G Protein-Coupled Estrogen Receptor Mediates Cell Proliferation through the cAMP/PKA/CREB Pathway in Murine Bone Marrow Mesenchymal Stem Cells


 , ,
, ,
Abstract
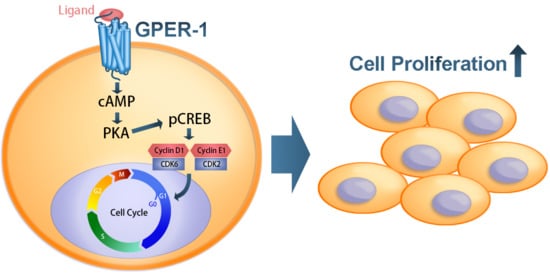
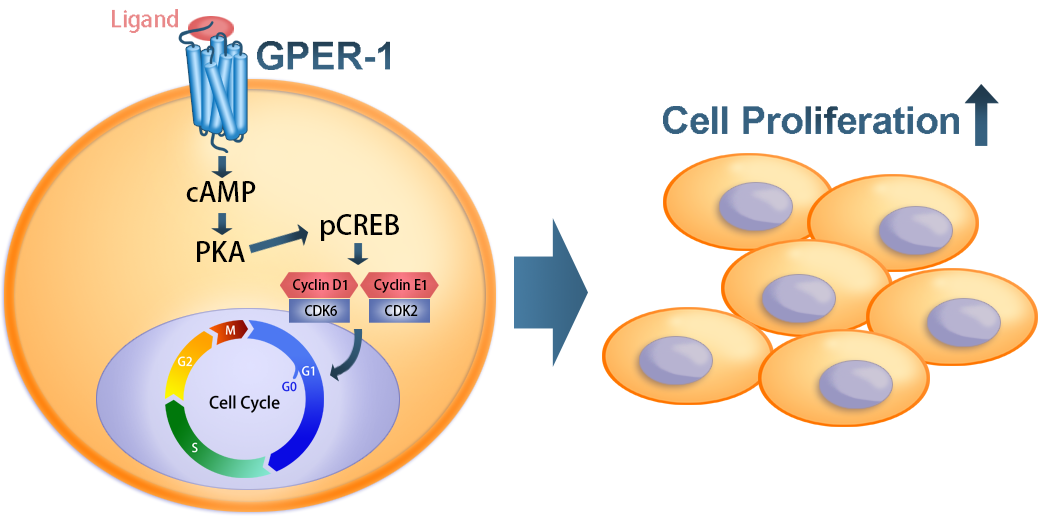
1. Introduction
2. Results
2.1. GPER-1 Promotes Osteogenic Cell Proliferation in Cultured Rat Tibia, as Determined from Ex Vivo Experiments
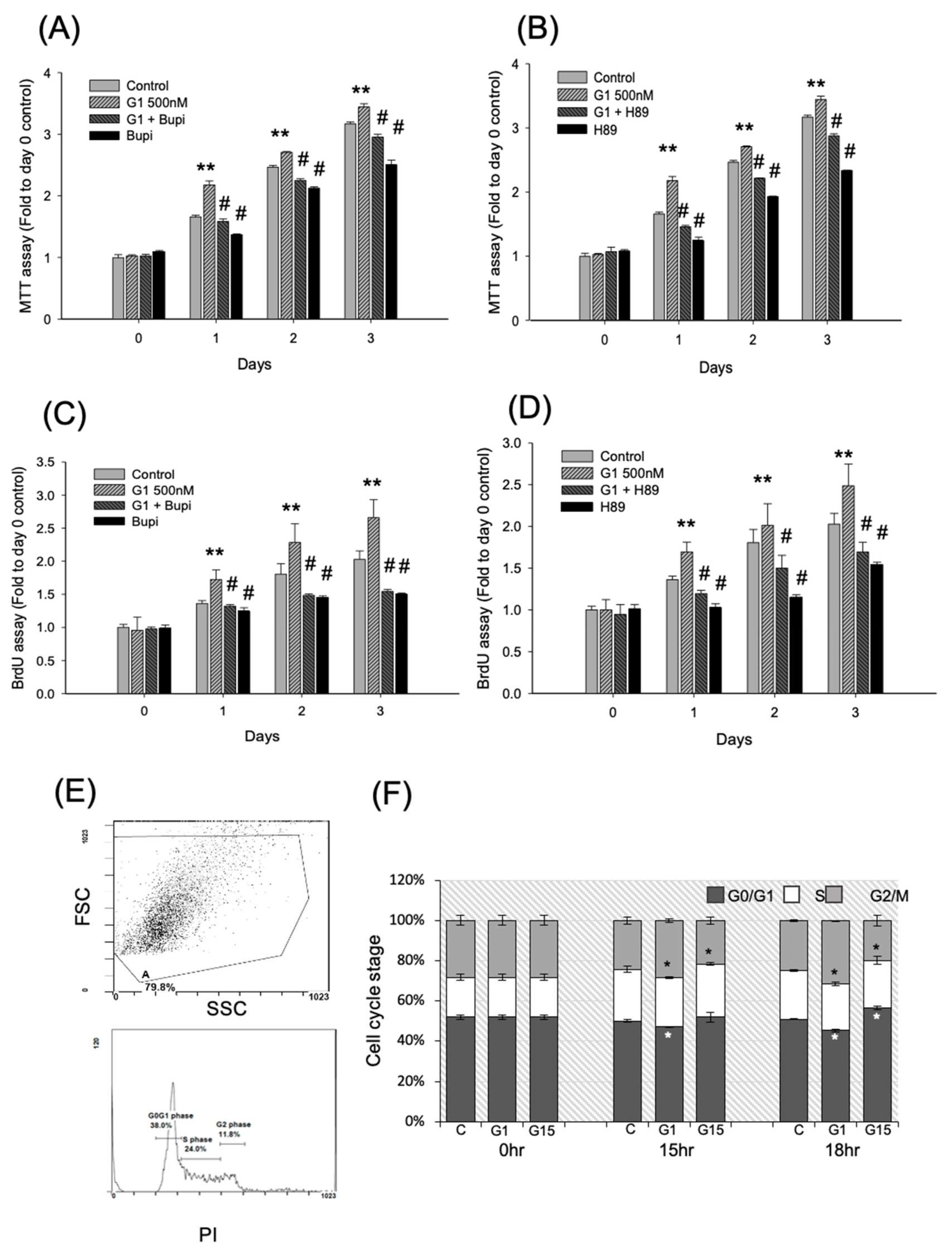
2.2. Activation of GPER-1 Promotes the Viability and Proliferation of Murine BMSCs
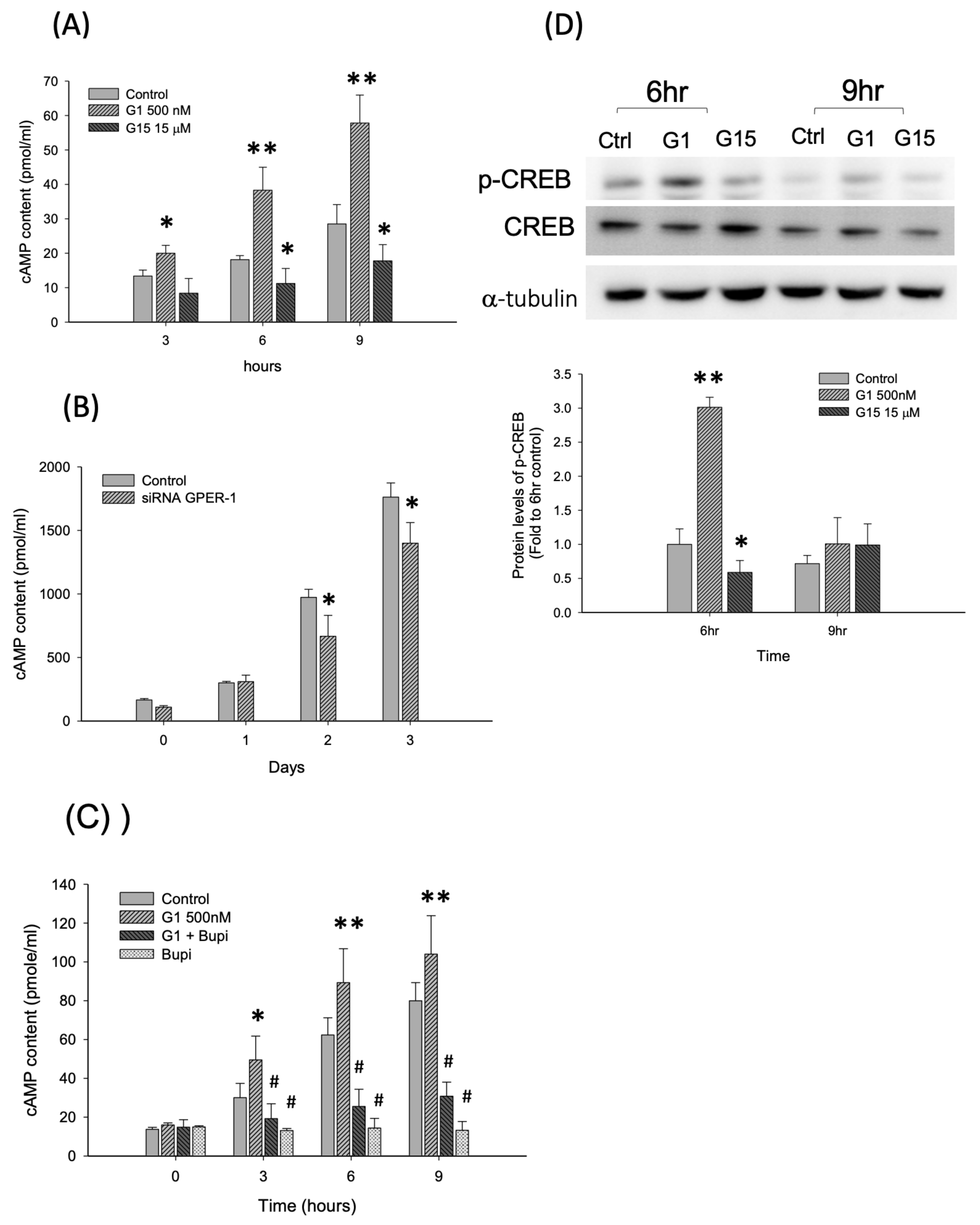
2.3. Activation of GPER-1 Promotes the Proliferation of Murine BMSCs Through cAMP/PKA/p-CREB Signaling
2.4. Activation of GPER-1 Promotes Cell Cycle Kinetics
2.5. Activation of GPER-1 Regulates Cell Cycle Regulators to Promote the Cell Cycle
3. Discussion
4. Materials and Methods
4.1. Cell Proliferation in Ex Vivo Experiments
4.2. Cell Culture
4.3. Drug Treatments
4.4. GPER-1 siRNA Transfection
4.5. MTT Assay
4.6. Cell Proliferation Examination (BrdU Assay)
4.7. cAMP Concentration Assay
4.8. Cell Cycle Kinetics Detected Through Flow Cytometry
4.9. RNA Extraction and Real-Time Polymerase Chain Reaction
4.10. Protein Extraction and Western Blotting
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manolagas, S. Sex Steroids and Bone. Recent Prog. Horm. Res. 2002, 57, 385–409. [Google Scholar] [CrossRef]
- Gay, C.V.; Weber, J.A. Regulation of differentiated osteoclasts. Crit. Rev. Eukaryot. Gene Expr. 2000, 10, 213–230. [Google Scholar] [CrossRef]
- Syed, F.A.; Mödder, U.I.L.; Fraser, D.G.; Spelsberg, T.C.; Rosen, C.J.; Krust, A.; Chambon, P.; Jameson, J.L.; Khosla, S. Skeletal effects of estrogen are mediated by opposing actions of classical and nonclassical estrogen receptor pathways. J. Bone Miner. Res. 2005, 20, 1992–2001. [Google Scholar] [CrossRef]
- Zhou, S.; Zilberman, Y.; Wassermann, K.; Bain, S.D.; Sadovsky, Y.; Gazit, D. Estrogen modulates estrogen receptor alpha and beta expression, osteogenic activity, and apoptosis in mesenchymal stem cells (MSCs) of osteoporotic mice. J. Cell. Biochem. Suppl. 2001, 81 (Suppl. 36), 144–155. [Google Scholar] [CrossRef]
- Samuels, A.; Perry, M.; Gibson, R.; Colley, S.M.; Tobias, J. Role of endothelial nitric oxide synthase in estrogen-induced osteogenesis. Bone 2001, 29, 24–29. [Google Scholar] [CrossRef]
- Windahl, S.H.; Saxon, L.; Börjesson, A.E.; Lagerquist, M.K.; Frenkel, B.; Henning, P.; Lerner, U.; Galea, G.L.; Meakin, L.B.; Engdahl, C.; et al. Estrogen receptor-α is required for the osteogenic response to mechanical loading in a ligand-independent manner involving its activation function 1 but not 2. J. Bone Miner. Res. 2013, 28, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.; Hajibeigi, A.; Long, M.; Hahner, L.; Gore, C.; Hsieh, J.-T.; Clegg, D.; Zerwekh, J.; Oz, O.K. GPR30 deficiency causes increased bone mass, mineralization, and growth plate proliferative activity in male mice. J. Bone Miner. Res. 2011, 26, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.S.; Brown, M. Estrogen Receptor Target Gene: An Evolving Concept. Mol. Endocrinol. 2006, 20, 1707–1714. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef]
- Barton, M. Not lost in translation: Emerging clinical importance of the G protein-coupled estrogen receptor GPER. Steroids 2016, 111, 37–45. [Google Scholar] [CrossRef]
- Khosla, S. Update on estrogens and the skeleton. J. Clin. Endocrinol. Metab. 2010, 95, 3569–3577. [Google Scholar] [CrossRef] [PubMed]
- Mårtensson, U.E.; Salehi, A.A.; Windahl, S.; Gomez, M.F.; Swärd, K.; Daszkiewicz-Nilsson, J.; Wendt, A.; Andersson, N.; Hellstrand, P.; Grände, P.-O.; et al. Deletion of the G Protein-Coupled Receptor 30 Impairs Glucose Tolerance, Reduces Bone Growth, Increases Blood Pressure, and Eliminates Estradiol-Stimulated Insulin Release in Female Mice. Endocrinology 2009, 150, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Iravani, M.; Lagerquist, M.K.; Karimian, E.; Chagin, A.S.; Ohlsson, C.; Savendahl, L. Effects of the selective GPER1 agonist G1 on bone growth. Endocr. Connect. 2019, 8, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Chagin, A.S.; Sävendahl, L. GPR30 Estrogen Receptor Expression in the Growth Plate Declines as Puberty Progresses. J. Clin. Endocrinol. Metab. 2007, 92, 4873–4877. [Google Scholar] [CrossRef]
- Heino, T.J.; Chagin, A.S.; Sävendahl, L. The novel estrogen receptor G-protein-coupled receptor 30 is expressed in human bone. J. Endocrinol. 2008, 197, R1–R6. [Google Scholar] [CrossRef]
- Niada, S.; Giannasi, C.; Ferreira, L.M.J.; Milani, A.; Arrigoni, E.; Brini, A.T. 17β-estradiol differently affects osteogenic differentiation of mesenchymal stem/stromal cells from adipose tissue and bone marrow. Differentiation 2016, 92, 291–297. [Google Scholar] [CrossRef]
- Bidwell, J.P.; Alvarez, M.B.; Hood, M.; Childress, P. Functional Impairment of Bone Formation in the Pathogenesis of Osteoporosis: The Bone Marrow Regenerative Competence. Curr. Osteoporos. Rep. 2013, 11, 117–125. [Google Scholar] [CrossRef]
- Filardo, E.J. Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: A novel signaling pathway with potential significance for breast cancer. J. Steroid Biochem. Mol. Boil. 2002, 80, 231–238. [Google Scholar] [CrossRef]
- Revankar, C.M. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Brailoiu, E.; Yerrum, S.; Shupp, H.A.; Slifker, M.J.; Cunliffe, H.E.; Black, M.A.; Donato, A.L.; Arterburn, J.B.; Oprea, T.I.; et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res. 2010, 70, 1184–1194. [Google Scholar] [CrossRef]
- Prossnitz, E.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Maggiolini, M.; Picard, D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J. Endocrinol. 2009, 204, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R. Estrogen-Induced Activation of Erk-1 and Erk-2 Requires the G Protein-Coupled Receptor Homolog, GPR30, and Occurs via Trans-Activation of the Epidermal Growth Factor Receptor through Release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Wallacides, A.; Chesnel, A.; Ajj, H.; Chillet, M.; Flament, S.; Dumond, H. Estrogens promote proliferation of the seminoma-like TCam-2 cell line through a GPER-dependent ERalpha36 induction. Mol. Cell. Endocrinol. 2012, 350, 61–71. [Google Scholar] [CrossRef]
- Holm, A.; Baldetorp, B.; Olde, B.; Leeb-Lundberg, L.F.; Leeb-Lundberg, L.F.; Nilsson, B.-O. The GPER1 Agonist G-1 Attenuates Endothelial Cell Proliferation by Inhibiting DNA Synthesis and Accumulating Cells in the S and G2 Phases of the Cell Cycle. J. Vasc. Res. 2011, 48, 327–335. [Google Scholar] [CrossRef]
- Olde, B.; Leeb-Lundberg, L.F. GPR30/GPER1: Searching for a role in estrogen physiology. Trends Endocrinol. Metab. 2009, 20, 409–416. [Google Scholar] [CrossRef]
- Zhao, B.; Xiong, Y.; Zhang, Y.; Jia, L.; Zhang, W.; Xu, X. Rutin promotes osteogenic differentiation of periodontal ligament stem cells through the GPR30-mediated PI3K/AKT/mTOR signaling pathway. Exp. Boil. Med. 2020, 245, 552–561. [Google Scholar] [CrossRef]
- Yang, F.; Lin, Z.W.; Huang, T.Y.; Chen, T.T.; Cui, J.; Li, M.Y.; Hua, Y. Ligustilide, a major bioactive component of Angelica sinensis, promotes bone formation via the GPR30/EGFR pathway. Sci. Rep. 2019, 9, 6991. [Google Scholar] [CrossRef]
- Lin, X.; Li, L.; Wu, S.; Tian, J.; Zheng, W. Activation of GPR30 promotes osteogenic differentiation of MC3T3-E1 cells: An implication in osteoporosis. IUBMB Life 2019, 71, 1751–1759. [Google Scholar] [CrossRef]
- Jia, B.; Gao, Y.; Li, M.; Shi, J.; Peng, Y.; Du, X.; Klocker, H.; Sampson, N.; Shen, Y.; Liu, M.; et al. GPR30 promotes prostate stromal cell activation via suppression of ER? Expression and its downstream signaling pathway. Endocrinology 2016, 157, 3023–3035. [Google Scholar] [CrossRef] [PubMed]
- Teplyuk, N.M.; Galindo, M.; Teplyuk, V.I.; Pratap, J.; Young, D.W.; Lapointe, D.; Javed, A.; Stein, J.L.; Lian, J.B.; Stein, G.S.; et al. Runx2 Regulates G Protein-coupled Signaling Pathways to Control Growth of Osteoblast Progenitors. J. Boil. Chem. 2008, 283, 27585–27597. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Ge, H.-F.; Zhang, C.; Chen, J.-Y.; Li, H.-H.; Fang, X.-Y.; Tan, L.; Liu, X.; Jia, Z.-C.; Feng, H.; et al. G protein-coupled estrogen receptor 1 negatively regulates the proliferation of mouse-derived neural stem/progenitor cells via extracellular signal-regulated kinase pathway. Brain Res. 2019, 1714, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Albanito, L.; Lappano, R.; Madeo, A.; Chimento, A.; Prossnitz, E.; Cappello, A.R.; Dolce, V.; Abonante, S.; Pezzi, V.; Maggiolini, M. Effects of Atrazine on Estrogen Receptor α– and G Protein–Coupled Receptor 30–Mediated Signaling and Proliferation in Cancer Cells and Cancer-Associated Fibroblasts. Environ. Health Perspect. 2015, 123, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Guo, R.; Qiao, Y.; Zhang, Y.; Lei, J.; Wang, X.; Li, L.; Hu, D. The G Protein–Coupled Receptor GPR30 Mediates the Nontranscriptional Effect of Estrogen on the Activation of PI3K/Akt Pathway in Endometrial Cancer Cells. Int. J. Gynecol. Cancer 2013, 23, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.-Y.; Weng, J.-R.; Hu, J.-L.; Wang, D.; Sargeant, A.M.; Chiu, C.-F. G15, a GPR30 antagonist, induces apoptosis and autophagy in human oral squamous carcinoma cells. Chem. Interact. 2013, 206, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Nijweide, P.J.; Burger, E.H.; Feyen, J.H. Cells of bone: Proliferation, differentiation, and hormonal regulation. Physiol. Rev. 1986, 66, 855–886. [Google Scholar] [CrossRef]
- Chuang, S.-C.; Chen, C.-H.; Fu, Y.-C.; Tai, I.-C.; Li, C.-J.; Chang, L.-F.; Ho, M.-L.; Chang, J.-K. Estrogen receptor mediates simvastatin-stimulated osteogenic effects in bone marrow mesenchymal stem cells. Biochem. Pharmacol. 2015, 98, 453–464. [Google Scholar] [CrossRef]
- Song, C.; Wang, J.; Song, Q.; Li, X.; Chen, Z.; Ma, Q.; Liu, Z.; Jia, H.; Dang, G. Simvastatin induces estrogen receptor-alpha (ER-α) in murine bone marrow stromal cells. J. Bone Miner. Metab. 2008, 26, 213–217. [Google Scholar] [CrossRef]
- Prossnitz, E.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef]
- Lin, B.C.; Suzawa, M.; Blind, R.; Tobias, S.C.; Bulun, S.E.; Scanlan, T.S.; A Ingraham, H. Stimulating the GPR30 estrogen receptor with a novel tamoxifen analogue activates SF-1 and promotes endometrial cell proliferation. Cancer Res. 2009, 69, 5415–5423. [Google Scholar] [CrossRef] [PubMed]
- Puzhko, S.; Goodyer, C.G.; Kerachian, M.A.; Canaff, L.; Misra, M.; Jüppner, H.; Bastepe, M.; Hendy, G.N. Parathyroid hormone signaling via Gαs is selectively inhibited by an NH2-terminally truncated Gαs: Implications for pseudohypoparathyroidism. J. Bone Miner. Res. 2011, 26, 2473–2485. [Google Scholar] [CrossRef] [PubMed]
- Minamizaki, T.; Yoshiko, Y.; Kozai, K.; Aubin, J.E.; Maeda, N. EP2 and EP4 receptors differentially mediate MAPK pathways underlying anabolic actions of prostaglandin E2 on bone formation in rat calvaria cell cultures. Bone 2009, 44, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Daniel, P.; Filiz, G.; Brown, D.; Hollande, F.; Gonzales, M.; D’Abaco, G.M.; Papalexis, N.; Phillips, W.; Malaterre, J.; Ramsay, R.G.; et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis 2014, 3, e108. [Google Scholar] [CrossRef] [PubMed]
- New, D.C.; Wong, Y.H. Molecular mechanisms mediating the G protein-coupled receptor regulation of cell cycle progression. J. Mol. Signal. 2007, 2, 2. [Google Scholar] [CrossRef]
- Chen, C.-H.; Ho, M.-L.; Chang, J.-K.; Hung, S.-H.; Wang, G.-J. Green tea catechin enhances osteogenesis in a bone marrow mesenchymal stem cell line. Osteoporos. Int. 2005, 16, 2039–2045. [Google Scholar] [CrossRef]
- Chang, J.-K.; Li, C.-J.; Wu, S.-C.; Yeh, C.-H.; Chen, C.-H.; Fu, Y.-C.; Wang, G.-J.; Ho, M.-L. Effects of anti-inflammatory drugs on proliferation, cytotoxicity and osteogenesis in bone marrow mesenchymal stem cells. Biochem. Pharmacol. 2007, 74, 1371–1382. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes. | Primers |
|---|---|
| Mouse Cyclin A | F: TGAATCACCACATGCTAT |
| R: TAACCTCCATTTCCCTAAG | |
| Mouse Cyclin B | F: TAGGTACTGGAAAAGGTA |
| R: GCTTCTCTTCTCTAACAT | |
| Mouse Cyclin D1 | F: GCGTACCCTGACACCAATCT |
| R: CTCTTCGCACTTCTGCTCCT | |
| Mouse Cyclin D2 | F: ACCTGTTGACCATCGAGGAG R: CCAAGAAACGGTCCAGGTAA |
| Mouse cyclin E1 | F: GGAAAATCAGACCACCCAGA R: AGACTTCGCACACCTCCATT |
| Mouse Cyclin E2 | F: TCTCAGGAGACGTTCATCCA R: ACAAAAGGCACCATCCAGTC |
| Mouse p27 | F: CAGAATCATAAGCCCCTGGA R: GGTCCTCAGAGTTTGCCTGA |
| Mouse Actin | F: CCAACCGTGAAAAGATGACC |
| R: ACCAGAGGCATACAGGGACA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, S.-C.; Chen, C.-H.; Chou, Y.-S.; Ho, M.-L.; Chang, J.-K. G Protein-Coupled Estrogen Receptor Mediates Cell Proliferation through the cAMP/PKA/CREB Pathway in Murine Bone Marrow Mesenchymal Stem Cells. Int. J. Mol. Sci. 2020, 21, 6490. https://doi.org/10.3390/ijms21186490
Chuang S-C, Chen C-H, Chou Y-S, Ho M-L, Chang J-K. G Protein-Coupled Estrogen Receptor Mediates Cell Proliferation through the cAMP/PKA/CREB Pathway in Murine Bone Marrow Mesenchymal Stem Cells. International Journal of Molecular Sciences. 2020; 21(18):6490. https://doi.org/10.3390/ijms21186490
Chicago/Turabian StyleChuang, Shu-Chun, Chung-Hwan Chen, Ya-Shuan Chou, Mei-Ling Ho, and Je-Ken Chang. 2020. "G Protein-Coupled Estrogen Receptor Mediates Cell Proliferation through the cAMP/PKA/CREB Pathway in Murine Bone Marrow Mesenchymal Stem Cells" International Journal of Molecular Sciences 21, no. 18: 6490. https://doi.org/10.3390/ijms21186490
APA StyleChuang, S.-C., Chen, C.-H., Chou, Y.-S., Ho, M.-L., & Chang, J.-K. (2020). G Protein-Coupled Estrogen Receptor Mediates Cell Proliferation through the cAMP/PKA/CREB Pathway in Murine Bone Marrow Mesenchymal Stem Cells. International Journal of Molecular Sciences, 21(18), 6490. https://doi.org/10.3390/ijms21186490