Small Molecules and Peptides Targeting Glial Cell Line-Derived Neurotrophic Factor Receptors for the Treatment of Neurodegeneration



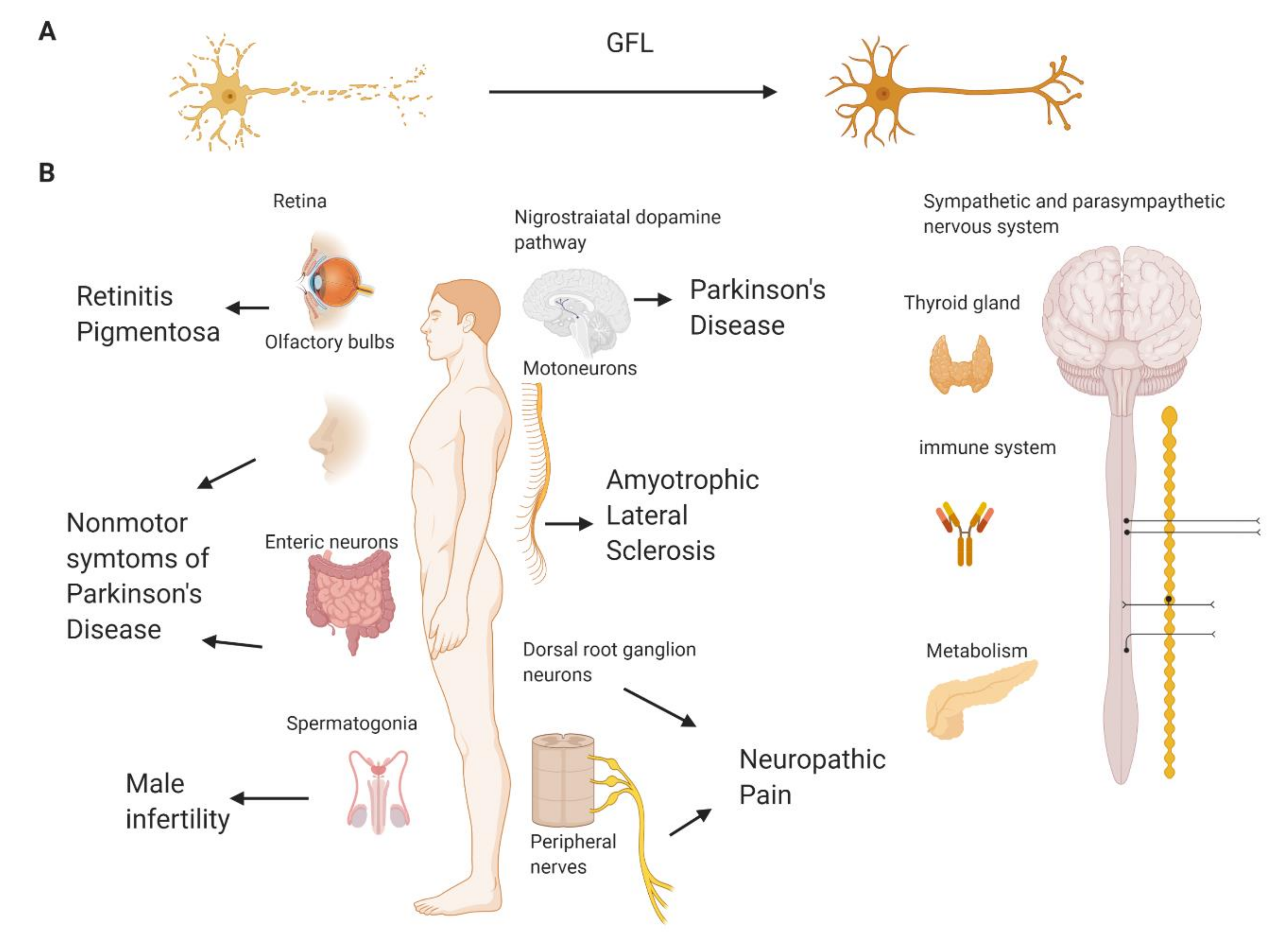{kind=link}
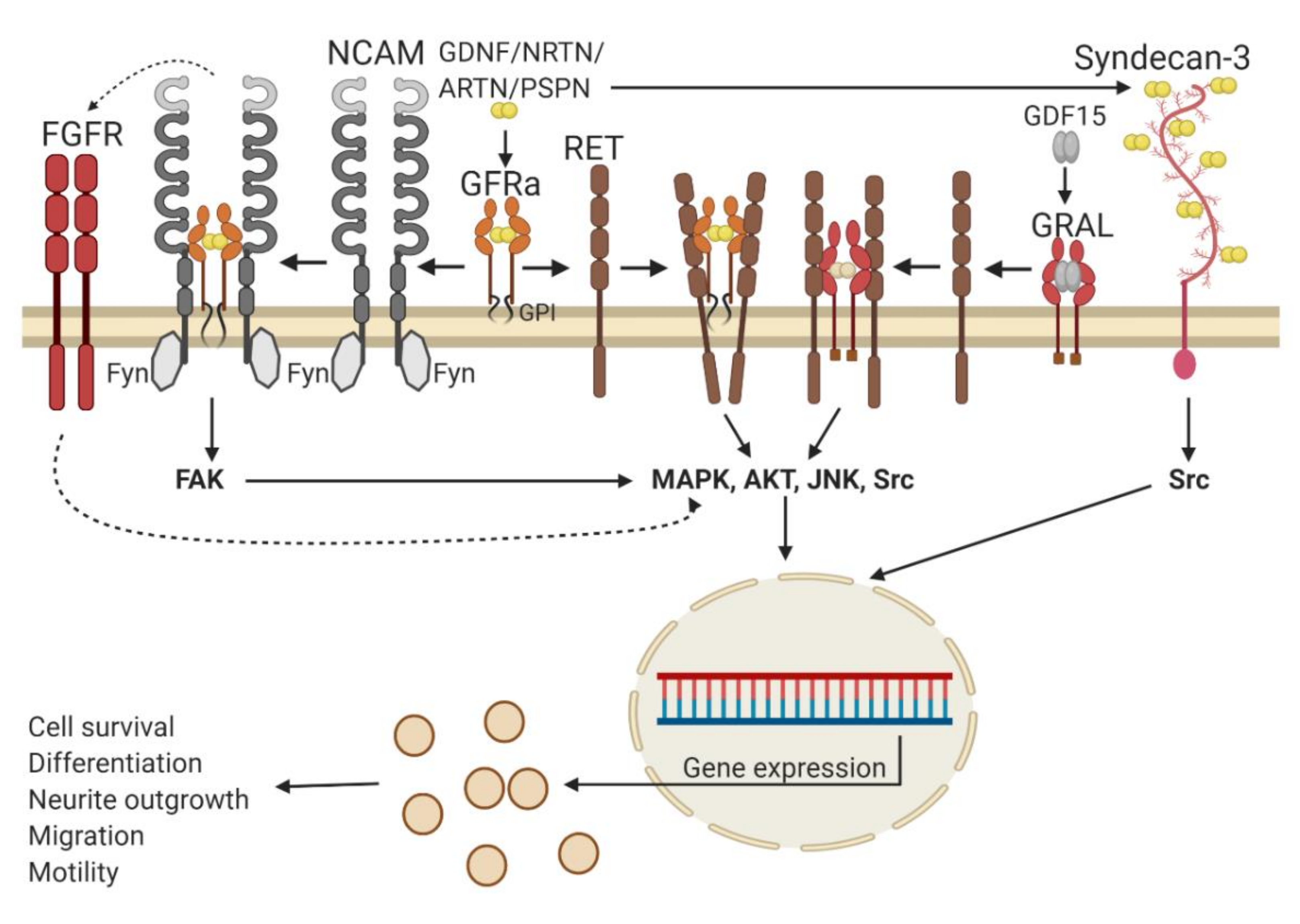{kind=link}
{kind=link}
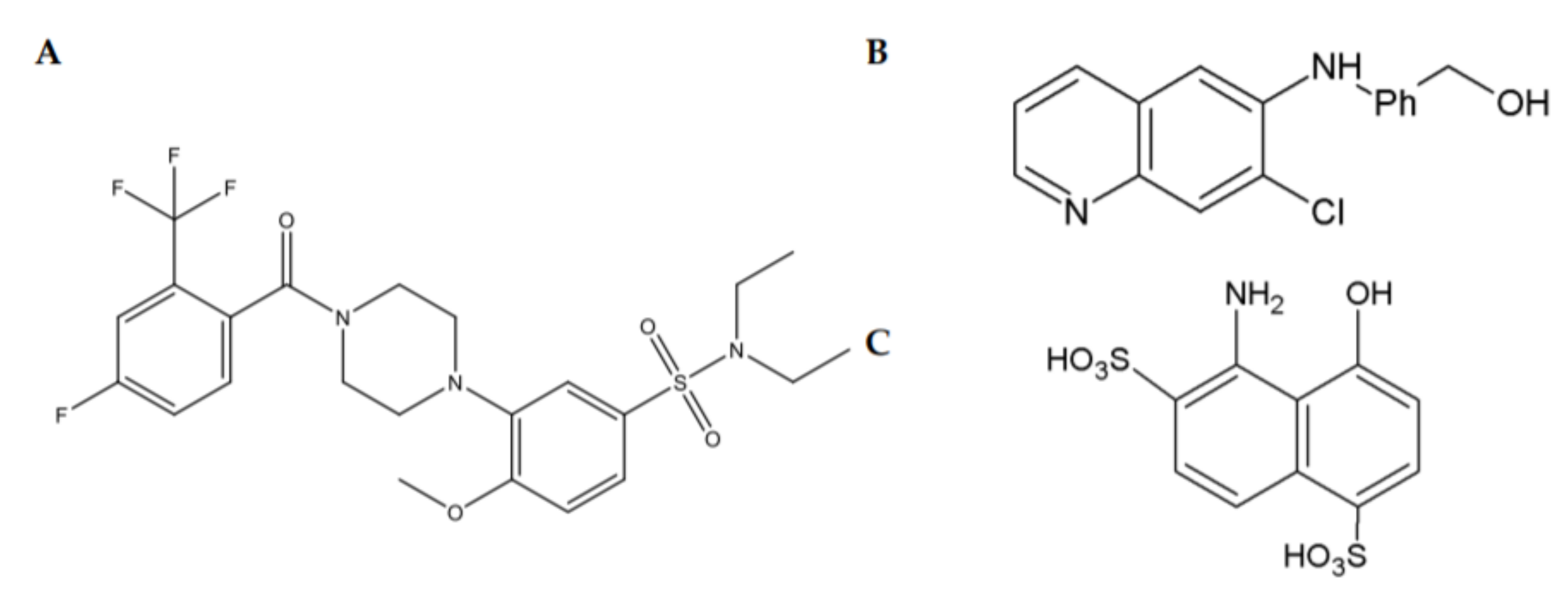{kind=link}
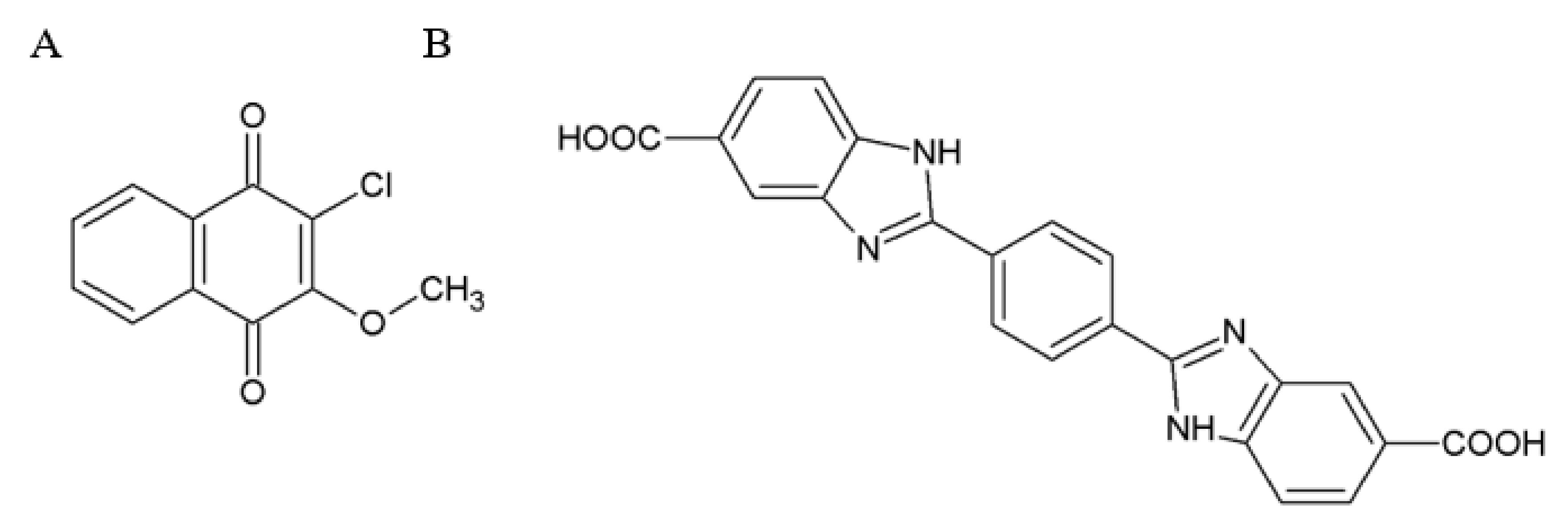{kind=link}
{kind=link}
Abstract
1. Introduction
2. Main Text
2.1. GFL Receptors and Signaling
2.2. Small Molecules Positively Modulating GFL Signalling
2.3. Small Molecules Targeting RET
2.4. Small Molecules Targeting GFRα Receptors
2.5. Peptides Targeting NCAM and RET
2.6. Peptides from the GDNF Proregion with an Unknown Mechanism of Action
2.7. Advantages and Limitations in the Use of Each Class of GFRα/RET and NCAM Modulators for the Management of Neurodegeneration
2.8. Other Possible Applications of Compounds Targeting GFL Receptors
3. Conclusions
4. Patents
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| ARTN | Artemin |
| BDNF | Brain-derived neurotrophic factor |
| CEREP | A panel of assays to evaluate potential toxicity of compounds to major systems and organs |
| CGRP | Calcitonin gene-related peptide |
| DM | Diabetes mellitus |
| FAK | Focal adhesion kinase |
| FGFR | Fibroblast growth factor receptor |
| GDNF | Glial cell line-derived neurotrophic factor |
| GFLs | Glial cell line-derived neurotrophic factor family ligands |
| GFRa | Glial cell line-derived neurotrophic factor family receptor alpha |
| GRAL | GDNF family receptor alpha-like protein |
| GDF15 | Growth differentiation factor-15 |
| IB4 | Isolectin B4 |
| NCAM | Neural cell adhesion molecule |
| NGF | Nerve growth factor |
| NP | Neuropathic pain |
| NRTN | Neurturin |
| PD | Parkinson’s disease |
| PSPN | Persephin |
| RET | REarranged in Transfection |
| SNL | Spinal nerve ligation |
| Trk | Tropomyosin receptor kinases |
References
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Saarma, M.; Goldman, A. Receptors identified for a weight regulator. Nature 2017, 550, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lindahl, M.; Hyvönen, M.E.; Parvinen, M.; De Rooij, D.G.; Hess, M.W.; Raatikainen-Ahokas, A.; Sainio, K.; Rauvala, H.; Lakso, M.; et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 2000, 287, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Pichel, J.G.; Shen, L.; Sheng, H.Z.; Granholm, A.-C.; Drago, J.; Grinberg, A.; Lee, E.J.; Huang, S.P.; Saarma, M.; Hoffer, B.J.; et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature 1996, 382, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chang, C.-C.; Sun, Z.; Madsen, D.; Zhu, H.; Padkjær, S.B.; Wu, X.; Huang, T.; Hultman, K.; Paulsen, S.J.; et al. GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat. Med. 2017, 23, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Emmerson, P.J.; Wang, F.; Du, Y.; Liu, Q.; Pickard, R.T.; Gonciarz, M.D.; Coskun, T.; Hamang, M.J.; Sindelar, D.K.; Ballman, K.K.; et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat. Med. 2017, 23, 1215–1219. [Google Scholar] [CrossRef]
- Mullican, S.E.; Lin-Schmidt, X.; Chin, C.-N.; Chavez, J.A.; Furman, J.L.; Armstrong, A.A.; Beck, S.C.; South, V.J.; Dinh, T.Q.; Cash-Mason, T.D.; et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 2017, 23, 1150–1157. [Google Scholar] [CrossRef]
- Hsu, J.-Y.; Crawley, S.; Chen, M.; Ayupova, D.A.; Lindhout, D.A.; Higbee, J.; Kutach, A.; Joo, W.; Gao, Z.; Fu, D.; et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 2017, 550, 255–259. [Google Scholar] [CrossRef]
- Lin, L.-F.H.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef]
- Nutt, J.G.; Burchiel, K.J.; Comella, C.L.; Jankovic, J.; Lang, A.E.; Laws, E.R., Jr.; Lozano, A.M.; Penn, R.D.; Simpson, R.K., Jr.; Stacy, M.; et al. Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 2003, 60, 69–73. [Google Scholar] [CrossRef]
- Gill, S.S.; Patel, N.K.; Hotton, G.R.; O’Sullivan, K.; McCarter, R.; Bunnage, M.; Brooks, D.J.; Svendsen, C.N.; Heywood, P. Direct brain infusion of glial cell line–derived neurotrophic factor in Parkinson disease. Nat. Med. 2003, 9, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Slevin, J.T.; Gash, D.M.; Smith, C.D.; Gerhardt, G.A.; Kryscio, R.; Chebrolu, H.; Walton, A.; Wagner, R.; Young, A.B. Unilateral intraputaminal glial cell line-derived neurotrophic factor in patients with Parkinson disease: Response to 1 year each of treatment and withdrawal. Neurosurg. Focus 2006, 106, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Whone, A.; Luz, M.; Boca, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019, 142, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Whone, A.L.; Boca, M.; Luz, M.; Woolley, M.; Mooney, L.; Dharia, S.; Broadfoot, J.; Cronin, D.; Schroers, C.; Barua, N.U.; et al. Extended Treatment with Glial Cell Line-Derived Neurotrophic Factor in Parkinson’s Disease. J. Parkinsons. Dis. 2019, 9, 301–313. [Google Scholar] [CrossRef]
- Horger, B.A.; Nishimura, M.C.; Armanini, M.P.; Wang, L.-C.; Poulsen, K.T.; Rosenblad, C.; Kirik, D.; Moffat, B.; Simmons, L.; Johnson, E.; et al. Neurturin exerts potent actions on survival and function of midbrain dopaminergic neurons. J. Neurosci. 1998, 18, 4929–4937. [Google Scholar] [CrossRef] [PubMed]
- Olanow, W.C.; Bartus, R.T.; Baumann, T.L.; Factor, S.; Boulis, N.; Stacy, M.; Turner, D.A.; Marks, W.; Larson, P.; Starr, P.A.; et al. Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease: A double-blind, randomized, controlled trial. Ann. Neurol. 2015, 78, 248–257. [Google Scholar] [CrossRef]
- Marks, W.J.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet. Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Mahato, A.K.; Sidorova, Y.A. Glial cell line-derived neurotrophic factors (GFLs) and small molecules targeting RET receptor for the treatment of pain and Parkinson’s disease. Cell Tissue Res. 2020, in press. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Volcho, K.P.; Salakhutdinov, N.F. Neuroregeneration in Parkinson’s disease: From proteins to small molecules. Curr. Neuropharmacol. 2019, 17, 268–287. [Google Scholar] [CrossRef]
- Kordower, J.H.; Olanow, C.W.; Dodiya, H.B.; Chu, Y.; Beach, T.G.; Adler, C.H.; Halliday, G.M.; Bartus, R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 2013, 136, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; De Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Chmielarz, P.; Er, Ş.; Konovalova, J.; Bandrés, L.; Hlushchuk, I.; Albert, K.; Panhelainen, A.; Luk, K.; Airavaara, M.; Domanskyi, A. GDNF/RET signaling pathway activation eliminates Lewy Body pathology in midbrain dopamine neurons. BioRxiv 2019, 752899. [Google Scholar] [CrossRef]
- Okkerse, P.; Hay, J.L.; Versage, E.; Tang, Y.; Galluppi, G.; Ravina, B.; Verma, A.; Williams, L.; Aycardi, E.; Groeneveld, G.J. Pharmacokinetics and pharmacodynamics of multiple doses of BG00010, a neurotrophic factor with anti-hyperalgesic effects, in patients with sciatica. Br. J. Clin. Pharmacol. 2016, 82, 108–117. [Google Scholar] [CrossRef]
- Rolan, P.E.; O’Neill, G.; Versage, E.; Rana, J.; Tang, Y.; Galluppi, G.; Aycardi, E. First-In-Human, Double-Blind, Placebo-Controlled, Randomized, Dose-Escalation Study of BG00010, a Glial Cell Line-Derived Neurotrophic Factor Family Member, in Subjects with Unilateral Sciatica. PLoS ONE 2015, 10, e0125034. [Google Scholar] [CrossRef]
- Backonja, M.; Williams, L.; Miao, X.; Katz, N.; Chen, C. Safety and efficacy of neublastin in painful lumbosacral radiculopathy. Pain 2017, 158, 1802–1812. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 225–238. [Google Scholar]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, e1310. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Li, W.; Brakefield, D.; Pan, Y.; Hunter, D.; Myckatyn, T.M.; Parsadanian, A. Muscle-derived but not centrally derived transgene GDNF is neuroprotective in G93A-SOD1 mouse model of ALS. Exp. Neurol. 2007, 203, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; McHugh, J.; Tork, C.; Shelley, B.; Hayes, A.; Bellantuono, I.; Aebischer, P.; Svendsen, C.N. Direct muscle delivery of GDNF with human mesenchymal stem cells improves motor neuron survival and function in a rat model of familial ALS. Mol. Ther. 2008, 16, 2002–2010. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, G.M.; Alkaslasi, M.; Vit, J.P.; Lawless, G.; Godoy, M.; Gowing, G.; Shelest, O.; Svendsen, C.N. Systemic injection of AAV9-GDNF provides modest functional improvements in the SOD1 G93A ALS rat but has adverse side effects. Gene Ther. 2017, 24, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H.; Glass, J.D.; Svendsen, C.N. Stem cell transplantation for amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2018, 31, 655–661. [Google Scholar] [CrossRef]
- Bespalov, M.M.; Sidorova, Y.A.; Tumova, S.; Ahonen-Bishopp, A.; Magalhães, A.C.; Kulesskiy, E.; Paveliev, M.; Rivera, C.; Rauvala, H.; Saarma, M. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J. Cell Biol. 2011, 192. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef]
- Touchard, E.; Heiduschka, P.; Berdugo, M.; Kowalczuk, L.; Bigey, P.; Chahory, S.; Gandolphe, C.; Jeanny, J.-C.; Behar-Cohen, F. Non-viral gene therapy for GDNF production in RCS rat: The crucial role of the plasmid dose. Gene Ther. 2012, 19, 886–898. [Google Scholar] [CrossRef]
- Dalkara, D.; Kolstad, K.D.; Guerin, K.I.; Hoffmann, N.V.; Visel, M.; Klimczak, R.R.; Schaffer, D.V.; Flannery, J.G. AAV Mediated GDNF Secretion From Retinal Glia Slows Down Retinal Degeneration in a Rat Model of Retinitis Pigmentosa. Mol. Ther. 2011, 19, 1602–1608. [Google Scholar] [CrossRef]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Hauck, S.M.; Kinkl, N.; Deeg, C.A.; Swiatek-de Lange, M.; Schöffmann, S.; Ueffing, M. GDNF Family Ligands Trigger Indirect Neuroprotective Signaling in Retinal Glial Cells. Mol. Cell. Biol. 2006, 26, 2746–2757. [Google Scholar] [CrossRef] [PubMed]
- Barak, S.; Ahmadiantehrani, S.; Logrip, M.L.; Ron, D. GDNF and alcohol use disorder. Addict. Biol. 2019, 24, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Carnicella, S.; Ron, D. GDNF—A potential target to treat addiction. Pharmacol. Ther. 2009, 122, 9–18. [Google Scholar] [CrossRef]
- Golden, J.P.; Milbrandt, J.; Johnson, E.M. Neurturin and persephin promote the survival of embryonic basal forebrain cholinergic neurons in vitro. Exp. Neurol. 2003, 184, 447–455. [Google Scholar] [CrossRef]
- Irala, D.; Bonafina, A.; Fontanet, P.A.; Alsina, F.C.; Paratcha, G.; Ledda, F. The GDNF-GFRα1 complex promotes the development of hippocampal dendritic arbors and spines via NCAM. Development 2016, 143, 4224–4235. [Google Scholar] [CrossRef]
- Revilla, S.; Ursulet, S.; Álvarez-López, M.J.; Castro-Freire, M.; Perpiñá, U.; García-Mesa, Y.; Bortolozzi, A.; Giménez-Llort, L.; Kaliman, P.; Cristòfol, R.; et al. Lenti-GDNF gene therapy protects against Alzheimer’s disease-like neuropathology in 3xTg-AD mice and MC65 cells. CNS Neurosci. Ther. 2014, 20, 961–972. [Google Scholar] [CrossRef]
- Laurikainen, A.; Hiltunen, J.O.; Thomas-Crusells, J.; Vanhatalo, S.; Arumäe, U.; Airaksinen, M.S.; Klinge, E.; Saarma, M. Neurturin is a neurotrophic factor for penile parasympathetic neurons in adult rat. J. Neurobiol. 2000, 43, 198–205. [Google Scholar] [CrossRef]
- Laurikainen, A.; Hiltunen, J.O.; Vanhatalo, S.; Klinge, E.; Saarma, M. Glial cell line-derived neurotrophic factor is expressed in penis of adult rat and retrogradely transported in penile parasympathetic and sensory nerves. Cell Tissue Res. 2000, 302, 321–329. [Google Scholar] [CrossRef]
- Hiltunen, P.H.; Airaksinen, M.S. Sympathetic cholinergic target innervation requires GDNF family receptor GFRα2. Mol. Cell. Neurosci. 2004, 26, 450–457. [Google Scholar] [CrossRef]
- Gianino, S. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development 2003, 130, 2187–2198. [Google Scholar] [CrossRef] [PubMed]
- Jokinen, V.; Sidorova, Y.; Viisanen, H.; Suleymanova, I.; Tiilikainen, H.; Li, Z.; Lilius, T.O.; Mätlik, K.; Anttila, J.E.; Airavaara, M.; et al. Differential Spinal and Supraspinal Activation of Glia in a Rat Model of Morphine Tolerance. Neuroscience 2018, 375, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Ji, R.R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef] [PubMed]
- Rickert, U.; Grampp, S.; Wilms, H.; Spreu, J.; Knerlich-Lukoschus, F.; Held-Feindt, J.; Lucius, R. Glial Cell Line-Derived Neurotrophic Factor Family Members Reduce Microglial Activation via Inhibiting p38MAPKs-Mediated Inflammatory Responses. J. Neurodegener. Dis. 2014, 2014, 369468. [Google Scholar] [CrossRef]
- Machado-Pereira, M.; Santos, T.; Ferreira, L.; Bernardino, L.; Ferreira, R. Anti-Inflammatory Strategy for M2 Microglial Polarization Using Retinoic Acid-Loaded Nanoparticles. Mediat. Inflamm. 2017, 2017, 6742427. [Google Scholar] [CrossRef]
- Andreou, K.E.; Soto, M.S.; Allen, D.; Economopoulos, V.; de Bernardi, A.; Larkin, J.R.; Sibson, N.R. Anti-inflammatory Microglia/Macrophages As a Potential Therapeutic Target in Brain Metastasis. Front. Oncol. 2017, 7, 251. [Google Scholar] [CrossRef]
- Strelau, J.; Strzelczyk, A.; Rusu, P.; Bendner, G.; Wiese, S.; Diella, F.; Altick, A.L.; von Bartheld, C.S.; Klein, R.; Sendtner, M.; et al. Progressive Postnatal Motoneuron Loss in Mice Lacking GDF-15. J. Neurosci. 2009, 29, 13640–13648. [Google Scholar] [CrossRef]
- Strelau, J.; Schober, A.; Sullivan, A.; Schilling, L.; Unsicker, K. Growth/differentiation factor-15 (GDF-15), a novel member of the TGF-β superfamily, promotes survival of lesioned mesencephalic dopaminergic neurons in vitro and in vivo and is induced in neurons following cortical lesioning. J. Neural. Transm. Suppl. 2003, 65, 197–203. [Google Scholar] [CrossRef]
- Strelau, J.; Sullivan, A.; Böttner, M.; Lingor, P.; Falkenstein, E.; Suter-Crazzolara, C.; Galter, D.; Jaszai, J.; Krieglstein, K.; Unsicker, K. Growth/Differentiation Factor-15/Macrophage Inhibitory Cytokine-1 Is a Novel Trophic Factor for Midbrain Dopaminergic Neurons In Vivo. J. Neurosci. 2000, 20, 8597–8603. [Google Scholar] [CrossRef]
- Emmerson, P.J.; Duffin, K.L.; Chintharlapalli, S.; Wu, X. GDF15 and Growth Control. Front. Physiol. 2018, 9, e1712. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tsai, V.W.W.; Nguyen, A.D.; Johnen, H.; Kuffner, T.; Shi, Y.C.; Lin, S.; Herzog, H.; Brown, D.A.; Breit, S.N.; et al. Macrophage inhibitory cytokine 1 (MIC-1/GDF15) decreases food intake, body weight and improves glucose tolerance in mice on normal & obesogenic diets. PLoS ONE 2012, 7, e34868. [Google Scholar] [CrossRef]
- Tsai, V.W.W.; Manandhar, R.; Jrøgensen, S.B.; Lee-Ng, K.K.M.; Zhang, H.P.; Marquis, C.P.; Jiang, L.; Husaini, Y.; Lin, S.; Sainsbury, A.; et al. The anorectic actions of the TGFβ cytokine MIC-1/GDF15 require an intact brainstem area postrema and nucleus of the solitary tract. PLoS ONE 2014, 9, e100370. [Google Scholar] [CrossRef] [PubMed]
- Andres, C.; Meyer, S.; Dina, O.A.; Levine, J.D.; Hucho, T. Quantitative automated microscopy (QuAM) elucidates growth factor specific signalling in pain sensitization. Mol. Pain 2010, 6, 98. [Google Scholar] [CrossRef]
- Ritter, J.M.; Flower, R.J.; Henderson, G.; Loke, Y.; MacEwan, D.; Rang, H. Rang & Dale’s Pharmacology, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2019; ISBN 9780702074486. [Google Scholar]
- Xiong, Y.; Walker, K.; Min, X.; Hale, C.; Tran, T.; Komorowski, R.; Yang, J.; Davda, J.; Nuanmanee, N.; Kemp, D.; et al. Long-acting MIC-1/GDF15 molecules to treat obesity: Evidence from mice to monkeys. Sci. Transl. Med. 2017, 9, eaan8732. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Sacramento, C.B.; Jouihan, H.; Ali, S.; Le Lay, J.; Oldham, S.; Bhagroo, N.; Boland, B.B.; Cann, J.; Chang, Y.; et al. Neurturin and a GLP-1 analogue act synergistically to alleviate diabetes in zucker diabetic fatty rats. Diabetes 2017, 66, 2007–2018. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Bartus, R.T.; Manfredsson, F.P.; Olanow, C.W.; Kordower, J.H. Long-term post-mortem studies following neurturin gene therapy in patients with advanced Parkinson’s disease. Brain 2020, 143, 960–975. [Google Scholar] [CrossRef]
- Fu, R.; Wang, L.-Q.; Chu, G.-L.; Zhou, L.-H. Involvement of phospholipase C-γ in the pro-survival role of glial cell line-derived neurotrophic factor in developing motoneurons in rat spinal cords. Mol. Med. Rep. 2012, 6, 805–810. [Google Scholar] [CrossRef][Green Version]
- Bespalov, M.M.; Saarma, M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol. Sci. 2007, 28, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Parkash, V.; Leppänen, V.-M.; Virtanen, H.; Jurvansuu, J.M.; Bespalov, M.M.; Sidorova, Y.A.; Runeberg-Roos, P.; Saarma, M.; Goldman, A. The structure of the glial cell line-derived neurotrophic factor-coreceptor complex: Insights into RET signaling and heparin binding. J. Biol. Chem. 2008, 283, 35164–35172. [Google Scholar] [CrossRef]
- Paratcha, G.; Ledda, F.; Ibáñez, C.F. The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 2003, 113, 867–879. [Google Scholar] [CrossRef]
- Nielsen, J.; Gotfryd, K.; Li, S.; Kulahin, N.; Soroka, V.; Rasmussen, K.K.; Bock, E.; Berezin, V. Role of Glial Cell Line-Derived Neurotrophic Factor (GDNF)-Neural Cell Adhesion Molecule (NCAM) Interactions in Induction of Neurite Outgrowth and Identification of a Binding Site for NCAM in the Heel Region of GDNF. J. Neurosci. 2009, 29, 11360–11376. [Google Scholar] [CrossRef] [PubMed]
- Trupp, M.; Arenas, E.; Fainzilber, M.; Nilsson, A.S.; Sieber, B.A.; Grigoriou, M.; Kilkenny, C.; Salazar-Grueso, E.; Pachnis, V.; Arumae, U.; et al. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature 1996, 381, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Tokugawa, K.; Yamamoto, K.; Nishiguchi, M.; Sekine, T.; Sakai, M.; Ueki, T.; Chaki, S.; Okuyama, S. XIB4035, a novel nonpeptidyl small molecule agonist for GFRα-1. Neurochem. Int. 2003, 42, 81–86. [Google Scholar] [CrossRef]
- Hedstrom, K.L.; Murtie, J.C.; Albers, K.; Calcutt, N.A.; Corfas, G. Treating small fiber neuropathy by topical application of a small molecule modulator of ligand-induced GFRα/RET receptor signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 2325–2330. [Google Scholar] [CrossRef] [PubMed]
- Jmaeff, S.; Sidorova, Y.; Nedev, H.; Saarma, M.; Saragovi, H.U. Small-molecule agonists of the RET receptor tyrosine kinase activate biased trophic signals that are influenced by the presence of GFRa1 co-receptors. J. Biol. Chem. 2020, 295, 6532–6542. [Google Scholar] [CrossRef]
- Höke, A. Augmenting glial cell-line derived neurotrophic factor signaling to treat painful neuropathies. Proc. Natl. Acad. Sci. USA 2014, 111, 2060–2061. [Google Scholar] [CrossRef]
- Runeberg-Roos, P.; Piccinini, E.; Penttinen, A.-M.; Mätlik, K.; Heikkinen, H.; Kuure, S.; Bespalov, M.M.; Peränen, J.; Garea-Rodríguez, E.; Fuchs, E.; et al. Developing therapeutically more efficient Neurturin variants for treatment of Parkinson’s disease. Neurobiol. Dis. 2016, 96, 335–345. [Google Scholar] [CrossRef]
- Jmaeff, S.; Sidorova, Y.; Lippiatt, H.; Barcelona, P.F.; Nedev, H.; Saragovi, L.M.; Hancock, M.A.; Saarma, M.; Saragovi, H.U. Small-molecule ligands that bind the RET receptor activate neuroprotective signals independent of but modulated by co-receptor GFRα1. Mol. Pharmacol. 2020, 98, 1–12. [Google Scholar] [CrossRef]
- Mahato, A.K.; Kopra, J.; Renko, J.; Visnapuu, T.; Korhonen, I.; Pulkkinen, N.; Bespalov, M.M.; Domanskyi, A.; Ronken, E.; Piepponen, T.P.; et al. Glial cell line–derived neurotrophic factor receptor Rearranged during transfection agonist supports dopamine neurons in Vitro and enhances dopamine release In Vivo. Mov. Disord. 2020, 35, 245–255. [Google Scholar] [CrossRef]
- Sidorova, Y.A.; Bespalov, M.M.; Wong, A.W.; Kambur, O.; Jokinen, V.; Lilius, T.O.; Suleymanova, I.; Karelson, G.; Rauhala, P.V.; Karelson, M.; et al. A Novel Small Molecule GDNF Receptor RET Agonist, BT13, Promotes Neurite Growth from Sensory Neurons in Vitro and Attenuates Experimental Neuropathy in the Rat. Front. Parmacology 2017, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Bespalov, M.M.; Sidorova, Y.A.; Suleymanova, I.; Thompson, J.; Kambur, O.; Viljami, J.; Lilius, T.; Karelson, G.; Puusepp, L.; Rauhala, P.; et al. Novel agonist of GDNF family ligand receptor RET for the treatment of experimental neuropathy. BioRxiv 2016. [Google Scholar] [CrossRef]
- Gardell, L.R.; Wang, R.; Ehrenfels, C.; Ossipov, M.H.; Rossomando, A.J.; Miller, S.; Buckley, C.; Cai, A.K.; Tse, A.; Foley, S.F.; et al. Multiple actions of systemic artemin in experimental neuropathy. Nat. Med. 2003, 9, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Rossomando, A.; Sah, D.W.Y.; Ossipov, M.H.; King, T.; Porreca, F. Artemin induced functional recovery and reinnervation after partial nerve injury. Pain 2014, 155, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Boucher, T.J.; Okuse, K.; Bennett, D.L.H.; Munson, J.B.; Wood, J.N.; McMahon, S.B. Potent Analgesic Effects of GDNF in Neuropathic Pain States. Science 2000, 290, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Viisanen, H.; Nuotio, U.; Kambur, O.; Mahato, A.K.; Jokinen, V.; Lilius, T.; Li, W.; Santos, H.A.; Karelson, M.; Rauhala, P.; et al. Novel RET agonist for the treatment of experimental neuropathies. Mol. Pain 2020, 16, 174480692095086. [Google Scholar] [CrossRef]
- Mahato, A.K.; Renko, J.; Kopra, J.; Visnapuu, T. GDNF receptor agonist supports dopamine neurons in vitro and protects their function in animal model of Parkinson’s. Abbreviated title: GDNF mimetic for Parkinson’s management. BioRxiv 2019, 540021. [Google Scholar] [CrossRef]
- Ivanova, L.; Tammiku-Taul, J.; García-Sosa, A.T.; Sidorova, Y.; Saarma, M.; Karelson, M. Molecular Dynamics Simulations of the Interactions between Glial Cell Line-Derived Neurotrophic Factor Family Receptor GFRα1 and Small-Molecule Ligands. ACS Omega 2018, 3, 11407–11414. [Google Scholar] [CrossRef]
- Ivanova, L.; Tammiku-Taul, J.; Sidorova, Y.; Saarma, M.; Karelson, M. Small-Molecule Ligands as Potential GDNF Family Receptor Agonists. ACS Omega 2018, 3, 1022–1030. [Google Scholar] [CrossRef]
- Ilieva, M.; Nielsen, J.; Korshunova, I.; Gotfryd, K.; Bock, E.; Pankratova, S.; Michel, T.M. Artemin and an Artemin-Derived Peptide, Artefin, Induce Neuronal Survival, and Differentiation Through Ret and NCAM. Front. Mol. Neurosci. 2019, 12, 47. [Google Scholar] [CrossRef]
- Immonen, T.; Alakuijala, A.; Hytönen, M.; Sainio, K.; Poteryaev, D.; Saarma, M.; Pasternack, M.; Sariola, H. A proGDNF-related peptide BEP increases synaptic excitation in rat hippocampus. Exp. Neurol. 2008, 210, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Bradley, L.H.; Fuqua, J.; Richardson, A.; Turchan-Cholewo, J.; Ai, Y.; Kelps, K.A.; Glass, J.D.; He, X.; Zhang, Z.; Grondin, R.; et al. Dopamine Neuron Stimulating Actions of a GDNF Propeptide. PLoS ONE 2010, 5, e9752. [Google Scholar] [CrossRef] [PubMed]
- Stenslik, M.J.; Potts, L.F.; Sonne, J.W.H.; Cass, W.A.; Turchan-Cholewo, J.; Pomerleau, F.; Huettl, P.; Ai, Y.; Gash, D.M.; Gerhardt, G.A.; et al. Methodology and effects of repeated intranasal delivery of DNSP-11 in a rat model of Parkinson’s disease. J. Neurosci. Methods 2015, 251, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Menacho, I.; Mologni, L.; McDonald, N.Q. Mechanisms of RET signaling in cancer: Current and future implications for targeted therapy. Cell. Signal. 2014, 26, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Amodru, V.; Taieb, D.; Guerin, C.; Romanet, P.; Paladino, N.; Brue, T.; Cuny, T.; Barlier, A.; Sebag, F.; Castinetti, F. MEN2-related pheochromocytoma: Current state of knowledge, specific characteristics in MEN2B, and perspectives. Endocrine 2020, 69, 496–503. [Google Scholar] [CrossRef]
- Yadav, L.; Pietilä, E.; Öhman, T.; Liu, X.; Mahato, A.K.; Sidorova, Y.; Lehti, K.; Saarma, M.; Varjosalo, M. PTPRA Phosphatase Regulates GDNF-Dependent RET Signaling and Inhibits the RET Mutant MEN2A Oncogenic Potential. iScience 2020, 23, 100871. [Google Scholar] [CrossRef]
- Runeberg-Roos, P.; Virtanen, H.; Saarma, M. RET(MEN 2B) is active in the endoplasmic reticulum before reaching the cell surface. Oncogene 2007, 26, 7909–7915. [Google Scholar] [CrossRef]
- Turconi, G.; Kopra, J.; Kulesskaya, N.; Vilenius, C.; Piepponen, P.T.; Andressoo, J. Chronic two-fold elevation of endogenous GDNF levels is safe and enhances motor and dopaminergic function in aged mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 831–842. [Google Scholar] [CrossRef]
- Manfredsson, F.P.; Tumer, N.; Erdos, B.; Landa, T.; Broxson, C.S.; Sullivan, L.F.; Rising, A.C.; Foust, K.D.; Zhang, Y.; Muzyczka, N.; et al. Nigrostriatal rAAV-mediated GDNF Overexpression Induces Robust Weight Loss in a Rat Model of Age-related Obesity. Mol. Ther. 2009, 17, 980–991. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Ai, Y.; Fischer, B.; Zhang, A.M.; Grondin, R.C.; Zhang, Z.; Gerhardt, G.A.; Gash, D.M. Point source concentration of GDNF may explain failure of phase II clinical trial. Exp. Neurol. 2006, 202, 497–505. [Google Scholar] [CrossRef]
- Piltonen, M.; Bespalov, M.M.; Ervasti, D.; Matilainen, T.; Sidorova, Y.A.; Rauvala, H.; Saarma, M.; Mannisto, P.T. Heparin-binding determinants of GDNF reduce its tissue distribution but are beneficial for the protection of nigral dopaminergic neurons. Exp. Neurol. 2009, 219, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, Y.A.; Mätlik, K.; Paveliev, M.; Lindahl, M.; Piranen, E.; Milbrandt, J.; Arumäe, U.; Saarma, M.; Bespalov, M.M. Persephin signaling through GFRα1: The potential for the treatment of Parkinson’s disease. Mol. Cell. Neurosci. 2010, 44, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lippoldt, E.K.; Elmes, R.R.; McCoy, D.D.; Knowlton, W.M.; McKemy, D.D. Artemin, a Glial Cell Line-Derived Neurotrophic Factor Family Member, Induces TRPM8-Dependent Cold Pain. J. Neurosci. 2013, 33, 12543–12552. [Google Scholar] [CrossRef]
- Lippoldt, E.K.; Ongun, S.; Kusaka, G.K.; McKemy, D.D. Inflammatory and neuropathic cold allodynia are selectively mediated by the neurotrophic factor receptor GFRα3. Proc. Natl. Acad. Sci. USA 2016, 113, 4506–4511. [Google Scholar] [CrossRef] [PubMed]
- Eslamboli, A.; Georgievska, B.; Ridley, R.M.; Baker, H.F.; Muzyczka, N.; Burger, C.; Mandel, R.J.; Annett, L.; Kirik, D. Continuous low-level glial cell line-derived neurotrophic factor delivery using recombinant adeno-associated viral vectors provides neuroprotection and induces behavioral recovery in a primate model of Parkinson’s disease. J. Neurosci. 2005, 25, 769–777. [Google Scholar] [CrossRef]
- Georgievska, B.; Kirik, D.; Björklund, A. Aberrant sprouting and downregulation of tyrosine hydroxylase in lesioned nigrostriatal dopamine neurons induced by long-lasting overexpression of glial cell line derived neurotrophic factor in the striatum by lentiviral gene transfer. Exp. Neurol. 2002, 177, 461–474. [Google Scholar] [CrossRef]
- Pfeiffer, R.F. Non-motor symptoms in Parkinson’s disease. Park. Relat. Disord. 2016, 22, S119–S122. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidorova, Y.A.; Saarma, M. Small Molecules and Peptides Targeting Glial Cell Line-Derived Neurotrophic Factor Receptors for the Treatment of Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 6575. https://doi.org/10.3390/ijms21186575
Sidorova YA, Saarma M. Small Molecules and Peptides Targeting Glial Cell Line-Derived Neurotrophic Factor Receptors for the Treatment of Neurodegeneration. International Journal of Molecular Sciences. 2020; 21(18):6575. https://doi.org/10.3390/ijms21186575
Chicago/Turabian StyleSidorova, Yulia A., and Mart Saarma. 2020. "Small Molecules and Peptides Targeting Glial Cell Line-Derived Neurotrophic Factor Receptors for the Treatment of Neurodegeneration" International Journal of Molecular Sciences 21, no. 18: 6575. https://doi.org/10.3390/ijms21186575
APA StyleSidorova, Y. A., & Saarma, M. (2020). Small Molecules and Peptides Targeting Glial Cell Line-Derived Neurotrophic Factor Receptors for the Treatment of Neurodegeneration. International Journal of Molecular Sciences, 21(18), 6575. https://doi.org/10.3390/ijms21186575