State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy

 , , , , ,
, , , , ,
Abstract
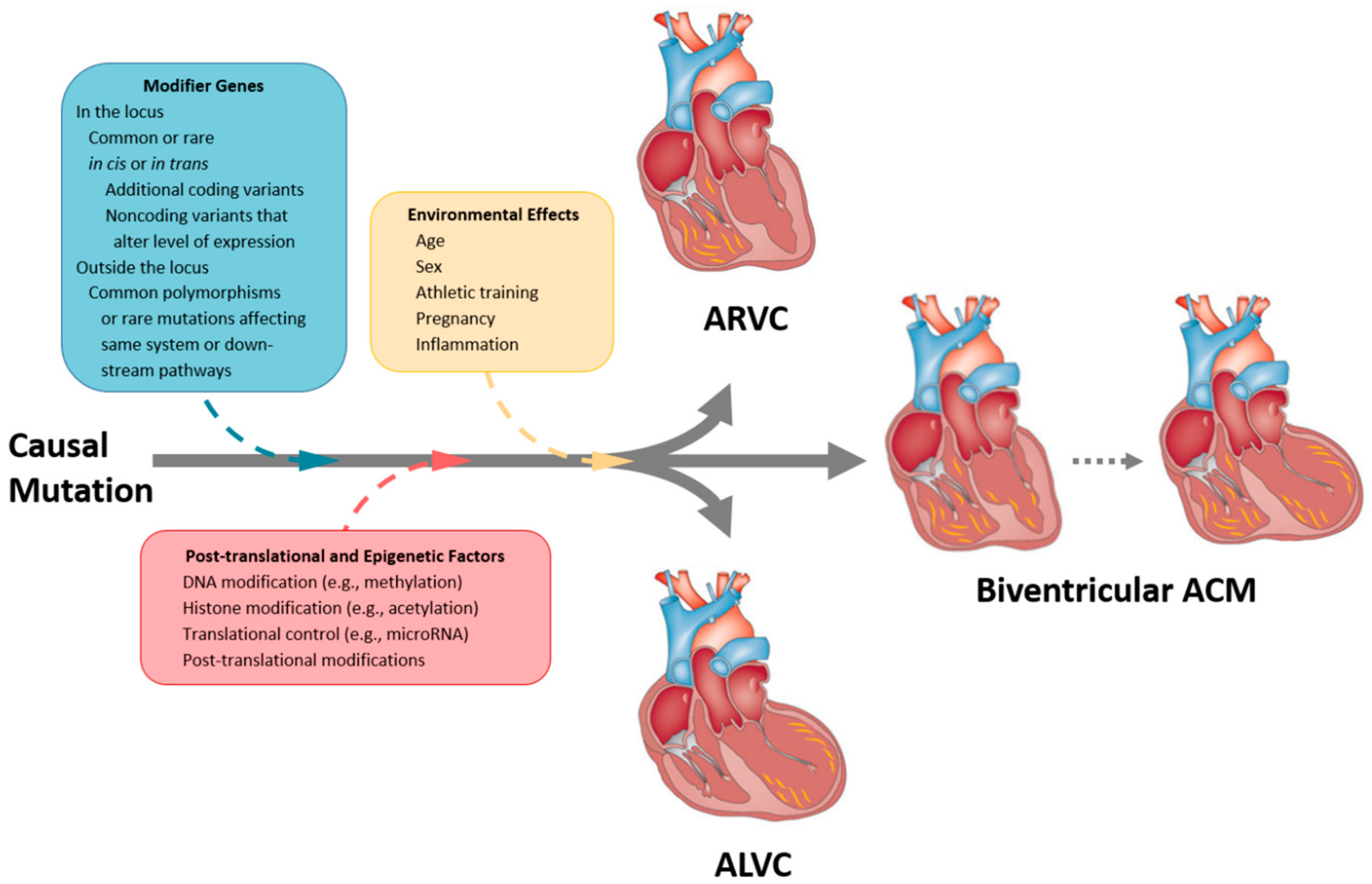
1. Introduction to Arrhythmogenic Cardiomyopathies: Evolving Concepts
2. Classification of Arrhythmogenic Cardiomyopathies
3. Genetic Basis of ACM
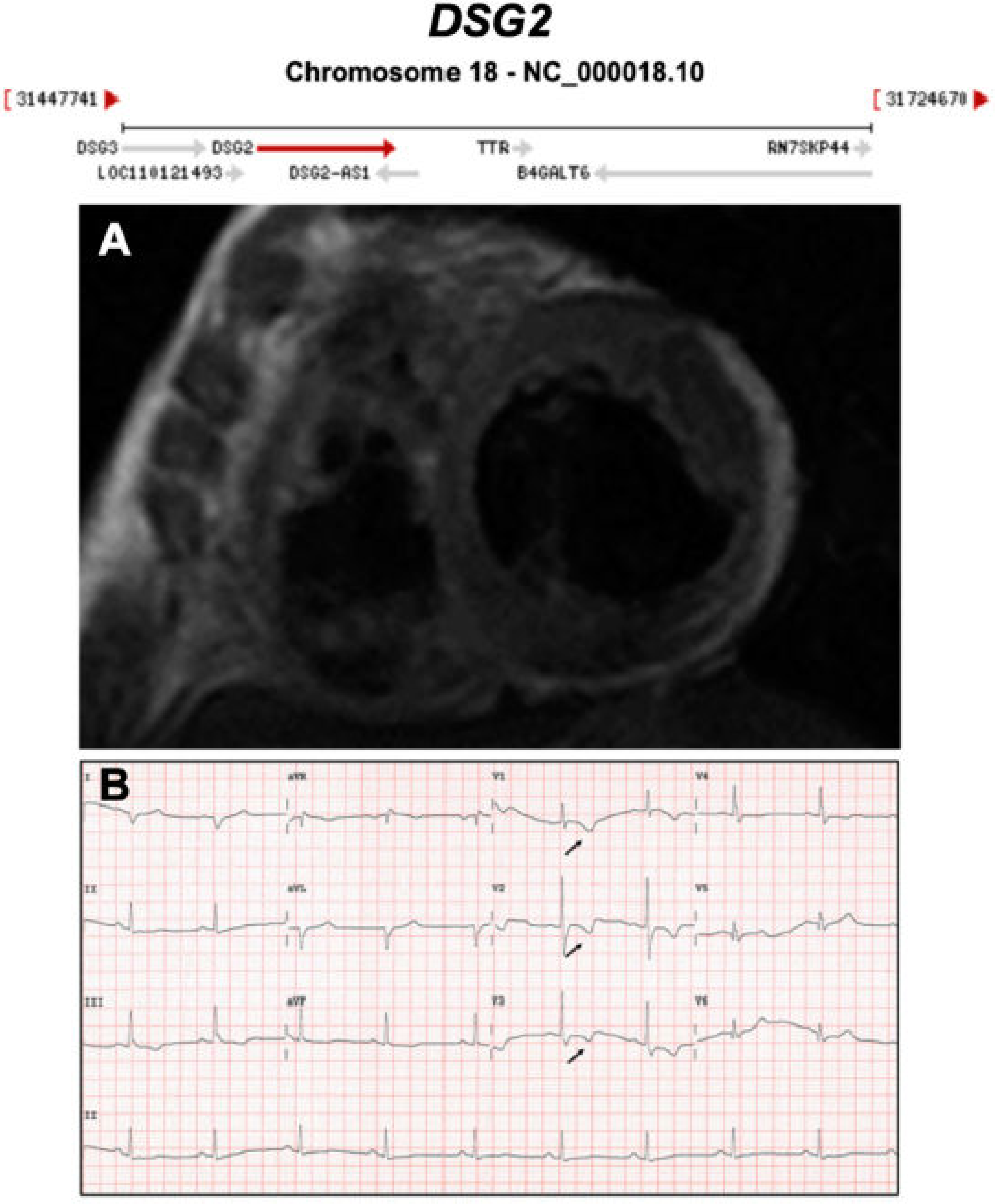
3.1. Desmosomal Genes
3.2. Non-Desmosomal Genes
3.2.1. Titin (TTN) (Encoded by TTN)
3.2.2. Lamin A/C (LMNA) (Encoded by LMNA)
3.2.3. Desmin (DES) (Encoded by DES)
3.2.4. Filamin C (FLNC) (Encoded by FLNC)
3.2.5. Tight Junction Protein 1 (TJP1) (Also Known as Zona Occludens 1) (Encoded by TJP1)
3.2.6. N-Cadherin (CDH2) (Also Known as Cadherin-2) (Encoded by CDH2)
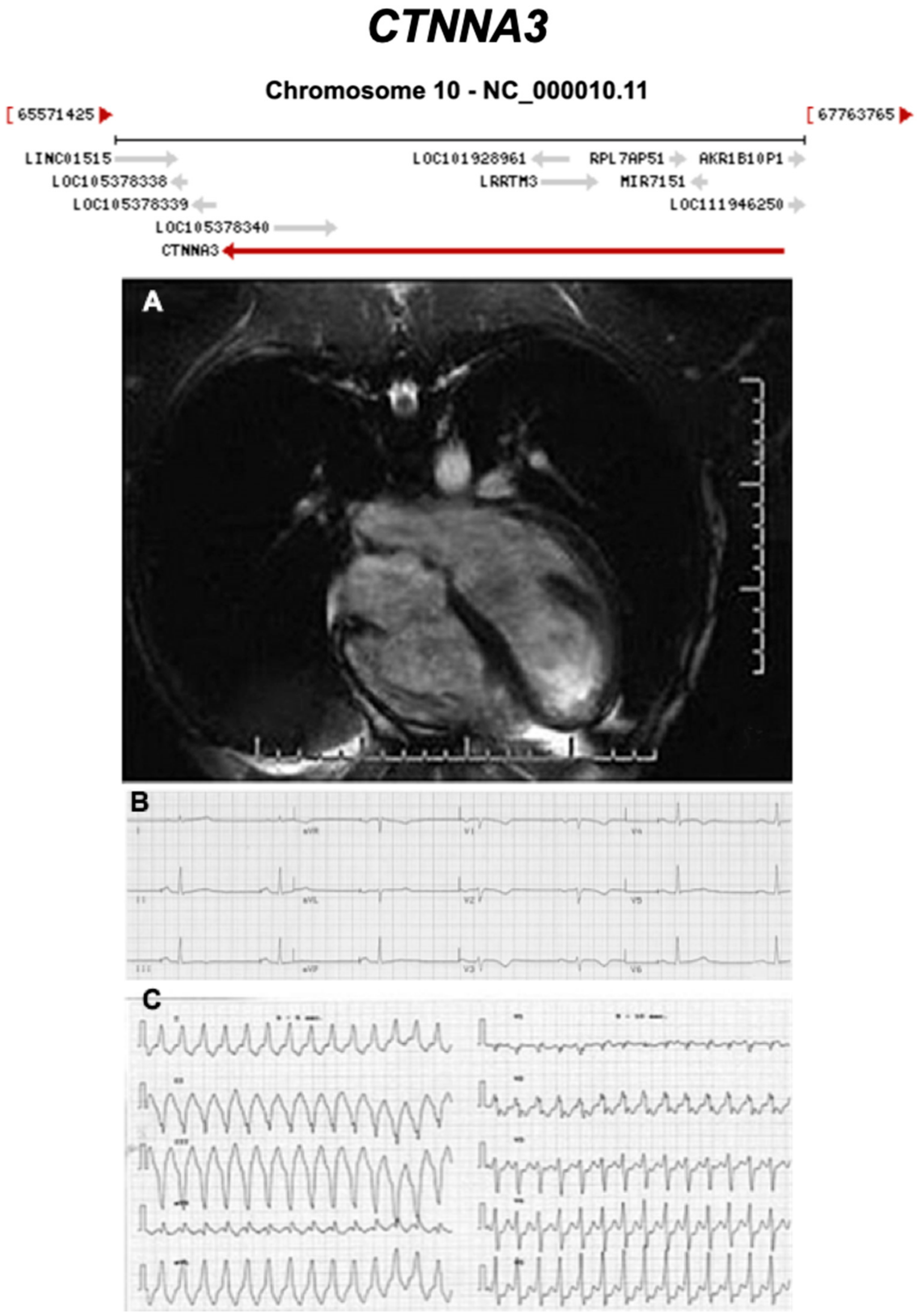
3.2.7. Catenin Alpha 3 (CTNNA3) (Encoded by CTNNA3)
3.2.8. Transmembrane 43 (Also Known as LUMA) (TMEM43) (Encoded by TMEM43)
3.2.9. Phospholamban (PLN) (Encoded by PLN)
3.2.10. Ryanodine Receptor 2 (RYR2) (Encoded by RYR2)
3.2.11. Voltage-Gated Sodium Channel (Nav1.5) (Encoded by SCN5A)
3.2.12. Transforming Growth Factor Beta 3 (TGFβ3) (Encoded by TGFB3)
3.2.13. RNA-Binding Motif Protein 20 (RBM20) (Encoded by RBM20)
3.2.14. BCL2-Associated Athanogene 3 (BAG3) (Encoded by BAG3)
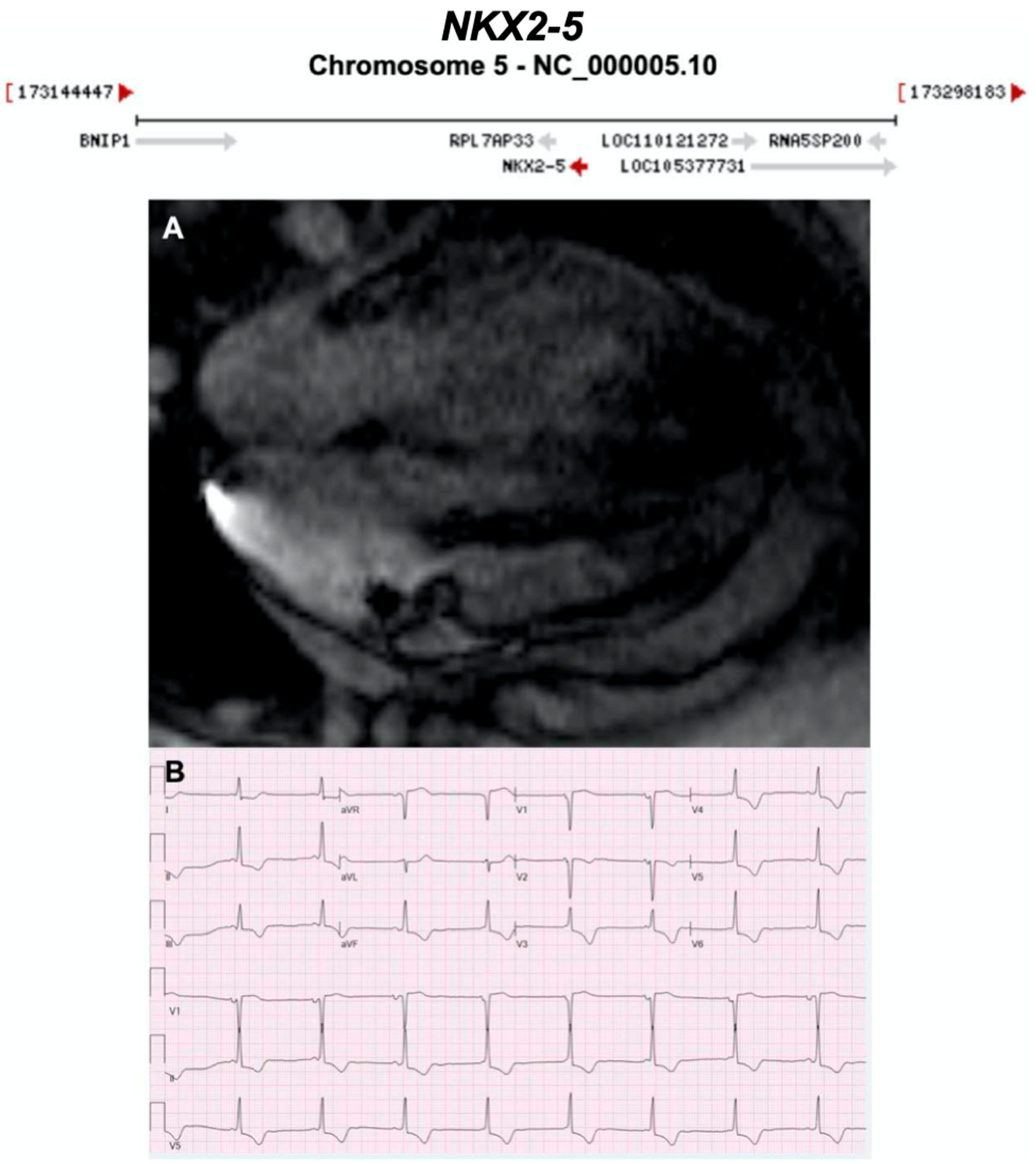
3.2.15. NK2 Homeobox 5 (NKX2-5) (Encoded by NKX2-5)
4. Effect Modifiers
4.1. Inflammation
4.2. Exercise
5. SCD Risk Prediction
6. Translational Perspectives and Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACM | Arrhythmogenic Cardiomyopathy |
| ACMG-AMP | American College of Medical Genetics and Genomics and the Association for Molecular Pathology |
| aDCM | Arrhythmogenic Dilated Cardiomyopathy |
| AD | Autosomal dominant |
| AF | Atrial Fibrillation |
| AHA | American Heart Association |
| AJ | Adherens Junction |
| ALVC | Arrhythmogenic Left Ventricular Cardiomyopathy |
| AP/MS | Affinity Purification and Mass Spectrometry |
| AR | Autosomal recessive |
| ARVC | Arrhythmogenic Right Ventricular Cardiomyopathy |
| ARVD | Arrhythmogenic Right Ventricular Dysplasia |
| ASD | Atrial Septal Defect |
| BiV | Biventricular |
| BivACM | Biventricular Arrhythmogenic Cardiomyopathy |
| BrS | Brugada syndrome |
| CMP | Cardiomyopathy |
| CMR | Cardiovascular magnetic resonance |
| CPVT | Catecholaminergic Polymorphic Ventricular Tachycardia |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DAD | Delayed After Depolarisations |
| DCM | Dilated Cardiomyopathy |
| ECG | Electrocardiogram |
| ENCODE | Encyclopedia of DNA Elements |
| ESC | European Society of Cardiology |
| FDG-PET | 18F-fluorodeoxyglucose positron emission tomography |
| G+ | Genotype negative |
| G- | Genotype negative |
| GSEA | Gene set enrichment analysis |
| GTEx | Genotype-Tissue expression project |
| GWAS | Genome-wide association studies |
| HCM | Hypertrophic Cardiomyopathy |
| HF | Heart failure |
| hiPSC-CMs | Human induced pluripotent stem cell cardiomyocytes |
| HNDC | Hypokinetic Non-dilated Cardiomyopathy |
| HRS | Heart Rhythm Society |
| ICD | Implantable Cardiac Defibrillator |
| ID | Intercalated Disc |
| ISFC | International Society and Federation of Cardiology |
| LBBB | Left Bundle Branch Block |
| LGE | Late gadolinium enhancement |
| LQTS | Long QT syndrome |
| LV | Left ventricle |
| LVEF | Left ventricular ejection fraction |
| MPRAs | Massive Parallel Reporter Assays |
| MR | Mitral Regurgitation |
| NSVT | Non-Sustained Ventricular Tachycardia |
| P+ | Phenotype positive |
| P- | Phenotype negative |
| PHTN | Pulmonary Hypertension |
| PVC | Premature Ventricular Contraction |
| RCM | Restrictive Cardiomyopathy |
| RV | Right ventricle |
| RVOT | Right ventricular outflow tract |
| RWMA | Regional wall motion abnormality |
| SAGE | Serial Analysis of Gene Expression |
| SC | Subcutaneous |
| SCD | Sudden Cardiac Death |
| SERCA | Sarcoplasmic reticulum Ca2+-ATPase |
| SR | Sarcoplasmic reticulum |
| TFC | Task Force Criteria |
| TV | Transvenous |
| TWI | T Wave Inversion |
| UTR | Untranslated regions |
| VT | Ventricular Tachycardia |
| VTA | Ventricular Tachyarrhythmia |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| WHO | World Health Organisation |
References
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B.; Medeiros-Domingo, A. Translating emerging molecular genetic insights into clinical practice in inherited cardiomyopathies. J. Mol. Med. 2018, 96, 993–1024. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the task force criteria. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef]
- Michalodimitrakis, M.; Papadomanolakis, A.; Stiakakis, J.; Kanaki, K. Left side right ventricular cardiomyopathy. Med. Sci. Law 2002, 42, 313–317. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-dominant arrhythmogenic cardiomyopathy: An under-recognized clinical entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef]
- Berte, B.; Denis, A.; Amraoui, S.; Yamashita, S.; Komatsu, Y.; Pillois, X.; Sacher, F.; Mahida, S.; Wielandts, J.Y.; Sellal, J.M.; et al. Characterization of the left-sided substrate in arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2015, 8, 1403–1412. [Google Scholar] [CrossRef]
- Roberts, W.C.; Ko, J.M.; Kuiper, J.J.; Hall, S.A.; Meyer, D.M. Some previously neglected examples of arrhythmogenic right ventricular dysplasia/cardiomyopathy and frequency of its various reported manifestations. Am. J. Cardiol. 2010, 106, 268–274. [Google Scholar] [CrossRef]
- Calkins, H. Arrhythmogenic right ventricular dysplasia. Curr. Probl. Cardiol. 2013, 38, 103–123. [Google Scholar] [CrossRef]
- Thiene, G.; Rizzo, S.; Pilichou, K.; Basso, C. Chapter 2—Arrhythmogenic cardiomyopathy: History and pathology. In Cardiac MRI in Diagnosis, Clinical Management, and Prognosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia; Abidov, A., Oliva, I.B., Marcus, F.I., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 5–33. [Google Scholar] [CrossRef]
- Corrado, D.; van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef] [PubMed]
- Brigden, W. Uncommon myocardial diseases: The non-coronary cardiomyopathies. Lancet 1957, 273, 1243–1249. [Google Scholar] [CrossRef]
- Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies. Br. Heart J. 1980, 44, 672–673. [CrossRef] [PubMed]
- Fontaine, G.; Guiraudon, G.; Frank, R.; Vedel, J.; Grosgogeat, Y.; Cabrol, C.; Facquet, J. Stimulation Studies and Epicardial Mapping in Ventricular Tachycardia: Study of Mechanisms and Selection for Surgery; University Park Press: Baltimore, MD, USA, 1997; pp. 334–350. [Google Scholar]
- Thiene, G.; Nava, A.; Corrado, D.; Rossi, L.; Pennelli, N. Right ventricular cardiomyopathy and sudden death in young people. N. Engl. J. Med. 1988, 318, 129–133. [Google Scholar] [CrossRef] [PubMed]
- McKenna, W.J.; Thiene, G.; Nava, A.; Fontaliran, F.; Blomstrom-Lundqvist, C.; Fontaine, G.; Camerini, F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task force of the working group myocardial and pericardial disease of the european society of cardiology and of the scientific council on cardiomyopathies of the international society and federation of cardiology. Br. Heart J. 1994, 71, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O’Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 world health organization/international society and federation of cardiology task force on the definition and classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef]
- Maron, B.J.; Towbin, J.A.; Thiene, G.; Antzelevitch, C.; Corrado, D.; Arnett, D.; Moss, A.J.; Seidman, C.E.; Young, J.B.; American Heart Association; et al. Contemporary definitions and classification of the cardiomyopathies: An American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; Quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef]
- Arbustini, E.; Narula, N.; Tavazzi, L.; Serio, A.; Grasso, M.; Favalli, V.; Bellazzi, R.; Tajik, J.A.; Bonow, R.O.; Fuster, V.; et al. The MOGE(S) classification of cardiomyopathy for clinicians. J. Am. Coll. Cardiol. 2014, 64, 304–318. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brooke, M.A.; Calkins, H.; Corrado, D.; Duru, F.; Green, K.J.; et al. Definition and treatment of arrhythmogenic cardiomyopathy: An updated expert panel report. Eur. J. Heart Fail. 2019, 21, 955–964. [Google Scholar] [CrossRef]
- Corrado, D.; Perazzolo Marra, M.; Zorzi, A.; Beffagna, G.; Cipriani, A.; Lazzari, M.; Migliore, F.; Pilichou, K.; Rampazzo, A.; Rigato, I.; et al. Diagnosis of arrhythmogenic cardiomyopathy: The padua criteria. Int. J. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Böhm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Khanji, M.Y.; Chahal, A.A.; Lopes, L.R.; Petersen, S.E. Cardiovascular magnetic resonance imaging volume criteria for arrhythmogenic right ventricular cardiomyopathy: Need for update? Eur. Heart J. 2020, 41, 1451. [Google Scholar] [CrossRef]
- Corrado, D.; Cipriani, A.; De Lazzari, M.; Marra, M.P. Right ventricular dilatation in arrhythmogenic right ventricular cardiomyopathy: Need for a revision of the 2010 international task force criteria. Eur. Heart J. 2020, 41, 1452–1453. [Google Scholar] [CrossRef] [PubMed]
- Protonotarios, N.; Tsatsopoulou, A.; Patsourakos, P.; Alexopoulos, D.; Gezerlis, P.; Simitsis, S.; Scampardonis, G. Cardiac abnormalities in familial palmoplantar keratosis. Br. Heart J. 1986, 56, 321–326. [Google Scholar] [CrossRef]
- McKoy, G.; Protonotarios, N.; Crosby, A.; Tsatsopoulou, A.; Anastasakis, A.; Coonar, A.; Norman, M.; Baboonian, C.; Jeffery, S.; McKenna, W.J. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 2000, 355, 2119–2124. [Google Scholar] [CrossRef]
- Carvajal-Huerta, L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J. Am. Acad. Dermatol. 1998, 39, 418–421. [Google Scholar] [CrossRef]
- Norgett, E.E.; Hatsell, S.J.; Carvajal-Huerta, L.; Ruiz Cabezas, J.-C.; Common, J.; Purkis, P.E.; Whittock, N.; Leigh, I.M.; Stevens, H.P.; Kelsell, D.P. Recessive mutation in desmoplakin disrupts desmoplakin—Intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum. Mol. Genet. 2000, 9, 2761–2766. [Google Scholar] [CrossRef]
- James, C.A.; Syrris, P.; van Tintelen, J.P.; Calkins, H. The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1393–1400. [Google Scholar] [CrossRef]
- Gandjbakhch, E.; Redheuil, A.; Pousset, F.; Charron, P.; Frank, R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2018, 72, 784–804. [Google Scholar] [CrossRef]
- Gene Validity Curation—ClinGen Knowledge Base | Clinical Genome Resource. Available online: https://www.clinicalgenome.org/ (accessed on 21 July 2020).
- Trenkwalder, T.; Deisenhofer, I.; Hadamitzky, M.; Schunkert, H.; Reinhard, W. Novel frame-shift mutation in PKP2 associated with arrhythmogenic right ventricular cardiomyopathy: A case report. BMC Med. Genet. 2015, 16, 117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, P.; Li, Z.; Yu, B.; Ma, F.; Li, X.; Wang, D.W. Distal myopathy induced arrhythmogenic right ventricular cardiomyopathy in a pedigree carrying novel DSG2 null variant. Int. J. Cardiol. 2020, 298, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Mattesi, G.; Zorzi, A.; Corrado, D.; Cipriani, A. Natural history of arrhythmogenic cardiomyopathy. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Gaido, L.; Battaglia, A.; Matta, M.; Giustetto, C.; Frea, S.; Imazio, M.; Richiardi, E.; Garberoglio, L.; Gaita, F. Phenotypic expression of ARVC: How 12 lead ECG can predict left or right ventricle involvement. A familiar case series and a review of literature. Int. J. Cardiol. 2017, 236, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.; Bratis, K.; Protonotarios, N.; Tsatsopoulou, A.; Papadopoulos, G. Cardiac magnetic resonance can early assess the presence and severity of heart involvement in Naxos disease. Int. J. Cardiol. 2012, 154, e19–e20. [Google Scholar] [CrossRef]
- Augusto, J.B.; Eiros, R.; Nakou, E.; Moura-Ferreira, S.; Treibel, T.A.; Captur, G.; Akhtar, M.M.; Protonotarios, A.; Gossios, T.D.; Savvatis, K.; et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: A comprehensive genotype-imaging phenotype study. Eur. Heart J. Cardiovasc. Imaging 2020, 21, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Graw, S.; Sinagra, G.; Barnes, C.; Slavov, D.; Brun, F.; Pinamonti, B.; Salcedo, E.E.; Sauer, W.; Pyxaras, S.; et al. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 2011, 124, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Quarta, G.; Syrris, P.; Ashworth, M.; Jenkins, S.; Alapi, K.Z.; Morgan, J.; Muir, A.; Pantazis, A.; McKenna, W.J.; Elliott, P.M. Mutations in the lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2012, 33, 1128–1136. [Google Scholar] [CrossRef]
- Koitka, K.; Dahiya, A.; Lo, A.; Scalia, G.M.; Atherton, J.J.; Prasad, S.B. Myofibrillar cardiomyopathy due to a novel desmin gene mutation: Complementary role of echocardiography, cardiac magnetic resonance, and genetic testing in delineating diagnosis. CASE 2017, 1, 28–33. [Google Scholar] [CrossRef]
- Hall, C.L.; Akhtar, M.M.; Sabater-Molina, M.; Futema, M.; Asimaki, A.; Protonotarios, A.; Dalageorgou, C.; Pittman, A.M.; Suarez, M.P.; Aguilera, B.; et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int. J. Cardiol. 2020, 307, 101–108. [Google Scholar] [CrossRef]
- De Bortoli, M.; Alex, V.P.; Poloni, G.; Calore, M.; Minervini, G.; Mazzotti, E.; Rigato, I.; Ebert, M.; Lorenzon, A.; Vazza, G.; et al. Whole-exome sequencing identifies pathogenic variants in TJP1 gene associated with arrhythmogenic cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e002123. [Google Scholar] [CrossRef] [PubMed]
- van Hengel, J.; Calore, M.; Bauce, B.; Dazzo, E.; Mazzotti, E.; De Bortoli, M.; Lorenzon, A.; Li Mura, I.E.A.; Beffagna, G.; Rigato, I.; et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, F.; Zorio, E.; Jimenez-Jaimez, J.; Salguero-Bodes, R.; Zwart, R.; Gonzalez-Lopez, E.; Molina, P.; Bermudez-Jimenez, F.; Delgado, J.F.; Braza-Boils, A.; et al. Clinical characteristics and determinants of the phenotype in TMEM43 arrhythmogenic right ventricular cardiomyopathy type 5. Heart Rhythm 2020, 17, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Te Rijdt, W.P.; Ten Sande, J.N.; Gorter, T.M.; van der Zwaag, P.A.; van Rijsingen, I.A.; Boekholdt, S.M.; van Tintelen, J.P.; van Haelst, P.L.; Planken, R.N.; de Boer, R.A.; et al. Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: Phenotypic insights from cardiovascular magnetic resonance imaging. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.; Medeiros-Domingo, A.; Gasperetti, A.; Breitenstein, A.; Steffel, J.; Guidetti, F.; Flammer, A.; Odening, K.; Ruschitzka, F.; Duru, F.; et al. Familial dilated cardiomyopathy associated with a novel heterozygous RYR2 early truncating variant. Cardiol. J. 2020. [Google Scholar] [CrossRef]
- Te Riele, A.S.J.M.; Agullo-Pascual, E.; James, C.A.; Leo-Macias, A.; Cerrone, M.; Zhang, M.; Lin, X.; Lin, B.; Sobreira, N.L.; Amat-Alarcon, N.; et al. Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non-canonical mechanisms for disease pathogenesis. Cardiovasc. Res. 2017, 113, 102–111. [Google Scholar] [CrossRef]
- Delmar, M.; McKenna, W.J. The cardiac desmosome and arrhythmogenic cardiomyopathies: From gene to disease. Circ. Res. 2010, 107, 700–714. [Google Scholar] [CrossRef]
- Najor, N.A. Desmosomes in human disease. Annu. Rev. Pathol. 2018, 13, 51–70. [Google Scholar] [CrossRef]
- Vermij, S.H.; Abriel, H.; van Veen, T.A.B. Refining the molecular organization of the cardiac intercalated disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef]
- Agullo-Pascual, E.; Cerrone, M.; Delmar, M. Arrhythmogenic cardiomyopathy and Brugada syndrome: Diseases of the connexome. FEBS Lett. 2014, 588, 1322–1330. [Google Scholar] [CrossRef]
- Coonar, A.S.; Protonotarios, N.; Tsatsopoulou, A.; Needham, E.W.; Houlston, R.S.; Cliff, S.; Otter, M.I.; Murday, V.A.; Mattu, R.K.; McKenna, W.J. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse nonepidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation 1998, 97, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; Edson, S.; Towbin, J.A. Genetics of arrhythmogenic right ventricular cardiomyopathy: A practical guide for physicians. J. Am. Coll. Cardiol. 2013, 61, 1945–1948. [Google Scholar] [CrossRef] [PubMed]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.; Murray, B.; te Riele, A.S.; van den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.G.; van der Zwaag, P.A.; van der Werf, C.; van der Smagt, J.J.; Noorman, M.; Bhuiyan, Z.A.; Wiesfeld, A.C.; Volders, P.G.; van Langen, I.M.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 2011, 123, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Alcalai, R.; Metzger, S.; Rosenheck, S.; Meiner, V.; Chajek-Shaul, T. A recessive mutation in desmoplakin causes arrhythmogenic right ventricular dysplasia, skin disorder, and woolly hair. J. Am. Coll. Cardiol. 2003, 42, 319–327. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A. Naxos disease and carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef]
- Protonotarios, A.; Anastasakis, A.; Panagiotakos, D.B.; Antoniades, L.; Syrris, P.; Vouliotis, A.; Stefanadis, C.; Tsatsopoulou, A.; McKenna, W.J.; Protonotarios, N. Arrhythmic risk assessment in genotyped families with arrhythmogenic right ventricular cardiomyopathy. Europace 2016, 18, 610–616. [Google Scholar] [CrossRef]
- Xu, Z.; Zhu, W.; Wang, C.; Huang, L.; Zhou, Q.; Hu, J.; Cheng, X.; Hong, K. Genotype-phenotype relationship in patients with arrhythmogenic right ventricular cardiomyopathy caused by desmosomal gene mutations: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 41387. [Google Scholar] [CrossRef]
- Pinamonti, B.; Dragos, A.M.; Pyxaras, S.A.; Merlo, M.; Pivetta, A.; Barbati, G.; Di Lenarda, A.; Morgera, T.; Mestroni, L.; Sinagra, G. Prognostic predictors in arrhythmogenic right ventricular cardiomyopathy: Results from a 10-year registry. Eur. Heart J. 2011, 32, 1105–1113. [Google Scholar] [CrossRef]
- Sarantitis, I.; Papanastasopoulos, P.; Manousi, M.; Baikoussis, N.G.; Apostolakis, E. The cytoskeleton of the cardiac muscle cell. Hell. J. Cardiol. 2012, 53, 367–379. [Google Scholar]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef] [PubMed]
- Granzier, H.L.; Labeit, S. The giant protein titin: A major player in myocardial mechanics, signaling, and disease. Circ. Res. 2004, 94, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P.; Tsai, E.J.; Hilfiker-Kleiner, D.; Kamiya, C.A.; Mazzarotto, F.; et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N. Engl. J. Med. 2016, 374, 233–241. [Google Scholar] [CrossRef]
- Brun, F.; Barnes, C.V.; Sinagra, G.; Slavov, D.; Barbati, G.; Zhu, X.; Graw, S.L.; Spezzacatene, A.; Pinamonti, B.; Merlo, M.; et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2014, 51, 669–676. [Google Scholar] [CrossRef]
- Forleo, C.; Carmosino, M.; Resta, N.; Rampazzo, A.; Valecce, R.; Sorrentino, S.; Iacoviello, M.; Pisani, F.; Procino, G.; Gerbino, A.; et al. Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS ONE 2015, 10, e0121723. [Google Scholar] [CrossRef]
- Kato, K.; Takahashi, N.; Fujii, Y.; Umehara, A.; Nishiuchi, S.; Makiyama, T.; Ohno, S.; Horie, M. LMNA cardiomyopathy detected in Japanese arrhythmogenic right ventricular cardiomyopathy cohort. J. Cardiol. 2016, 68, 346–351. [Google Scholar] [CrossRef]
- Chen, W.; Huo, J.; Ma, A.; Bai, L.; Liu, P. A novel mutation of the LMNA gene in a family with dilated cardiomyopathy, conduction system disease, and sudden cardiac death of young females. Mol. Cell. Biochem. 2013, 382, 307–311. [Google Scholar] [CrossRef]
- Marian, A.J.; Asatryan, B.; Wehrens, X.H.T. Genetic basis and molecular biology of cardiac arrhythmias in cardiomyopathies. Cardiovasc. Res. 2020, 116, 1600–1619. [Google Scholar] [CrossRef]
- Van Steensel, B.; Belmont, A.S. Lamina-associated domains: Links with chromosome architecture, heterochromatin, and gene repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef]
- Cheedipudi, S.M.; Matkovich, S.J.; Coarfa, C.; Hu, X.; Robertson, M.J.; Sweet, M.; Taylor, M.; Mestroni, L.; Cleveland, J.; Willerson, J.T.; et al. Genomic reorganization of lamin-associated domains in cardiac myocytes is associated with differential gene expression and DNA methylation in human dilated cardiomyopathy. Circ. Res. 2019, 124, 1198–1213. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, N.; Crasto, S.; Miragoli, M.; Bertero, A.; Paulis, M.; Kunderfranco, P.; Serio, S.; Forni, A.; Lucarelli, C.; Dal Ferro, M.; et al. The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat. Commun. 2019, 10, 2267. [Google Scholar] [CrossRef] [PubMed]
- Wahbi, K.; Ben Yaou, R.; Gandjbakhch, E.; Anselme, F.; Gossios, T.; Lakdawala, N.K.; Stalens, C.; Sacher, F.; Babuty, D.; Trochu, J.N.; et al. Development and validation of a new risk prediction score for life-threatening ventricular tachyarrhythmias in laminopathies. Circulation 2019, 140, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R.; Robbins, J. Desmin and cardiac disease: An unfolding story. Circ. Res. 2018, 122, 1324–1326. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Van Gelder, I.C.; Asimaki, A.; Suurmeijer, A.J.; Wiesfeld, A.C.; Jongbloed, J.D.; van den Wijngaard, A.; Kuks, J.B.; van Spaendonck-Zwarts, K.Y.; Notermans, N.; et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm 2009, 6, 1574–1583. [Google Scholar] [CrossRef]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.Á.; Álvarez, M.; López-Fernández, S.; et al. Novel desmin mutation p.Glu401Asp impairs filament formation, disrupts cell membrane integrity, and causes severe arrhythmogenic left ventricular cardiomyopathy/dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Brodehl, A.; Dieding, M.; Klauke, B.; Dec, E.; Madaan, S.; Huang, T.; Gargus, J.; Fatima, A.; Saric, T.; Cakar, H.; et al. The novel desmin mutant p.A120D impairs filament formation, prevents intercalated disk localization, and causes sudden cardiac death. Circ. Cardiovasc. Genet. 2013, 6, 615–623. [Google Scholar] [CrossRef]
- Hedberg, C.; Melberg, A.; Kuhl, A.; Jenne, D.; Oldfors, A. Autosomal dominant myofibrillar myopathy with arrhythmogenic right ventricular cardiomyopathy 7 is caused by a DES mutation. Eur. J. Hum. Genet. 2012, 20, 984–985. [Google Scholar] [CrossRef]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef]
- Levin, J.; Bulst, S.; Thirion, C.; Schmidt, F.; Bötzel, K.; Krause, S.; Pertl, C.; Kretzschmar, H.; Walter, M.C.; Giese, A.; et al. Divergent molecular effects of desmin mutations on protein assembly in myofibrillar myopathy. J. Neuropathol. Exp. Neurol. 2010, 69, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Strach, K.; Sommer, T.; Grohé, C.; Meyer, C.; Fischer, D.; Walter, M.C.; Vorgerd, M.; Reilich, P.; Bär, H.; Reimann, J.; et al. Clinical, genetic, and cardiac magnetic resonance imaging findings in primary desminopathies. Neuromuscul. Disord. NMD 2008, 18, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Fürst, D.O.; Goldfarb, L.G.; Kley, R.A.; Vorgerd, M.; Olivé, M.; van der Ven, P.F.M. Filamin C-related myopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.-A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are associated with familial restrictive cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Begay, R.L.; Graw, S.L.; Sinagra, G.; Asimaki, A.; Rowland, T.J.; Slavov, D.B.; Gowan, K.; Jones, K.L.; Brun, F.; Merlo, M.; et al. Filamin C truncation mutations are associated with arrhythmogenic dilated cardiomyopathy and changes in the cell-cell adhesion structures. JACC Clin. Electrophysiol. 2018, 4, 504–514. [Google Scholar] [CrossRef]
- Mayosi, B.M.; Fish, M.; Shaboodien, G.; Mastantuono, E.; Kraus, S.; Wieland, T.; Kotta, M.-C.; Chin, A.; Laing, N.; Ntusi, N.B.A.; et al. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10. [Google Scholar] [CrossRef]
- Turkowski, K.L.; Tester, D.J.; Bos, J.M.; Haugaa, K.H.; Ackerman, M.J. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit Heart Dis. 2017, 12, 226–235. [Google Scholar] [CrossRef]
- Kaufmann, U.; Zuppinger, C.; Waibler, Z.; Rudiger, M.; Urbich, C.; Martin, B.; Jockusch, B.M.; Eppenberger, H.; Starzinski-Powitz, A. The armadillo repeat region targets ARVCF to cadherin-based cellular junctions. J. Cell Sci. 2000, 113, 4121–4135. [Google Scholar]
- Goossens, S.; Janssens, B.; Bonné, S.; De Rycke, R.; Braet, F.; van Hengel, J.; van Roy, F. A unique and specific interaction between alphaT-catenin and plakophilin-2 in the area composita, the mixed-type junctional structure of cardiac intercalated discs. J. Cell Sci. 2007, 120, 2126–2136. [Google Scholar] [CrossRef]
- Li, J.; Goossens, S.; van Hengel, J.; Gao, E.; Cheng, L.; Tyberghein, K.; Shang, X.; De Rycke, R.; van Roy, F.; Radice, G.L. Loss of alphaT-catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J. Cell Sci. 2012, 125, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Merner, N.D.; Hodgkinson, K.A.; Haywood, A.F.M.; Connors, S.; French, V.M.; Drenckhahn, J.-D.; Kupprion, C.; Ramadanova, K.; Thierfelder, L.; McKenna, W.; et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am. J. Hum. Genet. 2008, 82, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Siragam, V.; Cui, X.; Masse, S.; Ackerley, C.; Aafaqi, S.; Strandberg, L.; Tropak, M.; Fridman, M.D.; Nanthakumar, K.; Liu, J.; et al. TMEM43 mutation p.S358L alters intercalated disc protein expression and reduces conduction velocity in arrhythmogenic right ventricular cardiomyopathy. PLoS ONE 2014, 9, e109128. [Google Scholar] [CrossRef] [PubMed]
- Baskin, B.; Skinner, J.R.; Sanatani, S.; Terespolsky, D.; Krahn, A.D.; Ray, P.N.; Scherer, S.W.; Hamilton, R.M. TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Hum. Genet. 2013, 132, 1245–1252. [Google Scholar] [CrossRef]
- Haywood, A.F.; Merner, N.D.; Hodgkinson, K.A.; Houston, J.; Syrris, P.; Booth, V.; Connors, S.; Pantazis, A.; Quarta, G.; Elliott, P.; et al. Recurrent missense mutations in TMEM43 (ARVD5) due to founder effects cause arrhythmogenic cardiomyopathies in the UK and Canada. Eur. Heart J. 2013, 34, 1002–1011. [Google Scholar] [CrossRef]
- McTiernan, C.F.; Frye, C.S.; Lemster, B.H.; Kinder, E.A.; Ogletree-Hughes, M.L.; Moravec, C.S.; Feldman, A.M. The human phospholamban gene: Structure and expression. J. Mol. Cell. Cardiol. 1999, 31, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Van der Zwaag, P.A.; van Rijsingen, I.A.W.; Asimaki, A.; Jongbloed, J.D.H.; van Veldhuisen, D.J.; Wiesfeld, A.C.P.; Cox, M.G.P.J.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.W.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Antzelevitch, C.; Burashnikov, A. Overview of basic mechanisms of cardiac arrhythmia. Card. Electrophysiol. Clin. 2011, 3, 23–45. [Google Scholar] [CrossRef]
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq transatlantic network of excellence to cure phospholamban-induced cardiomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724. [Google Scholar] [CrossRef]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Kannankeril, P.J.; Mitchell, B.M.; Goonasekera, S.A.; Chelu, M.G.; Zhang, W.; Sood, S.; Kearney, D.L.; Danila, C.I.; De Biasi, M.; Wehrens, X.H.; et al. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc. Natl. Acad. Sci. USA 2006, 103, 12179–12184. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, Z.A.; van den Berg, M.P.; van Tintelen, J.P.; Bink-Boelkens, M.T.; Wiesfeld, A.C.; Alders, M.; Postma, A.V.; van Langen, I.; Mannens, M.M.; Wilde, A.A. Expanding spectrum of human RYR2-related disease: New electrocardiographic, structural, and genetic features. Circulation 2007, 116, 1569–1576. [Google Scholar] [CrossRef] [PubMed]
- McNair, W.P.; Sinagra, G.; Taylor, M.R.; Di Lenarda, A.; Ferguson, D.A.; Salcedo, E.E.; Slavov, D.; Zhu, X.; Caldwell, J.H.; Mestroni, L.; et al. SCN5A mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. J. Am. Coll. Cardiol. 2011, 57, 2160–2168. [Google Scholar] [CrossRef]
- Asatryan, B. Cardiac sodium channel dysfunction and dilated cardiomyopathy: A contemporary reappraisal of pathophysiological concepts. J. Clin. Med. 2019, 8, 1029. [Google Scholar] [CrossRef]
- Beffagna, G.; Occhi, G.; Nava, A.; Vitiello, L.; Ditadi, A.; Basso, C.; Bauce, B.; Carraro, G.; Thiene, G.; Towbin, J.A.; et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc. Res. 2005, 65, 366–373. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Danieli, G.A.; Buja, G.; Daliento, L.; Fasoli, G.; Scognamiglio, R.; Corrado, D.; Thiene, G. The gene for arrhythmogenic right ventricular cardiomyopathy maps to chromosome 14q23-q24. Hum. Mol. Genet. 1994, 3, 959–962. [Google Scholar] [CrossRef]
- Overall, C.M.; Wrana, J.L.; Sodek, J. Independent regulation of collagenase, 72-kDa progelatinase, and metalloendoproteinase inhibitor expression in human fibroblasts by transforming growth factor-beta. J. Biol. Chem. 1989, 264, 1860–1869. [Google Scholar]
- van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef]
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic genotypes in familial dilated cardiomyopathy: Implications for genetic testing and clinical management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Parikh, V.N.; Caleshu, C.; Reuter, C.; Lazzeroni, L.C.; Ingles, J.; Garcia, J.; McCaleb, K.; Adesiyun, T.; Sedaghat-Hamedani, F.; Kumar, S.; et al. Regional variation in RBM20 causes a highly penetrant arrhythmogenic cardiomyopathy. Circ. Heart Fail. 2019, 12, e005371. [Google Scholar] [CrossRef] [PubMed]
- Hey, T.M.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Molgaard, H.; Moller, J.E.; Eiskjaer, H.; Mogensen, J. Pathogenic RBM20-variants are associated with a severe disease expression in male patients with dilated cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef] [PubMed]
- Brauch, K.M.; Karst, M.L.; Herron, K.J.; de Andrade, M.; Pellikka, P.A.; Rodeheffer, R.J.; Michels, V.V.; Olson, T.M. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 2009, 54, 930–941. [Google Scholar] [CrossRef]
- Dominguez, F.; Cuenca, S.; Bilinska, Z.; Toro, R.; Villard, E.; Barriales-Villa, R.; Ochoa, J.P.; Asselbergs, F.; Sammani, A.; Franaszczyk, M.; et al. Dilated cardiomyopathy due to BLC2-associated athanogene 3 (BAG3) mutations. J. Am. Coll. Cardiol. 2018, 72, 2471–2481. [Google Scholar] [CrossRef]
- Norton, N.; Li, D.; Rieder, M.J.; Siegfried, J.D.; Rampersaud, E.; Zuchner, S.; Mangos, S.; Gonzalez-Quintana, J.; Wang, L.; McGee, S.; et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 273–282. [Google Scholar] [CrossRef]
- Arimura, T.; Ishikawa, T.; Nunoda, S.; Kawai, S.; Kimura, A. Dilated cardiomyopathy-associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum. Mutat. 2011, 32, 1481–1491. [Google Scholar] [CrossRef]
- Feldman, A.M.; Begay, R.L.; Knezevic, T.; Myers, V.D.; Slavov, D.B.; Zhu, W.; Gowan, K.; Graw, S.L.; Jones, K.L.; Tilley, D.G.; et al. Decreased levels of BAG3 in a family with a rare variant and in idiopathic dilated cardiomyopathy. J. Cell. Physiol. 2014, 229, 1697–1702. [Google Scholar] [CrossRef]
- Franaszczyk, M.; Bilinska, Z.T.; Sobieszczanska-Malek, M.; Michalak, E.; Sleszycka, J.; Sioma, A.; Malek, L.A.; Kaczmarska, D.; Walczak, E.; Wlodarski, P.; et al. The BAG3 gene variants in polish patients with dilated cardiomyopathy: Four novel mutations and a genotype-phenotype correlation. J. Transl. Med. 2014, 12, 192. [Google Scholar] [CrossRef]
- Myers, V.D.; Gerhard, G.S.; McNamara, D.M.; Tomar, D.; Madesh, M.; Kaniper, S.; Ramsey, F.V.; Fisher, S.G.; Ingersoll, R.G.; Kasch-Semenza, L.; et al. Association of variants in BAG3 with cardiomyopathy outcomes in african american individuals. JAMA Cardiol. 2018, 3, 929–938. [Google Scholar] [CrossRef]
- Reamon-Buettner, S.M.; Borlak, J. NKX2-5: An update on this hypermutable homeodomain protein and its role in human congenital heart disease (CHD). Hum. Mutat. 2010, 31, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.W.; Silberbach, G.M.; Kavanaugh-McHugh, A.; Cottrill, C.; Zhang, Y.; Riggs, S.; Smalls, O.; Johnson, M.C.; Watson, M.S.; Seidman, J.G.; et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J. Clin. Investig. 1999, 104, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Qiu, X.B.; Li, R.G.; Qu, X.K.; Wang, J.; Xu, Y.J.; Liu, X.; Fang, W.Y.; Yang, Y.Q.; Liao, D.N. A novel NKX2-5 loss-of-function mutation predisposes to familial dilated cardiomyopathy and arrhythmias. Int. J. Mol. Med. 2015, 35, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Perera, J.L.; Johnson, N.M.; Judge, D.P.; Crosson, J.E. Novel and highly lethal NKX2.5 missense mutation in a family with sudden death and ventricular arrhythmia. Pediatric Cardiol. 2014, 35, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Sveinbjornsson, G.; Olafsdottir, E.F.; Thorolfsdottir, R.B.; Davidsson, O.B.; Helgadottir, A.; Jonasdottir, A.; Jonasdottir, A.; Bjornsson, E.; Jensson, B.O.; Arnadottir, G.A.; et al. Variants in NKX2-5 and FLNC cause dilated cardiomyopathy and sudden cardiac death. Circ. Genom. Precis. Med. 2018, 11, e002151. [Google Scholar] [CrossRef]
- James, C.A.; Calkins, H. Arrhythmogenic right ventricular cardiomyopathy: Progress toward personalized management. Annu. Rev. Med. 2019, 70, 1–18. [Google Scholar] [CrossRef]
- Judith, A.G.; Bhonsale, A.; Cynthia, A.J.; te Anneline, S.R.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld Ans, C.P.; Abhishek, C.S.; Kassamali, B.; et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Asimaki, A.; Tandri, H.; Duffy, E.R.; Winterfield, J.R.; Mackey-Bojack, S.; Picken, M.M.; Cooper, L.T.; Wilber, D.J.; Marcus, F.I.; Basso, C.; et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2011, 4, 743–752. [Google Scholar] [CrossRef]
- Campian, M.E.; Verberne, H.J.; Hardziyenka, M.; de Groot, E.A.A.; van Moerkerken, A.F.; van Eck-Smit, B.L.F.; Tan, H.L. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2079–2085. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 2019, 140, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Sabel, K.G.; Blomstrom-Lundqvist, C.; Olsson, S.B.; Enestrom, S. Arrhythmogenic right ventricular dysplasia in brother and sister: Is it related to myocarditis? Pediatr. Cardiol. 1990, 11, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Mavrogeni, S.; Protonotarios, N.; Tsatsopoulou, A.; Papachristou, P.; Sfendouraki, E.; Papadopoulos, G. Naxos disease evolution mimicking acute myocarditis: The role of cardiovascular magnetic resonance imaging. Int. J. Cardiol. 2013, 166, e14–e15. [Google Scholar] [CrossRef] [PubMed]
- Patrianakos, A.P.; Protonotarios, N.; Nyktari, E.; Pagonidis, K.; Tsatsopoulou, A.; Parthenakis, F.I.; Vardas, P.E. Arrhythmogenic right ventricular cardiomyopathy/dysplasia and troponin release. Myocarditis or the “hot phase” of the disease? Int. J. Cardiol. 2012, 157, e26–e28. [Google Scholar] [CrossRef] [PubMed]
- DeWitt, E.S.; Chandler, S.F.; Hylind, R.J.; Ladouceur, V.B.; Blume, E.D.; VanderPluym, C.; Powell, A.J.; Fynn-Thompson, F.; Roberts, A.E.; Sanders, S.P.; et al. Phenotypic manifestations of arrhythmogenic cardiomyopathy in children and adolescents. J. Am. Coll. Cardiol. 2019, 74, 346–358. [Google Scholar] [CrossRef]
- Protonotarios, A.; Wicks, E.; Ashworth, M.; Stephenson, E.; Guttmann, O.; Savvatis, K.; Sekhri, N.; Mohiddin, S.A.; Syrris, P.; Menezes, L.; et al. Prevalence of (18)F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. Int. J. Cardiol. 2019, 284, 99–104. [Google Scholar] [CrossRef]
- Lopez-Ayala, J.M.; Pastor-Quirante, F.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Gimeno, J.R. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm 2015, 12, 766–773. [Google Scholar] [CrossRef]
- Reichl, K.; Kreykes, S.E.; Martin, C.M.; Shenoy, C. Desmoplakin variant-associated arrhythmogenic cardiomyopathy presenting as acute myocarditis. Circ. Genom. Precis. Med. 2018, 11, e002373. [Google Scholar] [CrossRef]
- Chatterjee, D.; Fatah, M.; Akdis, D.; Spears, D.A.; Koopmann, T.T.; Mittal, K.; Rafiq, M.A.; Cattanach, B.M.; Zhao, Q.; Healey, J.S.; et al. An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur. Heart J. 2018, 39, 3932–3944. [Google Scholar] [CrossRef]
- Caforio, A.L.P.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.Y.; et al. Evidence from family studies for autoimmunity in arrhythmogenic right ventricular cardiomyopathy: associations of circulating anti-heart and anti-intercalated disk autoantibodies with disease severity and family history. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef]
- James, C.A.; Bhonsale, A.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol. 2013, 62, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Ruwald, A.-C.; Marcus, F.; Estes, N.A.M., 3rd; Link, M.; McNitt, S.; Polonsky, B.; Calkins, H.; Towbin, J.A.; Moss, A.J.; Zareba, W. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: Results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2015, 36, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.C.; Bhonsale, A.; te Riele, A.S.J.M.; Tichnell, C.; Murray, B.; Russell, S.D.; Tandri, H.; Tedford, R.J.; Judge, D.P.; Calkins, H.; et al. Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J. Am. Heart Assoc. 2014, 3, e001471. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Orgeron, G.; Tichnell, C.; Murray, B.; Crosson, J.; Monfredi, O.; Cadrin-Tourigny, J.; Tandri, H.; Calkins, H.; James, C.A. Impact of exercise restriction on arrhythmic risk among patients with arrhythmogenic right ventricular cardiomyopathy. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Chahal, A.; Reza, N.; Santangeli, P. Risk Stratification in Arrhythmogenic RV Cardiomyopathy/Dysplasia without an ICD. Available online: https://www.acc.org/latest-in-cardiology/articles/2019/02/26/14/20/risk-stratification-in-arrhythmogenic-rv-cardiomyopathy-dysplasia-without-an-icd (accessed on 21 July 2020).
- Hodgkinson, K.A.; Howes, A.J.; Boland, P.; Shen, X.S.; Stuckless, S.; Young, T.L.; Curtis, F.; Collier, A.; Parfrey, P.S.; Connors, S.P. Long-term clinical outcome of arrhythmogenic right ventricular cardiomyopathy in individuals with a p.S358L mutation in TMEM43 following implantable cardioverter defibrillator therapy. Circ. Arrhythm. Electrophysiol. 2016, 9. [Google Scholar] [CrossRef]
- Date, S.V. The rosetta stone method. Methods Mol. Biol. 2008, 453, 169–180. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kelly, M.A.; Caleshu, C.; Morales, A.; Buchan, J.; Wolf, Z.; Harrison, S.M.; Cook, S.; Dillon, M.W.; Garcia, J.; Haverfield, E.; et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: Recommendations by clingen’s inherited cardiomyopathy expert panel. Genet. Med. 2018, 20, 351–359. [Google Scholar] [CrossRef]
- Kato, S.; Kurzrock, R. An avatar for precision cancer therapy. Nat. Biotechnol. 2018, 36, 1053–1055. [Google Scholar] [CrossRef]
- Cho, G.S.; Lee, D.I.; Tampakakis, E.; Murphy, S.; Andersen, P.; Uosaki, H.; Chelko, S.; Chakir, K.; Hong, I.; Seo, K.; et al. Neonatal transplantation confers maturation of PSC-derived cardiomyocytes conducive to modeling cardiomyopathy. Cell Rep. 2017, 18, 571–582. [Google Scholar] [CrossRef]
- Hoorntje, E.T.; Te Rijdt, W.P.; James, C.A.; Pilichou, K.; Basso, C.; Judge, D.P.; Bezzina, C.R.; van Tintelen, J.P. Arrhythmogenic cardiomyopathy: Pathology, genetics, and concepts in pathogenesis. Cardiovasc. Res. 2017, 113, 1521–1531. [Google Scholar] [CrossRef]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Padron-Barthe, L.; Villalba-Orero, M.; Gomez-Salinero, J.M.; Dominguez, F.; Roman, M.; Larrasa-Alonso, J.; Ortiz-Sanchez, P.; Martinez, F.; Lopez-Olaneta, M.; Bonzon-Kulichenko, E.; et al. Severe cardiac dysfunction and death caused by arrhythmogenic right ventricular cardiomyopathy type 5 are improved by inhibition of glycogen synthase kinase-3beta. Circulation 2019, 140, 1188–1204. [Google Scholar] [CrossRef]
- Seyhan, A.A.; Carini, C. Are innovation and new technologies in precision medicine paving a new era in patients centric care? J. Transl. Med. 2019, 17, 114. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.E.; Carr, B.G. Virtually perfect? Telemedicine for covid-19. N. Engl. J. Med. 2020, 382, 1679–1681. [Google Scholar] [CrossRef] [PubMed]
- All of Us Research Program Investigators. The “All of Us” Research Program. N. Engl. J. Med. 2019, 381, 668–676. [Google Scholar] [CrossRef] [PubMed]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1957 | The Term ‘Cardiomyopathy’ First Introduced [13] |
| Bridgen describes cardiomyopathies as ‘uncommon, noncoronary myocardial diseases’ and describes some general features found. | |
| 1980 | WHO-ISFC Task Force Report on Definition and Classification of Cardiomyopathies [14] |
| Cardiomyopathies defined as ‘heart muscle diseases of unknown cause’. Classified into four morphological phenotypes: DCM, HCM, RCM, and Unclassified cardiomyopathy. | |
| 1977 | ARVD First Recognised and Described [15] |
| Fontaine et al. coin and first recognise ARVD when mapping and treating VT originating from the RV. They propose this new disease be termed ‘dysplasia’ due to the likely theory of a postnatal developmental disorder. | |
| 1982 | ARVD First Comprehensive Classical Description [3] |
| Marcus et al. comprehensively describe ARVD in a case series of 24 patients. | |
| 1988 | ARVC First Described [16] |
| Thiene et al. describe ARVC in group of young adults who died from SCD mostly during exercise. | |
| 1994 | ESC-ISFC Task Force Diagnostic Criteria for ARVD/C [17] |
| Proposed new criteria for the clinical diagnosis of ARVD/C using structural, histological, electrocardiographic, arrhythmic, and familial features. | |
| 1995 | Updated WHO-ISFC Task Force Report on Definition and Classification of Cardiomyopathies [18] |
| Cardiomyopathies re-defined as ‘diseases of the myocardium associated with cardiac dysfunction’—broadening the definition to include known causes of myocardial disease. Classification split into two groups: primary (intrinsic to myocardium) and specific (secondary to external processes). Fifth phenotype added to primary group: ARVC. | |
| 2006 | AHA Statement on Contemporary Definitions and Classifications of the Cardiomyopathies [19] |
| The AHA propose a new aetiological-based categorisation split into primary (involving only the heart) and secondary (generalised multiorgan involvement) cardiomyopathies. These are subdivided into genetic, mixed, and acquired forms—importantly genetic forms are given their own designation for the first time. Ion channel disease added to the classification. | |
| 2008 | ESC Working Group on Myocardial and Pericardial Diseases: Classification of the Cardiomyopathies: A Position Statement * [20] |
| Cardiomyopathies defined as ‘A myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease and congenital heart disease sufficient to cause the observed myocardial abnormality.’ ESC divides cardiomyopathies into five known morphological phenotypes: (1) DCM, (2) HCM, (3) RCM, (4) ARVC, and (5) unclassified cardiomyopathies, which are further categorised into familial and nonfamilial groups. | |
| 2010 | Revised Task Force Diagnostic Criteria for ARVC [5] |
| Proposed an updated diagnostic scheme for ARVC based on (1) functional and structural characterization of the right ventricle, (2) histopathological characterisation, (3) repolarisation, (4) depolarisation abnormalities, (5) arrhythmias, and (6) family history and genotype. | |
| 2013 | MOGE(S) Classification of Cardiomyopathies [21] |
| Proposed a descriptive genotype-phenotype nosology system based on five attributes: Morphofunctional characteristics, Organ involvement, Genetic/familial inheritance pattern, AEtiological annotation, Functional Status of patients. | |
| 2019 | 2019 Definition and Treatment of Arrhythmogenic Cardiomyopathy: an Updated Expert Panel Report [22] |
| A consensus statement compiled following a moderated roundtable discussion of an international group of experts in 2017, Athens, Greece, which defined arrhythmogenic cardiomyopathy as a ‘family of diseases that feature structural myocardial abnormalities (identified by macro- and microscopic pathological examination besides cardiac imaging) and ventricular arrhythmia’. Terms include ARVC, ALVC, aDCM, isolated non-ischaemic scar and hypokinetic, non-dilated left ventricle. Fundamental aspects include arrhythmia, electrical abnormalities, structural abnormalities (important not essential chamber dimensions or contractility but tissue characterisation is important), heritability (family history, genetic aetiology, cardiocutaneous, neuromuscular features), exclusion of phenocopies (such as sarcoidosis, myocarditis, PHTN, and congenital abnormalities). | |
| 2019 | 2019 HRS Expert Consensus Statement On evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy [1] |
| New definition and classification proposed for ARVC (now referred to as ACM) defined as an ‘arrhythmogenic heart muscle disorder not explained by ischemic, hypertensive, or valvular heart disease.’ This is broad and include classical ARVC, ALVC, arrhythmogenic biventricular cardiomyopathy, as well as other cardiomyopathies such as amyloidosis, Chagas, sarcoidosis, myocarditis, HCM. | |
| 2020 | Diagnosis of Arrhythmogenic Cardiomyopathy: The Padua Criteria [23] |
| These criteria build on the 2010 TFC multi-parametric approach to include biventricular and ALVC involvement. Of note, introduction of new LV ECG criteria and tissue characterisation by CMR. |
| Gene | Protein Type | Frequency * | Predominant Inheritance Pattern | Predominant Ventricular Disease ** | OMIM Entry | Exon Location; Exon Count | Remarks | Gene-Disease Validity Classification *** |
|---|---|---|---|---|---|---|---|---|
| PKP2 | Desmosome | 20–45% | AD | RV, BIV | ARVC9 | 12p11.21; 14 | Classical ARVC, AR also reported | Definitive for ARVC |
| DSG2 | Desmosome | 4–15% | AD | RV, LV, BIV | ARVC10 | 18q12.1; 16 | Frequent LV involvement, AR also reported | Definitive for ARVC |
| DSP | Desmosome | 1–13% | AD | LV, BIV | ARVC8 | 6p24.3; 24 | Cardiocutaneous Syndrome AR (Carvajal), can also have cardiocutaneous with AD | Definitive for ARVC |
| DSC2 | Desmosome | 1–7% | AR | RV, BIV | ARVC11 | 18q12.1; 18 | Cardiocutaneous AR | Definitive for ARVC |
| JUP | Desmosome | 0–1% | AD and AR | RV, BIV | ARVC12 | 17q21.2; 19 | Cardiocutaneous syndrome AR (Naxos) | Definitive for ARVC |
| TTN | Sarcomere | 18% | AD | RV, LV, BIV | - | 2q31.2; 365 | DCM | Limited for ARVC |
| LMNA | Nuclear Intermediate Filament | 3–4% | AD | LV, BiV | - | 1q22; 17 | DCM, Lipodystrophies, Myopathies | Limited for ARVC |
| DES | Cytoplasmic Intermediate Filament | <1% | AD | LV, BIV | ARVC7 | 2q35; 9 | Myofibrillar myopathy, DCM | Moderate for ARVC |
| FLNC | Actin cross-link | 0–3% | AD | LV, BIV | - | 7q32.1; 48 | High propensity for arrhythmia, SCD, structural abnormalities, LGE and HF. Should consider ICD as a primary prevention. | - |
| TJP1 | Intercalated Disc | 0–4% [31] | AD | - | 15q13.1; 33 | Limited for ARVC | ||
| CDH2 | Intercalated Disc | 0–2% | AD | RV, BIV | - | 18q12.1; 19 | - | Limited for ARVC |
| CTNNA3 | Intercalated Disc | <1% | AD | RV, BIV | ARVC13 | 10q21.3; 27 | Low penetrance | Limited for ARVC |
| TMEM43 | Nuclear Envelope | <1% | AD | RV, BIV | ARVC5 | 3p25.1; 13 | Founder variant in Newfoundland, SCD | Definitive for ARVD 5 |
| PLN | Calcium Regulation | 0–12% | AD | LV, BIV | - | 6q22.31; 2 | Founder mutation in Netherlands. High risk of SCD—may consider ICD as primary prevention | Definitive for CMP |
| RYR2 | Calcium Regulation | 9% | AD | Exon 3 deletion DCM | ARVC2 | 1q43; 107 | CPVT | Refuted for ARVC, Definitive for CPVT |
| Conflicting evidence of misdiagnosis CPVT as ARVC with the exception of exon 3 deletions associated with structural abnormalities. | ||||||||
| SCN5A | Sodium Channel | 0–2% | AD | LV, BIV | - | 3p22.2; 29 | BrS, LQTS Type 3, AF | Limited for ARVC, Definitive for BrS |
| TGFB3 | Cytokine | Unknown | AD | RV | ARVC1 | 14q24.3; 8 | - | Limited for ARVC |
| RBM20 | Splicing Factor | Unknown | AD | LV | - | 10q25.2; 16 | High risk of SCD—considered primary prevention by ICD | Definitive for DCM |
| BAG3 | Chaperone | Unknown | AD | LV | - | 10q26.11; 4 | - | |
| NKX2-5 | Homeobox | Unknown | AD | - | 5q35.1; 3 | - |
| Epidemiology and screening of ACM | Highly variable |
| Under-studied, underdiagnosed | |
| GWAS may be helpful to establish associations between patients’ genotype and phenotype; and to test existing genetic risk scores from other cardiac phenotypes with ACM phenotypes for associations. Moreover, this could be potentially developed into a screening test | |
| Clinical heterogeneity | Inter and intra-familial variability (even with some mutations) |
| Deep phenotyping with bipolar and unipolar voltage maps of RV, RVOT, LV, and with phenocopies | |
| Mechanisms of ACM | Pathophysiology and disease progression |
| Myocarditis as a possible trigger and predisposition to arrhythmias | |
| Animal models may be used to study the effect of protein mutations on ACM phenotypes, e.g., titin mutation and its link to heart failure | |
| Genetics | 40% undefined genetically |
| Genotype-phenotype correlation | |
| More precise definition of clinical phenotype | |
| GWAS and phenotype-genotype for effect modifiers? | |
| Desmosome | Study extra-cardiac desmosome expression such as buccal cells and skin |
| Inflammation and myocarditis | Investigate role of inflammation in animals and humans |
| Discern if ACM patients more susceptible to myocarditis | |
| Is inflammation primary or secondary? | |
| Is myocarditis a trigger for arrhythmogenicity? | |
| Is myocarditis an acute phase of ACM? | |
| Predicting SCD risk | Refine the criteria for ICD implantation in ACM patients e.g., balancing the benefits and risks of ICD, specifying the type of ICD (TV vs. SC) |
| Validation Hopkins Primary Prevention ARVC SCD risk calculator | |
| Evaluating blood biomarkers longitudinal studies | |
| Systematic follow-up of G+/P+, G+/P+, G−/P− understand evolution of arrhythmia | |
| Use of non-ICD therapies to manage arrhythmia risk | |
| Other therapies | Trials using standard anti-HF regimens in ACM (β-blockers, Angiotensin-converting enzyme, Angiotensin II receptor blocker, Mineralocorticoid receptor antagonist, Angiotensin receptor neprilysin inhibitor) |
| Disease-specific pathways to identify novel targets | |
| Skin and hair for testing novel therapies | |
| Precision medicine with the use of ‘avatars’ for testing therapies | |
| Establish trials with surrogate endpoints to expedite drug delivery |
| DNA level | Genetic interaction mapping |
| DNA/protein interactions | |
| DNA accessibility assays | |
| RNA level | Microarrays |
| SAGE (Serial Analysis of Gene Expression) | |
| RNA sequencing | |
| MPRAs (Massive Parallel Reporter Assays) | |
| STARR-sequencing (similar to MPRA) | |
| Perturb-sequencing (couples CRISPR with gene knockdown single gene expression) | |
| Protein level | Yeast two-hybrid systems (bait with prey originally based on transcription factor GAL4) |
| AP/MS (Affinity Purification and Mass Spectrometry) | |
| Deep mutational scanning | |
| Loss of functional techniques | Mutagenesis (deletional or insertional mutagenesis laborious pre-CRISPR) |
| RNAi (RNA interference using s20 base dbRNA transfection using siRNA or virally-delivered short hairpin RNAs | |
| CRISPR | |
| Functional annotation of genes | Genomic annotation |
| Rosetta Stone approach [150] | |
| Bioinformatics | Clustering |
| Principal component analysis | |
| Machine learning | |
| Artificial neural networks | |
| Support vector machines (supervised machine learning) | |
| Gene ontology (DAVID) | |
| Gene set enrichment analysis (GSEA) | |
| Pathway analysis—Ingenuity | |
| Pathway Studio—Ariadne Genomics | |
| Consortia | ENCODE (Encyclopaedia of DNA Elements) |
| GTEx (Genotype-Tissue expression project) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, V.; Asatryan, B.; Siripanthong, B.; Munroe, P.B.; Tiku-Owens, A.; Lopes, L.R.; Khanji, M.Y.; Protonotarios, A.; Santangeli, P.; Muser, D.; et al. State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 6615. https://doi.org/10.3390/ijms21186615
Patel V, Asatryan B, Siripanthong B, Munroe PB, Tiku-Owens A, Lopes LR, Khanji MY, Protonotarios A, Santangeli P, Muser D, et al. State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. International Journal of Molecular Sciences. 2020; 21(18):6615. https://doi.org/10.3390/ijms21186615
Chicago/Turabian StylePatel, Viraj, Babken Asatryan, Bhurint Siripanthong, Patricia B. Munroe, Anjali Tiku-Owens, Luis R. Lopes, Mohammed Y. Khanji, Alexandros Protonotarios, Pasquale Santangeli, Daniele Muser, and et al. 2020. "State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy" International Journal of Molecular Sciences 21, no. 18: 6615. https://doi.org/10.3390/ijms21186615
APA StylePatel, V., Asatryan, B., Siripanthong, B., Munroe, P. B., Tiku-Owens, A., Lopes, L. R., Khanji, M. Y., Protonotarios, A., Santangeli, P., Muser, D., Marchlinski, F. E., Brady, P. A., & Chahal, C. A. A. (2020). State of the Art Review on Genetics and Precision Medicine in Arrhythmogenic Cardiomyopathy. International Journal of Molecular Sciences, 21(18), 6615. https://doi.org/10.3390/ijms21186615