Agent-Based Modeling of T Cell Receptor Cooperativity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
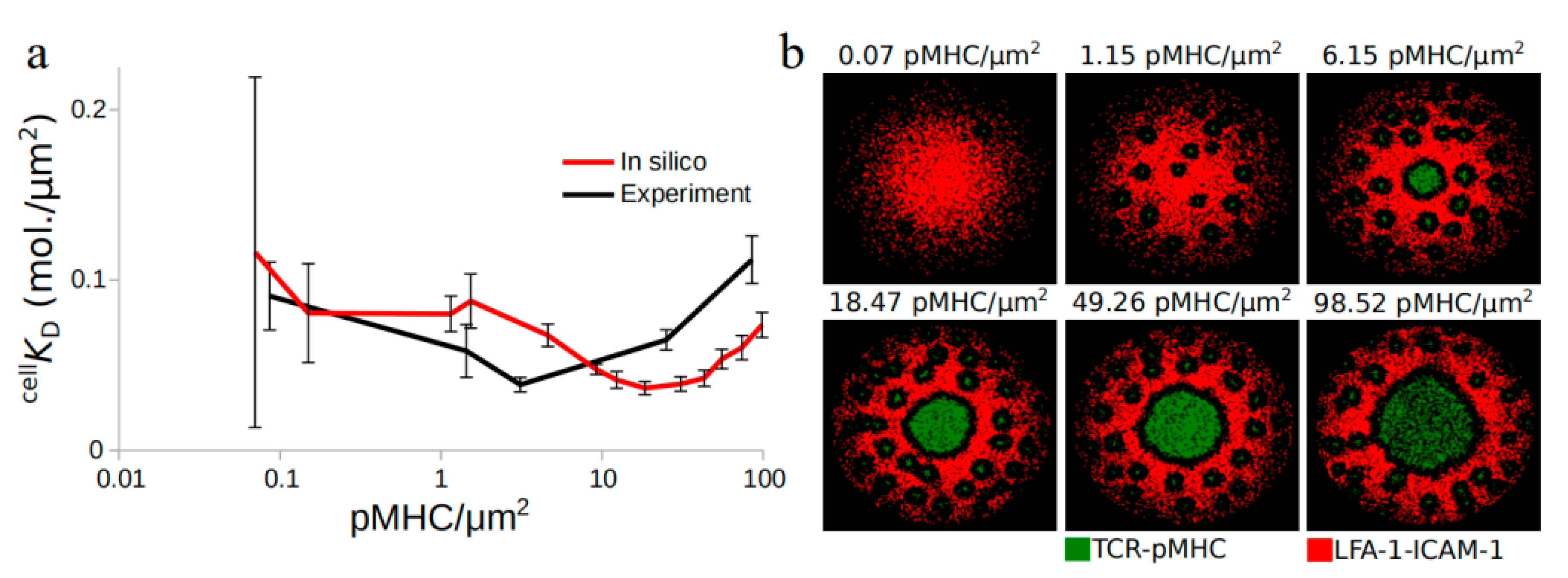
2.1. TCR Affinity Modulation during Antigen Recognition
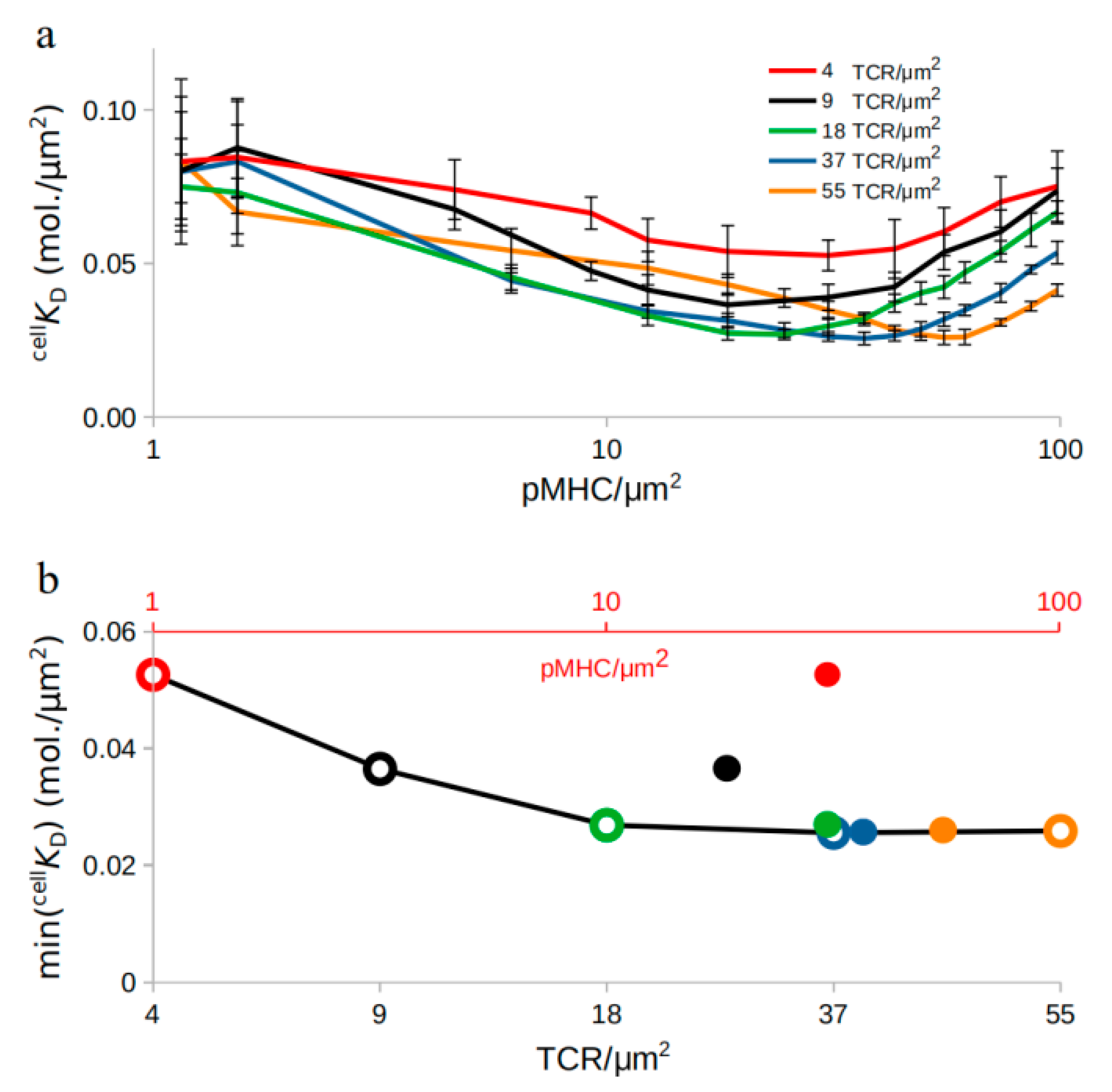
2.2. TCR-pMHC Affinity Is also Affected by the TCR Density
2.3. Centripetal Transport of TCR-pMHC as a Possible Mechanism for Affinity Modulation
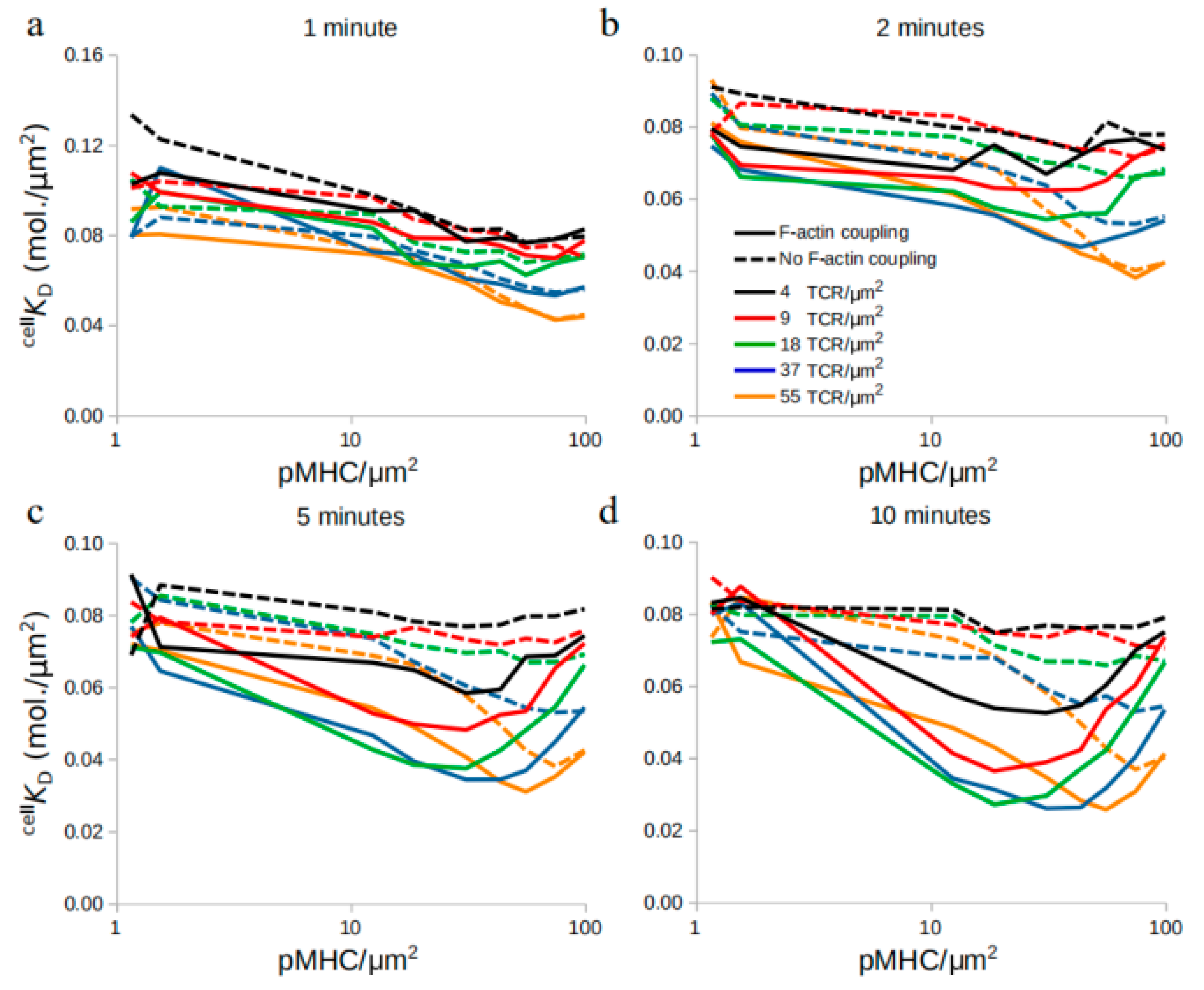
2.4. Affinity Dynamics Depend on the Time of Measurement
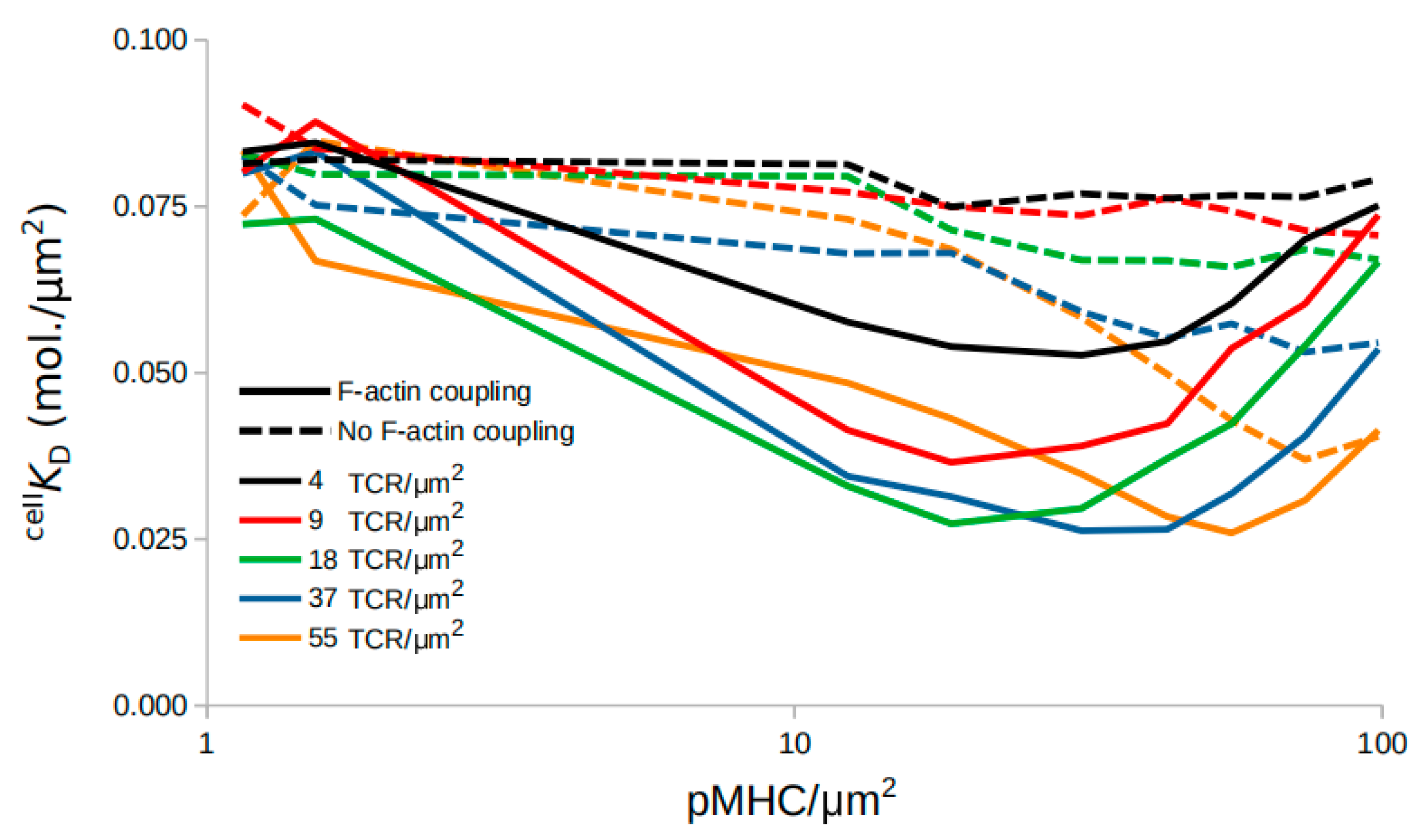
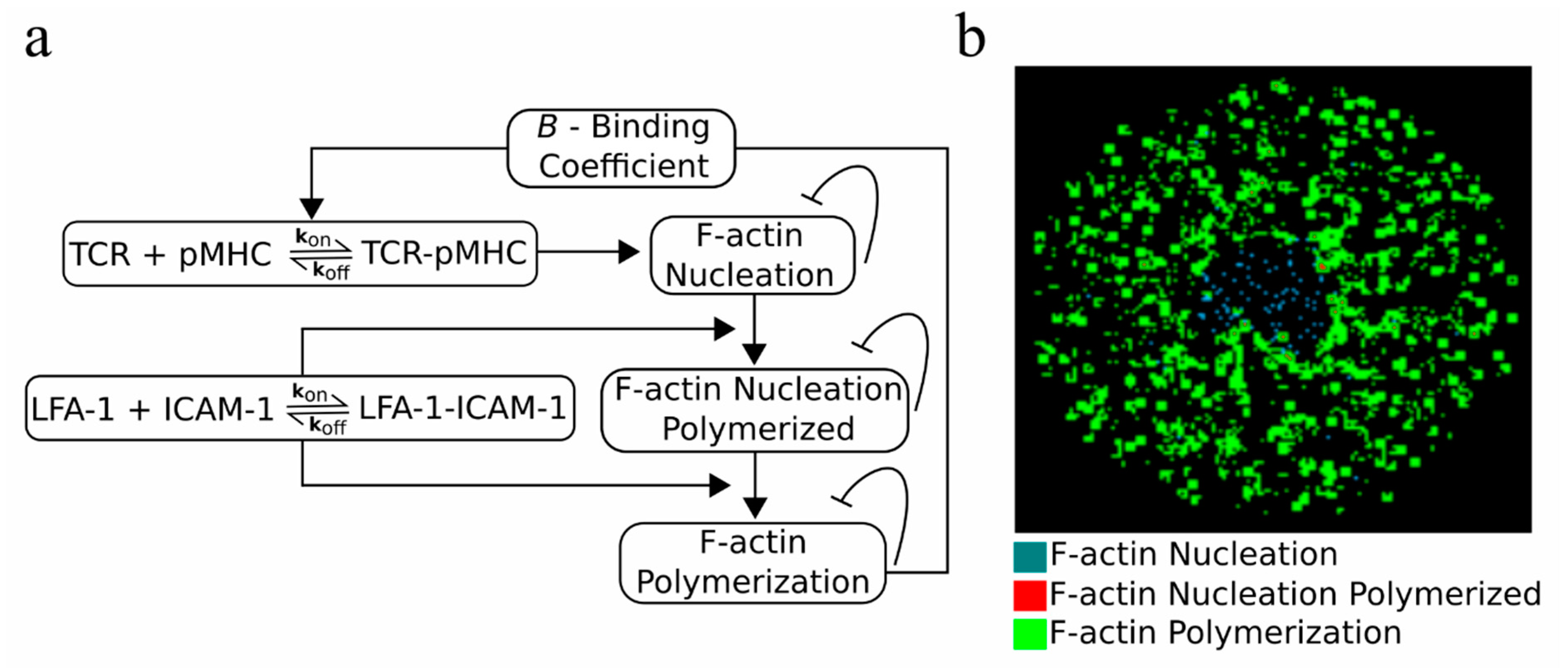
2.5. An F-actin Foci Model
2.6. Modulation of TCR-pMHC Association Rate by F-actin Foci
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Availability
Abbreviations
| APC | Antigen Presenting cell |
| B | Binding coefficient |
| IS | Immunological synapse |
| LFA-1 | Leukocyte function-associated antigen-1 |
| MCC | Moth cytochrome C (88-103) peptide |
| NFAT | Nuclear factor of activated T-cells |
| pMHC | Antigen peptide bound to a major histocompatibility complex |
| SBS | Size-based segregation |
| SLB | Supported lipid bilayer |
| cSMAC | Central supramolecular activation cluster |
| TCR | T cell receptor |
References
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The immunological synapse: A molecular machine controlling T cell activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, K. High sensitivity, low affinity—Paradox of T-cell receptor recognition. Curr. Opin. Immunol. 1994, 6, 9–12. [Google Scholar] [CrossRef]
- Schamel, W.W.; Risueño, R.M.; Minguet, S.; Ortíz, A.R.; Alarcón, B. A conformation-and avidity-based proofreading mechanism for the TCR–CD3 complex. Trends Immunol. 2006, 27, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Siller-Farfán, J.A.; Dushek, O. Molecular mechanisms of T cell sensitivity to antigen. Immunol. Rev. 2018, 285, 194–205. [Google Scholar] [CrossRef]
- Huang, J.; Brameshuber, M.; Zeng, X.; Xie, J.; Li, Q.J.; Chien, Y.H.; Valitutti, S.; Davis, M.M. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4+ T cells. Immunity 2013, 39, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Chervin, A.S.; Kranz, D.M. T-cell receptor binding affinities and kinetics: Impact on T-cell activity and specificity. Immunology 2009, 126, 165–176. [Google Scholar] [CrossRef] [PubMed]
- DeMond, A.L.; Mossman, K.D.; Starr, T.; Dustin, M.L.; Groves, J.T. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys. J. 2008, 94, 3286–3292. [Google Scholar] [CrossRef]
- Yi, J.; Wu, X.S.; Crites, T.; Hammer, J.A. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol. Biol. Cell 2012, 23, 834–852. [Google Scholar] [CrossRef]
- Murugesan, S.; Hong, J.; Yi, J.; Li, D.; Beach, J.R.; Shao, L.; Meinhardt, J.; Madison, G.; Wu, X.; Betzig, E.; et al. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J. Cell Biol. 2016, 215, 383–399. [Google Scholar] [CrossRef]
- Comrie, W.A.; Babich, A.; Burkhardt, J.K. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J. Cell Biol. 2015, 208, 475–491. [Google Scholar] [CrossRef]
- Pielak, R.M.; O’Donoghue, G.P.; Lin, J.J.; Alfieri, K.N.; Fay, N.C.; Low-Nam, S.T.; Groves, J.T. Early T cell receptor signals globally modulate ligand: Receptor affinities during antigen discrimination. Proc. Natl. Acad. Sci. USA 2017, 114, 12190–12195. [Google Scholar] [CrossRef] [PubMed]
- Martin-Blanco, N.; Blanco, R.; Alda-Catalinas, C.; Bovolenta, E.; Oeste, C.; Palmer, E.; Schamel, W.; Lythe, G.; Molina-París, C.; Castro, M.; et al. A window of opportunity for cooperativity in the T Cell Receptor. Nat. Commun. 2018, 9, 2618. [Google Scholar] [CrossRef] [PubMed]
- Molnár, E.; Deswal, S.; Schamel, W.W. Pre-clustered TCR complexes. FEBS Lett. 2010, 584, 4832–4837. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Alarcón, B. TCR nanoclusters as the framework for transmission of conformational changes and cooperativity. Front. Immunol. 2012, 3, 115. [Google Scholar] [CrossRef]
- Schamel, W.W.; Alarcón, B. Organization of the resting TCR in nanoscale oligomers. Immunol. Rev. 2013, 251, 13–20. [Google Scholar] [CrossRef]
- Sherman, E.; Barr, V.; Manley, S.; Patterson, G.; Balagopalan, L.; Akpan, I.; Regan, C.K.; Merrill, R.K.; Sommers, C.L.; Lippincott-Schwartz, J.; et al. Functional nanoscale organization of signaling molecules downstream of the T cell antigen receptor. Immunity 2011, 35, 705–720. [Google Scholar] [CrossRef]
- Pageon, S.V.; Tabarin, T.; Yamamoto, Y.; Ma, Y.; Nicovich, P.R.; Bridgeman, J.S.; Cohnen, A.; Benzing, C.; Gao, Y.; Crowther, M.D.; et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl. Acad. Sci. USA 2016, 113, 5454–5463. [Google Scholar] [CrossRef]
- Varma, R.; Campi, G.; Yokosuka, T.; Saito, T.; Dustin, M.L. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 2006, 25, 117–127. [Google Scholar] [CrossRef]
- Mossman, K.D.; Campi, G.; Groves, J.T.; Dustin, M.L. Altered TCR signaling from geometrically repatterned immunological synapses. Science 2005, 310, 1191–1193. [Google Scholar] [CrossRef]
- Campi, G.; Varma, R.; Dustin, M.L. Actin and agonist MHC–peptide complex–dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 2005, 202, 1031–1036. [Google Scholar] [CrossRef]
- Springer, T.A.; Dustin, M.L. Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 2012, 24, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Faull, R.J.; Ginsberg, M.H. Inside-out signaling through integrins. J. Am. Soc. Nephrol. 1996, 7, 1091–1097. [Google Scholar] [PubMed]
- Changeux, J.P.; Edelstein, S.J. Allosteric mechanisms of signal transduction. Science 2005, 308, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- Schamel, W.W.; Alarcon, B.; Höfer, T.; Minguet, S. The allostery model of TCR regulation. J. Immunol. 2017, 198, 47–52. [Google Scholar] [CrossRef]
- Kumari, S.; Depoil, D.; Martinelli, R.; Judokusumo, E.; Carmona, G.; Gertler, F.B.; Kam, L.C.; Carman, C.V.; Burkhardt, J.K.; Irvine, D.J.; et al. Actin foci facilitate activation of the phospholipase C-γ in primary T lymphocytes via the WASP pathway. eLife 2015, 4, 4953. [Google Scholar] [CrossRef]
- Beemiller, P.; Jacobelli, J.; Krummel, M.F. Integration of the movement of signaling microclusters with cellular motility in immunological synapses. Nat. Immunol. 2012, 13, 787–795. [Google Scholar] [CrossRef]
- Carrizosa, E.; Gomez, T.S.; Labno, C.M.; Dehring, D.A.K.; Liu, X.; Freedman, B.D.; Billadeau, D.D.; Burkhardt, J.K. Hematopoietic lineage cell-specific protein 1 is recruited to the immunological synapse by IL-2-inducible T cell kinase and regulates phospholipase Cγ1 Microcluster dynamics during T cell spreading. J. Immunol. 2009, 183, 7352–7361. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Braiman, A.; Barda-Saad, M.; Sommers, C.L.; Samelson, L.E. Recruitment and activation of PLCγ1 in T cells: A new insight into old domains. EMBO J. 2006, 25, 774–784. [Google Scholar] [CrossRef]
- Forrest, S.; Beauchemin, C. Computer immunology. Immunol. Rev. 2007, 216, 176–197. [Google Scholar] [CrossRef]
- Metzcar, J.; Wang, Y.; Heiland, R.; Macklin, P. A review of cell-based computational modeling in cancer biology. JCO Clin. Cancer Inform. 2019, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Igoshin, O.A.; Cotter, C.R.; Shimkets, L.J. Agent-Based modeling reveals possible mechanisms for observed aggregation cell behaviors. Biophys. J. 2018, 115, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Griffié, J.; Peters, R.; Owen, D.M. An agent-based model of molecular aggregation at the cell membrane. PLoS ONE 2020, 15, e0226825. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, T.; Figge, M.T. Dimensionality of Motion and Binding Valency Govern Receptor–Ligand Kinetics As Revealed by Agent-Based Modeling. Front. Immunol. 2017, 8, 1692. [Google Scholar] [CrossRef] [PubMed]
- Sugár, I.P.; Das, J.; Jayaprakash, C.; Sealfon, S.C. Multiscale modeling of complex formation and CD80 depletion during immune synapse development. Biophys. J. 2017, 112, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.; Meyer-Hermann, M. Cell transmembrane receptors determine tissue pattern stability. Phys. Rev. Lett. 2008, 101, 148102. [Google Scholar] [CrossRef]
- Miller, J.; Parker, M.; Bourret, R.B.; Giddings, M.C. An agent-based model of signal transduction in bacterial chemotaxis. PLoS ONE 2010, 5, e9454. [Google Scholar] [CrossRef]
- Pennisi, M.; Rajput, A.M.; Toldo, L.; Pappalardo, F. Agent based modeling of Treg-Teff cross regulation in relapsing-remitting multiple sclerosis. BMC Bioinform. 2013, 14, 1–14. [Google Scholar] [CrossRef]
- Gong, C.; Milberg, O.; Wang, B.; Vicini, P.; Narwal, R.; Roskos, L.; Popel, A.S. A computational multiscale agent-based model for simulating spatio-temporal tumour immune response to PD1 and PDL1 inhibition. J. R. Soc. Interface 2017, 14, 20170320. [Google Scholar] [CrossRef]
- Siokis, A.; Robert, P.A.; Demetriou, P.; Dustin, M.L.; Meyer-Hermann, M. F-Actin-Driven CD28-CD80 Localization in the Immune Synapse. Cell Rep. 2018, 24, 1151–1162. [Google Scholar] [CrossRef]
- Labrecque, N.; Whitfield, L.S.; Obst, R.; Waltzinger, C.; Benoist, C.; Mathis, D. How much TCR does a T cell need? Immunity 2001, 15, 71–82. [Google Scholar] [CrossRef]
- Hetrick, B.; Han, M.S.; Helgeson, L.A.; Nolen, B.J. Small molecules CK-666 and CK-869 inhibit actin-related protein 2/3 complex by blocking an activating conformational change. Chem. Biol. 2013, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Minguet, S.; Swamy, M.; Alarcón, B.; Luescher, I.F.; Schamel, W.W. Full activation of the T cell receptor requires both clustering and conformational changes at CD3. Immunity 2007, 26, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martín, N.; Risueño, R.M.; Morreale, A.; Zaldívar, I.; Fernández-Arenas, E.; Herranz, F.; Ortiz, A.R.; Alarcón, B. Cooperativity between T cell receptor complexes revealed by conformational mutants of CD3ε. Sci. Signal. 2009, 2, ra43. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, T.M.; Bieler, J.G.; Edidin, M.; Schneck, J.P. Increased TCR avidity after T cell activation: A mechanism for sensing low-density antigen. Immunity 2001, 14, 135–143. [Google Scholar] [CrossRef]
- Dixon, J.F.; Law, J.L.; Favero, J.J. Activation of human T lymphocytes by crosslinking of anti-CD3 monoclonal antibodies. J. Leukoc. Biol. 1989, 46, 214–220. [Google Scholar] [CrossRef]
- Dustin, M.L.; Springer, T.A. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 1989, 341, 619. [Google Scholar] [CrossRef]
- Dong, G.; Kalifa, R.; Nath, P.R.; Gelkop, S.; Isakov, N. TCR crosslinking promotes Crk adaptor protein binding to tyrosine-phosphorylated CD3ζ chain. Biochem. Biophys. Res. Commun. 2017, 488, 541–546. [Google Scholar] [CrossRef]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siokis, A.; Robert, P.A.; Meyer-Hermann, M. Agent-Based Modeling of T Cell Receptor Cooperativity. Int. J. Mol. Sci. 2020, 21, 6473. https://doi.org/10.3390/ijms21186473
Siokis A, Robert PA, Meyer-Hermann M. Agent-Based Modeling of T Cell Receptor Cooperativity. International Journal of Molecular Sciences. 2020; 21(18):6473. https://doi.org/10.3390/ijms21186473
Chicago/Turabian StyleSiokis, Anastasios, Philippe A. Robert, and Michael Meyer-Hermann. 2020. "Agent-Based Modeling of T Cell Receptor Cooperativity" International Journal of Molecular Sciences 21, no. 18: 6473. https://doi.org/10.3390/ijms21186473
APA StyleSiokis, A., Robert, P. A., & Meyer-Hermann, M. (2020). Agent-Based Modeling of T Cell Receptor Cooperativity. International Journal of Molecular Sciences, 21(18), 6473. https://doi.org/10.3390/ijms21186473