, M.P. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol. Metab. 2014, 25, 89–98. [Google Scholar] [CrossRef]
- Martínez-Levy, G.A.; Cruz-Fuentes, C.S. Genetic and Epigenetic Regulation of the Brain-Derived Neurotrophic Factor in the Central Nervous System. Yale J. Boil. Med. 2014, 87, 173–186. [Google Scholar]
- Tsankova, N.M.; Berton, O.; Renthal, W.; Kumar, A.; Neve, R.L.; Nestler, E.J. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006, 9, 519–525. [Google Scholar] [CrossRef]
- Fuchikami, M.; Morinobu, S.; Segawa, M.; Okamoto, Y.; Yamawaki, S.; Ozaki, N.; Inoue, T.; Kusumi, I.; Koyama, T.; Tsuchiyama, K.; et al. DNA Methylation Profiles of the Brain-Derived Neurotrophic Factor (BDNF) Gene as a Potent Diagnostic Biomarker in Major Depression. PLoS ONE 2011, 6, e23881. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-J.; Kim, J.-M.; Lee, J.-Y.; Kim, S.-Y.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Kim, H.-R.; Shin, M.-G.; Yoon, J.-S. BDNF promoter methylation and suicidal behavior in depressive patients. J. Affect. Disord. 2013, 151, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Chiavetto, L.B.; Bagnardi, V.; Zanardini, R.; Molteni, R.; Nielsen, M.G.; Placentino, A.; Giovannini, C.; Rillosi, L.; Ventriglia, M.; Riva, M.A.; et al. Serum and plasma BDNF levels in major depression: A replication study and meta-analyses. World J. Boil. Psychiatry 2010, 11, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Kurita, M.; Nishino, S.; Kato, M.; Numata, Y.; Sato, T. Plasma Brain-Derived Neurotrophic Factor Levels Predict the Clinical Outcome of Depression Treatment in a Naturalistic Study. PLoS ONE 2012, 7, e39212. [Google Scholar] [CrossRef]
- Lee, B.-H.; Kim, H.; Park, S.-H.; Kim, Y.-K. Decreased plasma BDNF level in depressive patients. J. Affect. Disord. 2007, 101, 239–244. [Google Scholar] [CrossRef]
- Tripp, A.; Oh, H.; Guilloux, J.-P.; Martinowich, K.; Lewis, D.A.; Sibille, E. Brain-Derived Neurotrophic Factor Signaling and Subgenual Anterior Cingulate Cortex Dysfunction in Major Depressive Disorder. Am. J. Psychiatry 2012, 169, 1194–1202. [Google Scholar] [CrossRef]
- Kim, J.-M.; Stewart, R.J.; Glozier, N.; Prince, M.; Kim, S.-W.; Yang, S.-J.; Shin, I.-S.; Yoon, J.-S. Physical health, depression and cognitive function as correlates of disability in an older Korean population. Int. J. Geriatr. Psychiatry 2005, 20, 160–167. [Google Scholar] [CrossRef]
- Kim, J.-M.; Stewart, R.J.; Kang, H.-J.; Kim, S.Y.; Kim, S.-W.; Shin, I.-S.; Park, M.-S.; Kim, H.-R.; Shin, M.-G.; Cho, K.-H.; et al. A longitudinal study of BDNF promoter methylation and genotype with poststroke depression. J. Affect. Disord. 2013, 149, 93–99. [Google Scholar] [CrossRef]
- Jin, H.-J.; Pei, L.; Li, Y.-N.; Zheng, H.; Yang, S.; Wan, Y.; Mao, L.; Xia, Y.-P.; He, Q.-W.; Li, M.; et al. Alleviative effects of fluoxetine on depressive-like behaviors by epigenetic regulation of BDNF gene transcription in mouse model of post-stroke depression. Sci. Rep. 2017, 7, 14926. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-Derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef]
- Crowder, R.J.; Freeman, R.S. Phosphatidylinositol 3-Kinase and Akt Protein Kinase Are Necessary and Sufficient for the Survival of Nerve Growth Factor-Dependent Sympathetic Neurons. J. Neurosci. 1998, 18, 2933–2943. [Google Scholar] [CrossRef] [PubMed]
- Han, B.H.; Holtzman, D.M. BDNF Protects the Neonatal Brain from Hypoxic-Ischemic Injury In Vivo via the ERK Pathway. J. Neurosci. 2000, 20, 5775–5781. [Google Scholar] [CrossRef] [PubMed]
- Makar, T.K.; Trisler, D.; Sura, K.; Sultana, S.; Patel, N.; Bever, C.T. Brain derived neurotrophic factor treatment reduces inflammation and apoptosis in experimental allergic encephalomyelitis. J. Neurol. Sci. 2008, 270, 70–76. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
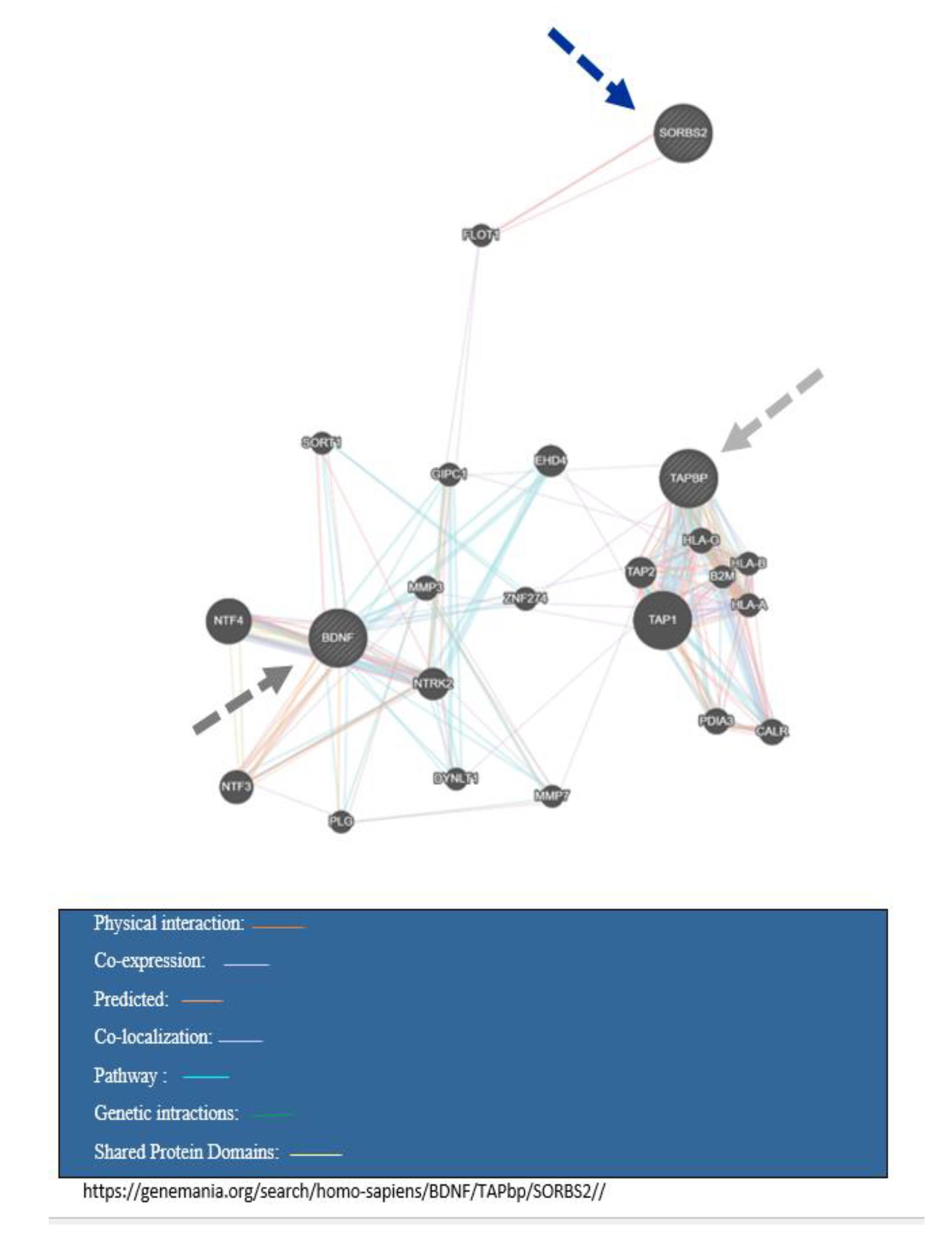{kind=link}
| Authors | Gene | Outcome | Tissue Type | Study | Population | Groups | Definition | Method |
|---|---|---|---|---|---|---|---|---|
| Cheng (2018) | TAPBP | Obesity | WBC | Case-Control | 62 years old and older | Obese/overweight: (n = 5) Normal weight: (n = 5) | BMI < 25 kg m−2, overweight, 25 ≤ BMI < 30 kg m−2, and obese, BMI ≥ 30 kg m−2 | Epigenome-wide DNA methylation was analyzed using the Infinium Human Methylation450 (HM450) BeadChip (Illumina, San Diego, CA, USA) |
| Murphy (2017) | TAPBP | Depression | Tissue brain | Case-Control | Adults | Tissue (n = 75) from two regions of the cortex (BA11, n = 40 BA25, n = 35) | Psychological autopsy method | Differential DNA methylation across the PSORS1C3-associated DMR (spanning a region) |
| Martin (2019) | TAPBP | Obesity | Cord blood | Case-Control | 18 years and older | Pre-pregnancy obesity in 187 mother-female and 173 mother-male offsprings | Infant birth weight and sex. | differential methylation at 6148 CpG sites (FDR) using Illumina umanMethylation450k BeadChip |
| Rhee (2017) | SORBS2 | Obesity | WBC | Case-Control | Twins Children | Obese children = 6 Normal body weight = 6 | Normal weight, BMI < 25 kg m−2, Obese and overweight (n = 5, BMI ≥ 25 kg m−2) versus Normal weight group (n = 5, BMI < 25 kg m−2) | Illumina Human HT-12 v4 Expression BeadChip Kit, with 47,318 probes. |
| Zhu (2019) | SORBS2 | Depression | WBC | Case-Control | Twins 18 and older | 79 monozygotic twin pairs discordant | MDD diagnoses were determined using the Structured Clinical Interview for DSM-IV Research Version (SCID-4-RV) | Integrative DNA methylome and transcriptome analysis Infinium HumanMethylationEPIC BeadChip (Illumina Inc., CA, USA) |
| Keller (2010) | SORBS2 | Obesity | Adipose tissue | Case-Control | 18 and older | Men (N¼39) and women (N¼66) | Differential methylation analysis using InfiniumHumanMethylation450 BeadChips | |
| Perroud (2013) | BDNF | Bipolar | WBC | Case-Control | 18 years and older | Control = 52 Bipolar disorder = 115 | 1.Suicidal or para-suicidal behaviors 2. Severe impulse control disorders 3. Anger problems 4. Receiving psychopharmacological treatment 5. Fulfilling DSM-IV (diagnostic and statistical manual of mental disorders, 4th edition) | Selected region/gene/high resolution melting method |
| Januar (2015) | BDNF | Depression | Buccal tissue | Case-Control | 65 years | Depressive = 251 Non-depressive = 773 | Diagnostic and Statistical Manual of Mental Disorders-IV criteria and using the Mini International Neuropsychiatric Interview (MIN) | Sequenom Mass ARRAY (San Diego, CA, USA) |
| Voisin (2015) | BDNF | Obesity | WBC | Case-Control | 14–16 years | Two sub-groups of healthy young Caucasians from two different age ranges | Lean: BMI < 25 Overweight: 25 ≤ BMI < 30 Obese: BMI ≥ 30 | Genome-wide Illumina Infinium human Methylation450 Bead Chip (Illumina) |
| Gardner (2015) | BDNF | Obesity | WBC | Case-Control | Children | 32 non-obese and 32 obese African-American children aged 5–6 years. | Normal weight (BMI-for-age percentile 5th–<85th) or obese (BMI for-age percentile ≥ 95th). | Methylation-sensitive restriction enzyme digestion 2.qRTPCR |
| Biological Process (GO) | Molecular Function (GO) | Cellular Component (GO) | KEGG Pathways | Super Pathway | Ref. | |
|---|---|---|---|---|---|---|
| TAPBP | Antigen processing and presentation of peptide antigen via MHC class I Antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent Antigen processing and presentation of exogenous peptide antigen Antigen processing and presentation of endogenous peptide antigen Antigen processing and presentation of endogenous peptide antigen via MHC class I | TAP binding Peptide Antigen Binding Peptide Binding MHC protein binding Peptide Antigen-Transporting ATPase Activity | Phagocytic Vesicle Membrane Integral Component of Lumenal Side of Endoplasmic Reticulum Membrane Integral Component of Endoplasmic Reticulum Membrane MHC Class I Protein Complex | Antigen processing and presentation Herpes simplex infection HTLV-I infection Graft-versus-host disease | Antigen Processing-Cross presentation ER-Phagosome pathway Immune response Antigen presentation by MHC class I Human cytomegalovirus infection Human immunodeficiency virus 1 infection Class I MHC mediated antigen processing and presentation | https://string-db.org/cgi/network.pl?taskId=BgKUv6snBF5M |
| SORBS2 | Developmental cell growt Molecular Function (GO) Protein kinase binding Mitogen-activated protein kinase binding Ephrin receptor binding Phosphotyrosine residue binding receptor tyrosine kinase binding | Protein Kinase Binding Mitogen-Activated Protein Kinase Binding Ephrin Receptor Binding Phosphotyrosine Residue Binding R Binding | Contractile Fiber Part Myofibril Actin Cytoskeleton Focal Adhesion Podosome | Chronic myeloid leukemia Bacterial invasion of epithelial cells ErbB signaling pathway Shigellosis Neurotrophin signaling pathway | Notch signaling pathway Actin filament organization Biological process Cell growth involved in cardiac muscle cell development | https://string-db.org/cgi/network.pl?taskId=kpCKo7eP0PWT |
| BDNF | Neurotrophin TRK receptor signaling pathway Transmembrane receptor protein tyrosine kinase signaling pathway Regulation of neuron death Regulation of cell deathNegative regulation of neuron death | Neurotrophin Binding Neurotrophin Receptor Binding Receptor Binding Activity Nerve Growth Factor Binding Cellular Component (Go) | Neuron Projection Cytoplasmic Vesicle Postsynaptic Membrane Dendrite Axon | Neurotrophin signaling pathway MAPK signaling pathway PI3K-Akt signaling pathway Cocaine addiction | Cellular apoptosis pathway mitochondrial apoptosis Apoptotic Pathways in Synovial Fibroblasts p53 Mediated Apoptosis DHA Signaling Telomerase Components in Cell Signaling PPAR Pathway Rac1 Pathway Glioma Invasiveness Actin-Based Motility by Rho Family GTPases ERK5 Signaling eIF2 Pathway Rap1 Pathway Nuclear Receptor Activation by Vitamin-A Paxillin Interactions Ras Pathway GPCR Pathway Pancreatic Adenocarcinoma Breast Cancer Regulation by Stathmin1 NFAT in Immune Response Estrogen Pathway ERK Signaling Rho Family GTPases MAPK Signaling Molecular Mechanisms of Cancer ILK Signaling GSK3 Signaling Nanog in Mammalian ESC Pluripotency 3-3-14 Induced Intracellular Signaling eNOS Signaling CREB Pathway IP3 Pathway Activation of PKC through GPCR Intracellular Calcium Signaling BDNF-TrkB Signaling ERK Pathway in Huntingtons Disease Follicle Stimulating Hormone (FSH) signaling pathway | https://string-db.org/cgi/network.pl?taskId=MU4AHU3o8Jwe |
| Genes | Term | p-Value | Adjusted p-Value | Odds Ratio | Combined Score |
|---|---|---|---|---|---|
| TAPBP | Antigen Presentation: Folding, assembly and peptide loading of class I MHC_Homo sapiens_R-HSA-983170 | 0.00125 | 1 | 800 | 5347.706 |
| ER-Phagosome pathway_Homo sapiens_R-HSA-1236974 | 0.00325 | 1 | 307.6923 | 1762.805 | |
| Antigen processing-Cross presentation_Homo sapiens_R-HSA-1236975 | 0.0041 | 1 | 243.9024 | 1340.679 | |
| Class I MHC mediated antigen processing & presentation_Homo sapiens_R-HSA-983169 | 0.01525 | 1 | 65.57377 | 274.3072 | |
| Adaptive Immune System_Homo sapiens_R-HSA-1280218 | 0.0381 | 1 | 26.24672 | 85.76236 | |
| Immune System_Homo sapiens_R-HSA-168256 | 0.07735 | 1 | 12.92825 | 33.08879 | |
| SORBS2 | Extracellular vesicles in the crosstalk of cardiac cells WP4300 | 9.50 × 10−4 | 0.44839 | 1052.632 | 7325.339 |
| BDNF | ERK Pathway in Huntington’s Disease WP3853 | 7.00 × 10−4 | 0.330392 | 1428.571 | 10377.79 |
| Follicle Stimulating Hormone (FSH) signaling pathway WP2035 | 0.00135 | 0.318593 | 740.7407 | 4894.571 | |
| BDNF-TrkB Signaling WP3676 | 0.0017 | 0.267461 | 588.2353 | 3751.263 | |
| Synaptic signaling pathways associated with autism spectrum disorder WP4539 | 0.0025 | 0.294995 | 400 | 2396.593 | |
| Prader-Willi and Angelman Syndrome WP3998 | 0.00305 | 0.287915 | 327.8689 | 1899.223 | |
| MECP2 and Associated Rett Syndrome WP3584 | 0.0031 | 0.243863 | 322.5806 | 1863.345 | |
| Spinal Cord Injury WP2431 | 0.0059 | 0.397824 | 169.4915 | 869.9687 | |
| Brain-Derived Neurotrophic Factor (BDNF) signaling pathway WP2380 | 0.0072 | 0.424795 | 138.8889 | 685.2341 | |
| Sudden Infant Death Syndrome (SIDS) Susceptibility Pathways WP706 | 0.0079 | 0.414306 | 126.5823 | 612.7726 | |
| MAPK Signaling Pathway WP382 | 0.0123 | 0.580555 | 81.30081 | 357.5744 | |
| PI3K-Akt Signaling Pathway WP4172 | 0.017 | 0.729449 | 58.82353 | 239.6794 |
| Authors | Reporting Score | External Validity | Internal Validity-Bias | Internal Validity-Confounding | Black and Dwaon Score |
|---|---|---|---|---|---|
| Cheng, et al. (2018) [26] | 6 | 2 | 1 | 2 | 11 |
| Murphy, et al. (2017) [27] | 5 | 1 | 0 | 0 | 6 |
| Martin, et al. (2019) [28] | 7 | 2 | 1 | 3 | 13 |
| Rhee, et al. (2017) [29] | 7 | 2 | 0 | 0 | 9 |
| Zhu, et al. (2019) [30] | 6 | 2 | 1 | 2 | 11 |
| Keller, et al. (2010) [31] | 4 | 0 | 1 | 0 | 4 |
| Perroud, et al. (2013) [32] | 7 | 1 | 1 | 0 | 9 |
| Januar, et al. (2015) [33] | 4 | 2 | 1 | 1 | 8 |
| Voisin, et al. (2015) [34] | 7 | 2 | 0 | 2 | 11 |
| Gardner, et al. (2015) [35] | 6 | 1 | 1 | 2 | 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gharipour, M.; Barekatain, M.; Sung, J.; Emami, N.; Sadeghian, L.; Dianatkhah, M.; Sarrafzadegan, N.; Jahanfar, S. The Epigenetic Overlap between Obesity and Mood Disorders: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 6758. https://doi.org/10.3390/ijms21186758
Gharipour M, Barekatain M, Sung J, Emami N, Sadeghian L, Dianatkhah M, Sarrafzadegan N, Jahanfar S. The Epigenetic Overlap between Obesity and Mood Disorders: A Systematic Review. International Journal of Molecular Sciences. 2020; 21(18):6758. https://doi.org/10.3390/ijms21186758
Chicago/Turabian StyleGharipour, Mojgan, Majid Barekatain, Johoon Sung, Naghmeh Emami, Ladan Sadeghian, Minoo Dianatkhah, Nizal Sarrafzadegan, and Shayesteh Jahanfar. 2020. "The Epigenetic Overlap between Obesity and Mood Disorders: A Systematic Review" International Journal of Molecular Sciences 21, no. 18: 6758. https://doi.org/10.3390/ijms21186758
APA StyleGharipour, M., Barekatain, M., Sung, J., Emami, N., Sadeghian, L., Dianatkhah, M., Sarrafzadegan, N., & Jahanfar, S. (2020). The Epigenetic Overlap between Obesity and Mood Disorders: A Systematic Review. International Journal of Molecular Sciences, 21(18), 6758. https://doi.org/10.3390/ijms21186758