Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
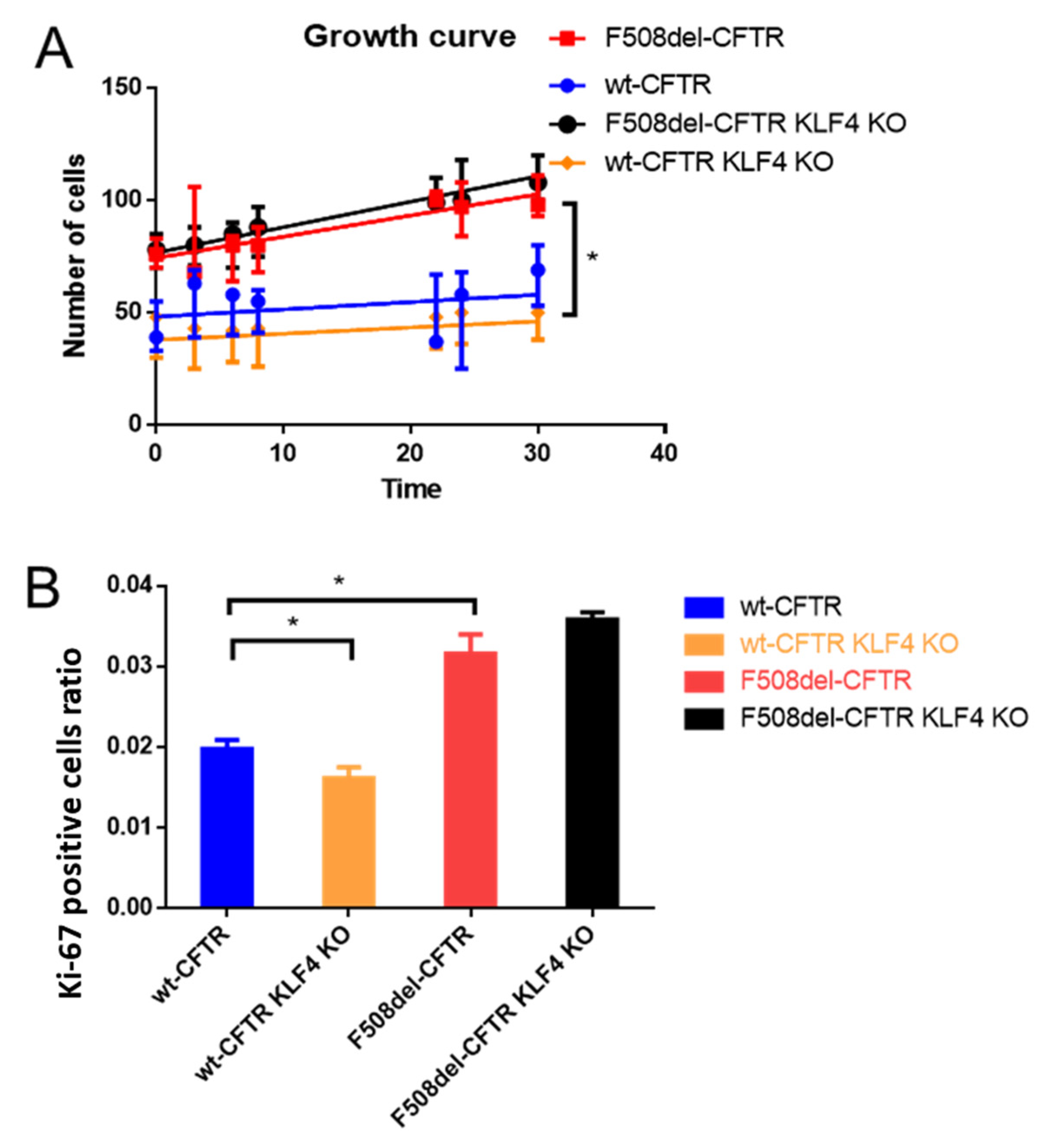
2.1. KLF4 KO Impact on Proliferation
2.2. KLF4 KO Impact on TEER and Wound Healing
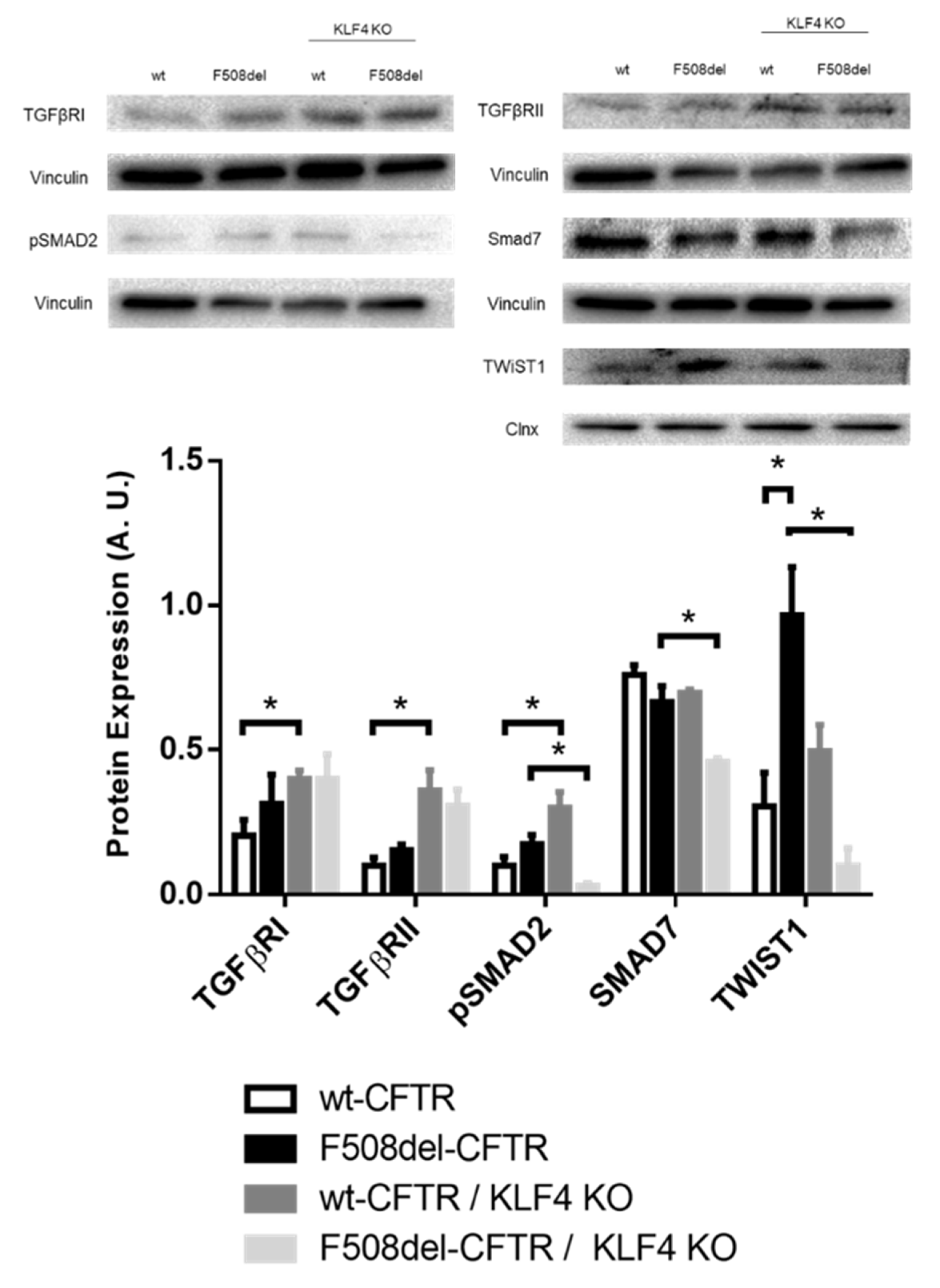
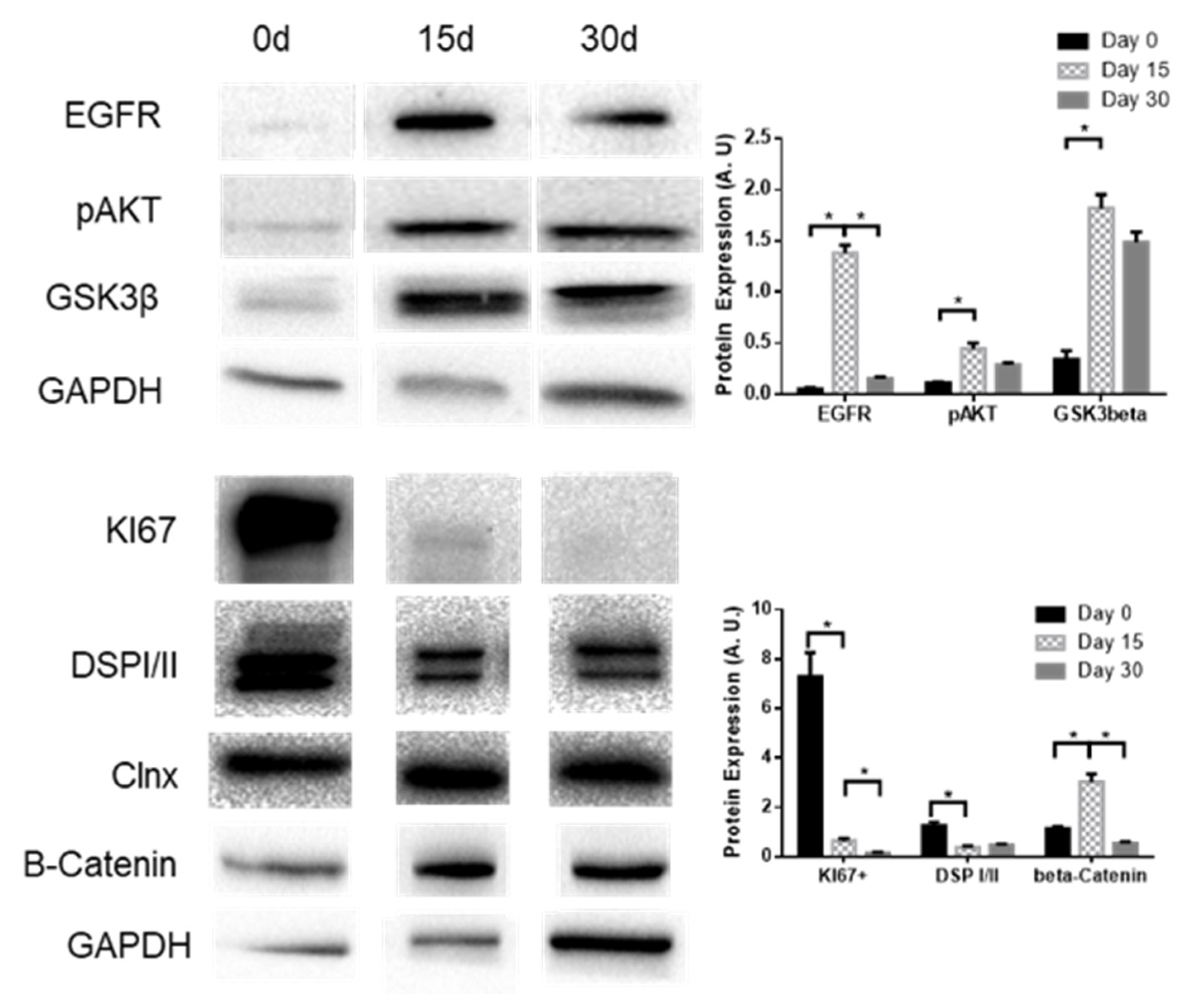
2.3. KLF4 KO Impact on Differentiation Markers
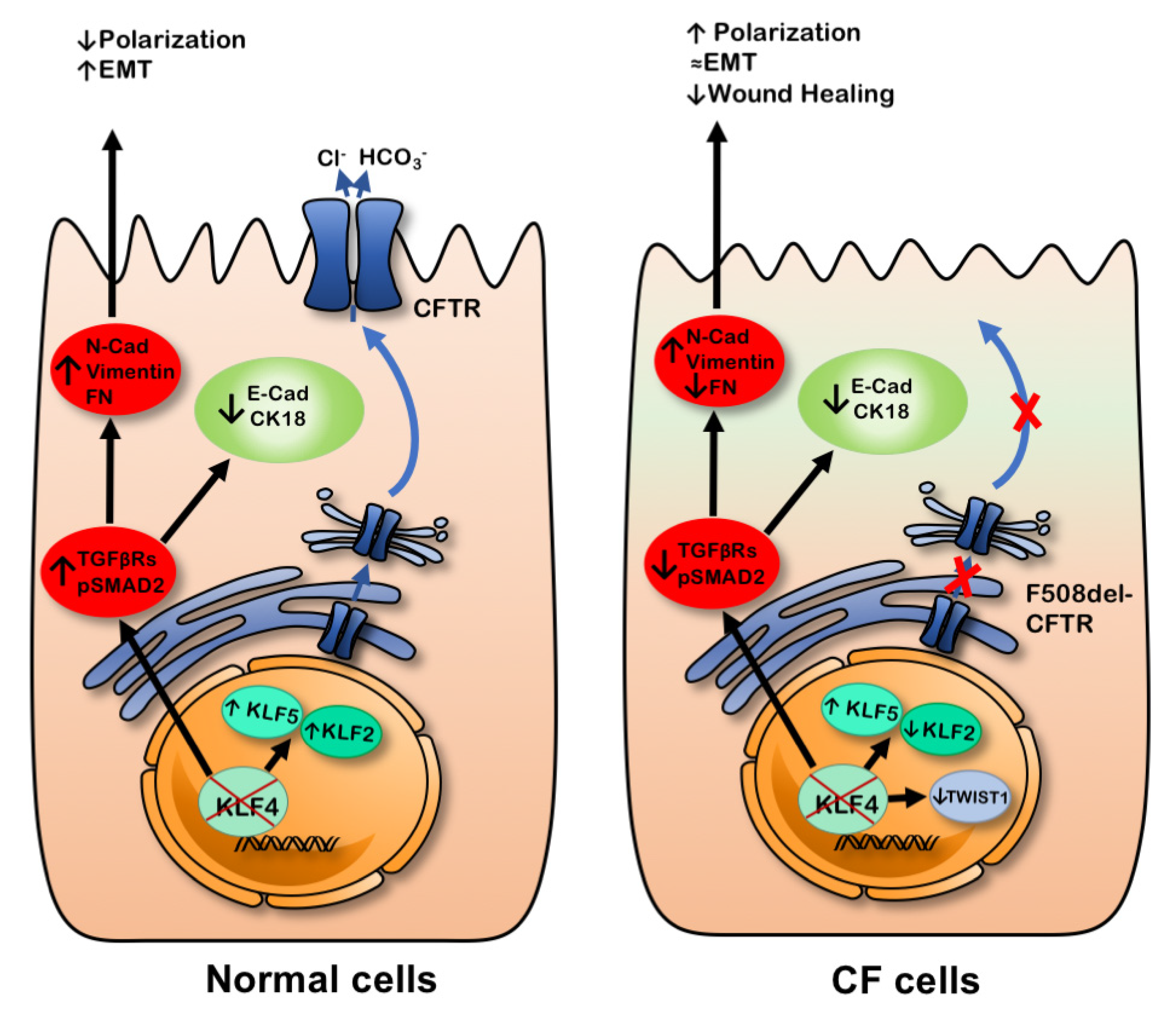
3. Discussion
4. Materials and Methods
4.1. Chemicals, Antibodies, and Primers
4.2. Cell Lines
4.3. TEER Measurements
4.4. KLF4 KO Generation
4.5. Western Blot (WB)
4.6. RT-qPCR
4.7. Growth Curves
4.8. Immunostaining
4.9. Wound Healing
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CF | Cystic fibrosis |
| CFBE | Cystic fibrosis bronchial epithelial |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CK | Cytokeratin |
| Clnx | Calnexin |
| Cx | Connexin |
| DSPI/II | Desmoplakin I/II |
| E-Cad | Epithelial cadherin |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial to mesenchymal transition |
| F508del | Deletion of phenylalanine at position 508 |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GSK3β | Glycogen synthase kinase 3 beta |
| KLF | Kruppel-like factors |
| KO | Knock out |
| PM | Plasma membrane |
| TEER | Transepithelial electrical resistance |
| TF | Transcription factor |
| TGF | Transforming growth factor |
| WB | Western blot |
| wt | Wild-type |
| ZO-1 | Zonula occludens-1 |
References
- Zolin, A.; Orenti, A.; Naehrlich, L.; van Rens, J. Ecfspr Annual Report 2017; European Cystic Fibrosis Society: Karup, Denmark, 2017. [Google Scholar]
- Davis, P.B. Cystic Fibrosis Since 1938. Am. J. Respir. Crit. Care Med. 2006, 173, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Amaral, M. Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol. Cell. Boil. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.E.; Cohen, J.C. Developmental Paradigm for Early Features of Cystic Fibrosis. Pediatr. Pulmonol. 2005, 40, 371–377. [Google Scholar] [CrossRef]
- Lesimple, P.; Liao, J.; Robert, R.; Gruenert, D.C.; Hanrahan, J.W. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 2010, 588, 1195–1209. [Google Scholar] [CrossRef] [PubMed]
- Hajj, R.; Lesimple, P.; Nawrocki-Raby, B.; Birembaut, P.; Puchelle, E.; Coraux, C. Human airway surface epithelial regeneration is delayed and abnormal in cystic fibrosis. J. Pathol. 2007, 211, 340–350. [Google Scholar] [CrossRef]
- Rout-Pitt, N.; Farrow, N.R.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 136. [Google Scholar] [CrossRef]
- Amaral, M.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Boil. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Liu, Y.-N.; Abou-Kheir, W.; Yin, J.J.; Fang, L.; Hynes, P.; Casey, O.; Hu, N.; Wan, Y.; Seng, V.; Sheppard-Tillman, H.; et al. Critical and Reciprocal Regulation of KLF4 and SLUG in Transforming Growth Factor β-Initiated Prostate Cancer Epithelial-Mesenchymal Transition. Mol. Cell. Boil. 2011, 32, 941–953. [Google Scholar] [CrossRef]
- Farrugia, M.K.; Vanderbilt, D.B.; Salkeni, M.A.; Ruppert, J.M. Kruppel-like Pluripotency Factors as Modulators of Cancer Cell Therapeutic Responses. Cancer Res. 2016, 76, 1677–1682. [Google Scholar] [CrossRef]
- Kabir, F.L.; Ambalavanan, N.; Liu, G.; Li, P.; Solomon, G.M.; Lal, C.V.; Mazur, M.; Halloran, B.; Szul, T.; Gerthoffer, W.T.; et al. MicroRNA-145 Antagonism Reverses TGF-β Inhibition of F508del CFTR Correction in Airway Epithelia. Am. J. Respir. Crit. Care Med. 2018, 197, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Crespin, S.; Bacchetta, M.; Saab, J.B.; Tantilipikorn, P.; Bellec, J.; Dudez, T.; Nguyen, T.; Chanson, M.; Lacroix, J.; Huang, S.; et al. Cx26 regulates proliferation of repairing basal airway epithelial cells. Int. J. Biochem. Cell Boil. 2014, 52, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.M.; Ott, C.J.; Leir, S.-H.; Gosalia, N.; Song, L.; London, D.; Furey, T.S.; Cotton, C.U.; Crawford, G.E.; Harris, A. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax 2011, 67, 385–391. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mutolo, M.J.; Leir, S.-H.; Fossum, S.L.; Browne, J.A.; Harris, A. A transcription factor network represses CFTR gene expression in airway epithelial cells. Biochem. J. 2018, 475, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Ray, G.; Kerschner, J.L.; Hao, S.; Perez, A.; Drumm, M.L.; Browne, J.A.; Leir, S.-H.; Longworth, M.; Harris, A. Functional genomics analysis of human colon organoids identifies key transcription factors. Physiol. Genom. 2020, 52, 234–244. [Google Scholar] [CrossRef]
- Rymut, S.M.; Corey, D.A.; Valerio, D.M.; Erokwu, B.O.; Flask, C.A.; Kelley, T.J.; Hodges, C.A. Improved Growth Patterns in Cystic Fibrosis Mice after Loss of Histone Deacetylase 6. Sci. Rep. 2017, 7, 3676. [Google Scholar] [CrossRef]
- Saavedra, M.T.; Patterson, A.D.; West, J.D.; Randell, S.H.; Riches, D.W.; Malcolm, K.C.; Cool, C.D.; Nick, J.A.; Dinarello, C.A. Abrogation of Anti-Inflammatory Transcription Factor LKLF in Neutrophil-Dominated Airways. Am. J. Respir. Cell Mol. Boil. 2008, 38, 679–688. [Google Scholar] [CrossRef]
- Typпaeв, K. Transcription Factor KLF2 and Its Role in the Regulation of Inflammatory Processes. Biochemistry (Moscow) 2020, 85, 54–67. [Google Scholar] [CrossRef]
- Sousa, L.; Pankonien, I.; Clarke, L.A.; Silva, I.; Kunzelmann, K.; Amaral, M. KLF4 Acts as a wt-CFTR Suppressor through an AKT-Mediated Pathway. Cells 2020, 9, 1607. [Google Scholar] [CrossRef]
- Chen, X.; Whitney, E.M.; Gao, S.Y.; Yang, V.W. Transcriptional Profiling of Krüppel-like Factor 4 Reveals a Function in Cell Cycle Regulation and Epithelial Differentiation. J. Mol. Boil. 2003, 326, 665–677. [Google Scholar] [CrossRef]
- Rowland, B.D.; Peeper, D.S. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 2005, 6, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Hao, B.; Zhan, Y.; Luo, G. Krüppel-like factor 4 expression in solid tumor prognosis: A meta-analysis. Clin. Chim. Acta 2018, 485, 50–59. [Google Scholar] [CrossRef]
- Chen, H.-F.; Wu, K.-J. Endothelial Transdifferentiation of Tumor Cells Triggered by the Twist1-Jagged1-KLF4 Axis: Relationship between Cancer Stemness and Angiogenesis. Stem Cells Int. 2015, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Katz, J.P.; Perreault, N.; Goldstein, B.G.; Actman, L.; McNally, S.R.; Silberg, D.G.; Furth, E.E.; Kaestner, K.H. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology 2005, 128, 935–945. [Google Scholar] [CrossRef]
- Lin, Z.-S.; Chu, H.-C.; Yen, Y.-C.; Lewis, B.C.; Chen, Y.-W. Krüppel-Like Factor 4, a Tumor Suppressor in Hepatocellular Carcinoma Cells Reverts Epithelial Mesenchymal Transition by Suppressing Slug Expression. PLoS ONE 2012, 7, e43593. [Google Scholar] [CrossRef] [PubMed]
- Yori, J.L.; Seachrist, D.D.; Johnson, E.; Lozada, K.L.; Abdul-Karim, F.W.; Chodosh, L.A.; Schiemann, W.P.; Keri, R.A. Krüppel-like Factor 4 Inhibits Tumorigenic Progression and Metastasis in a Mouse Model of Breast Cancer. Neoplasia 2011, 13, 601–610. [Google Scholar] [CrossRef]
- Li, L.; Yu, S.; Wu, Q.; Dou, N.; Li, Y.; Gao, Y. KLF4-Mediated CDH3 Upregulation Suppresses Human Hepatoma Cell Growth and Migration via GSK-3β Signaling. Int. J. Boil. Sci. 2019, 15, 953–961. [Google Scholar] [CrossRef]
- Tang, J.M.; Zhong, G.; Wu, J.; Chen, H.; Jia, Y. SOX2 recruits KLF4 to regulate nasopharyngeal carcinoma proliferation via PI3K/AKT signaling. Oncogenesis 2018, 7, 61. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C.; Gu, Q.; Sims, M.; Gu, W.; Pfeffer, L.; Yue, J. KLF4 Promotes Angiogenesis by Activating VEGF Signaling in Human Retinal Microvascular Endothelial Cells. PLoS ONE 2015, 10, e0130341. [Google Scholar] [CrossRef]
- Mai, J.; Zhong, Z.-Y.; Chen, X.-X.; Xiang, Y.-Q.; Li, X.; Zhang, H.-L.; Deng, R.; Zhu, X.-F. Abstract 4651: Polo-like kinase 1 phosphorylates and stabilizes KLF4 to promote tumorigenesis in nasopharyngeal carcinoma. Tumor Biol. 2019, 9, 3541–3554. [Google Scholar]
- Zhou, H.; Liu, Y.; Zhu, R.; Ding, F.; Wan, Y.; Li, Y.; Liu, Z. FBXO32 suppresses breast cancer tumorigenesis through targeting KLF4 to proteasomal degradation. Oncogene 2017, 36, 3312–3321. [Google Scholar] [CrossRef]
- Villarreal, G.; Zhang, Y.; Larman, H.B.; Gracia-Sancho, J.; Koo, A.; Garcia-Cardena, G. Defining the regulation of KLF4 expression and its downstream transcriptional targets in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 391, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Brembeck, F.H. The tissue-dependent Keratin 19 gene transcription is regulated by GKLF/KLF4 and Sp1. J. Boil. Chem. 2000, 275, 28230–28239. [Google Scholar] [CrossRef]
- Jean, J.-C.; George, E.; Kaestner, K.H.; Brown, L.A.S.; Spira, A.; Joyce-Brady, M. Transcription Factor Klf4, Induced in the Lung by Oxygen at Birth, Regulates Perinatal Fibroblast and Myofibroblast Differentiation. PLoS ONE 2013, 8, e54806. [Google Scholar] [CrossRef] [PubMed]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Ou, L.; Shi, Y.; Dong, W.; Liu, C.; Schmidt, T.J.; Nagarkatti, P.; Nagarkatti, M.; Fan, D.; Ai, W. Kruppel-like factor KLF4 facilitates cutaneous wound healing by promoting fibrocyte generation from myeloid-derived suppressor cells. J. Investig. Dermatol. 2015, 135, 1425–1434. [Google Scholar] [CrossRef]
- Djalilian, A.R.; McGaughey, D.M.; Patel, S.; Seo, E.Y.; Yang, C.; Cheng, J.; Tomić, M.; Sinha, S.; Ishida-Yamamoto, A.; Segre, J.A. Connexin 26 regulates epidermal barrier and wound remodeling and promotes psoriasiform response. J. Clin. Investig. 2006, 116, 1243–1253. [Google Scholar] [CrossRef]
- Tiwari, N.; Meyer-Schaller, N.; Arnold, P.; Antoniadis, H.; Pachkov, M.; van Nimwegen, E.; Christofori, G. Klf4 Is a Transcriptional Regulator of Genes Critical for Emt, Including Jnk1 (Mapk8). PLoS ONE 2013, 8, e57329. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Boil. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Noh, M.-G.; Oh, S.-J.; Ahn, E.-J.; Kim, Y.-J.; Jung, T.-Y.; Jung, S.; Kim, K.K.; Lee, J.-H.; Lee, K.-H.; Moon, K.-S. Prognostic significance of E-cadherin and N-cadherin expression in Gliomas. BMC Cancer 2017, 17, 583. [Google Scholar] [CrossRef]
- Walters, M.S.; Gomi, K.; Ashbridge, B.; Moore, M.A.S.; Arbelaez, V.; Heldrich, J.; Ding, B.-S.; Rafii, S.; Staudt, M.R.; Crystal, R.G. Generation of a human airway epithelium derived basal cell line with multipotent differentiation capacity. Respir. Res. 2013, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Johns, D.C.; Geiman, D.E.; Marban, E.; Dang, D.T.; Hamlin, G.; Sun, R.; Yang, V.W. Kruppel-Like Factor 4 (Gut-Enriched Kruppel-Like Factor) Inhibits Cell Proliferation by Blocking G1/S Progression of the Cell Cycle. J. Biol. Chem. 2001, 276, 30423–30428. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Nandan, M.O.; Chanchevalap, S.; Dalton, W.B.; Hisamuddin, I.M.; Yang, V.W. Krüppel-like factors 4 and 5: The yin and yang regulators of cellular proliferation. Cell Res. 2005, 15, 92–96. [Google Scholar] [CrossRef]
- Agbo, K.C.; Huang, J.Z.; Ghaleb, A.M.; Williams, J.L.; Shroyer, K.R.; Bialkowska, A.B.; Yang, V.W. Loss of the Krüppel-like factor 4 tumor suppressor is associated with epithelial-mesenchymal transition in colorectal cancer. J. Cancer Metastasis Treat. 2019, 2019, 77. [Google Scholar] [CrossRef] [PubMed]
- Xia, E.; Bhandari, A.; Shen, Y.; Zhou, X.; Wang, O. lncRNA LINC00673 induces proliferation, metastasis and epithelial-mesenchymal transition in thyroid carcinoma via Kruppel-like factor 2. Int. J. Oncol. 2018, 53, 1927–1938. [Google Scholar] [CrossRef]
- Tiwari, A.; Loughner, C.L.; Swamynathan, S.; Swamynathan, S.K. KLF4 Plays an Essential Role in Corneal Epithelial Homeostasis by Promoting Epithelial Cell Fate and Suppressing Epithelial–Mesenchymal Transition. Investig. Opthalmol. Vis. Sci. 2017, 58, 2785–2795. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, L.; Wang, X.; Li, Y.; Liu, Z.; Ye, W.; Huang, W.; Lin, G.; Liu, H.; Zhang, J.; et al. Overexpression of Krüppel-Like Factor 4 Suppresses Migration and Invasion of Non-Small Cell Lung Cancer Through c-Jun-NH2-Terminal Kinase/Epithelial-Mesenchymal Transition Signaling Pathway. Front. Pharmacol. 2020, 10, 1512. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Z.; Zhang, X.; Yang, S.; Lin, X.; Yang, X.; Lin, X.; Shi, J.; Wang, S.; Zhao, W.; et al. Klf4 reduces stemness phenotype, triggers mesenchymal-epithelial transition (MET)-like molecular changes, and prevents tumor progression in nasopharygeal carcinoma. Oncotarget 2017, 8, 93924–93941. [Google Scholar] [CrossRef]
- Brauer, P.R.; Kim, J.H.; Ochoa, H.J.; Stratton, E.R.; Black, K.M.; Rosencrans, W.; Stacey, E.; Hagos, E.G. Krüppel-like factor 4 mediates cellular migration and invasion by altering RhoA activity. Cell Commun. Adhes. 2018, 24, 1–10. [Google Scholar] [CrossRef]
- Yori, J.L.; Johnson, E.; Zhou, G.; Jain, M.K.; Keri, R.A. Krüppel-like Factor 4 Inhibits Epithelial-to-Mesenchymal Transition through Regulation of E-cadherin Gene Expression. J. Boil. Chem. 2010, 285, 16854–16863. [Google Scholar] [CrossRef]
- Ruan, Y.C.; Wang, Y.; Da Silva, N.; Kim, B.; Diao, R.Y.; Hill, E.; Brown, D.; Chan, H.C.; Breton, S. CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J. Cell Sci. 2014, 127, 4396–4408. [Google Scholar] [CrossRef] [PubMed]
- Castellani, S.; Guerra, L.; Favia, M.; Di Gioia, S.; Casavola, V.; Conese, M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: Role of ezrin and the RhoA/ROCK pathway. Lab. Investig. 2012, 92, 1527–1540. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chen, X.; Zhang, W.; Li, J.; Xu, R.; Wang, T.C.; Ai, W.; Liu, C. Krüppel-like Factor 4 Regulates Intestinal Epithelial Cell Morphology and Polarity. PLoS ONE 2012, 7, e32492. [Google Scholar] [CrossRef]
- Peters-Hall, J.R.; Brown, K.J.; Pillai, D.K.; Tomney, A.; Garvin, L.M.; Wu, X.; Rose, M.C. Quantitative Proteomics Reveals an Altered Cystic FibrosisIn VitroBronchial Epithelial Secretome. Am. J. Respir. Cell Mol. Boil. 2015, 53, 22–32. [Google Scholar] [CrossRef]
- Badaoui, M.; Zoso, A.; Idris, T.; Bacchetta, M.; Simonin, J.; Lemeille, S.; Wehrle-Haller, B.; Chanson, M. Vav3 Mediates Pseudomonas aeruginosa Adhesion to the Cystic Fibrosis Airway Epithelium. Cell Rep. 2020, 32, 107842. [Google Scholar] [CrossRef]
- Fujimoto, S.; Hayashi, R.; Hara, S.; Sasamoto, Y.; Harrington, J.; Tsujikawa, M.; Nishida, K. KLF4 prevents epithelial to mesenchymal transition in human corneal epithelial cells via endogenous TGF-β2 suppression. Regen. Ther. 2019, 11, 249–257. [Google Scholar] [CrossRef]
- Yan, J.; Du, F.; Li, S.-D.; Yuan, Y.; Jiang, J.-Y.; Li, S.; Li, X.-Y.; Du, Z.-X. AUF1 modulates TGF-β signal in renal tubular epithelial cells via post-transcriptional regulation of Nedd4L expression. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1865, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Nicola, T.; Kabir, F.L.; Coric, T.; Wall, S.B.; Zhang, W.; James, M.; MacEwen, M.; Ren, C.; Halloran, B.; Ambalavanan, N.; et al. CFTR dysfunction increases endoglin and TGF-β signaling in airway epithelia. Physiol. Rep. 2019, 7, e13977. [Google Scholar] [CrossRef]
- Korrodi-Gregorio, L.; Silva, J.V.; Santos-Sousa, L.; Freitas, M.J.; Felgueiras, J.; Fardilha, M. TGF-β cascade regulation by PPP1 and its interactors -impact on prostate cancer development and therapy. J. Cell. Mol. Med. 2014, 18, 555–567. [Google Scholar] [CrossRef]
- Meng, J.; Chen, S.; Han, J.-X.; Qian, B.; Wang, X.-R.; Zhong, W.-L.; Qin, Y.; Zhang, H.; Gao, W.-F.; Lei, Y.-Y.; et al. Twist1 Regulates Vimentin through Cul2 Circular RNA to Promote EMT in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 4150–4162. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, L.; Pankonien, I.; Simões, F.B.; Chanson, M.; Amaral, M.D. Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 6717. https://doi.org/10.3390/ijms21186717
Sousa L, Pankonien I, Simões FB, Chanson M, Amaral MD. Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. International Journal of Molecular Sciences. 2020; 21(18):6717. https://doi.org/10.3390/ijms21186717
Chicago/Turabian StyleSousa, Luís, Ines Pankonien, Filipa B. Simões, Marc Chanson, and Margarida D. Amaral. 2020. "Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis" International Journal of Molecular Sciences 21, no. 18: 6717. https://doi.org/10.3390/ijms21186717
APA StyleSousa, L., Pankonien, I., Simões, F. B., Chanson, M., & Amaral, M. D. (2020). Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. International Journal of Molecular Sciences, 21(18), 6717. https://doi.org/10.3390/ijms21186717