Deciphering SARS-CoV-2 Virologic and Immunologic Features

 , , , and
, , , and
Abstract
1. Introduction
2. Viral Description
2.1. Unravelling SARS-CoV-2 Genome and Viral Protein Features
2.2. Inside the SARS-CoV-2V Life Cycle
2.2.1. Entry and Fusion
2.2.2. Release and Replication
2.2.3. Assembly
2.2.4. Budding and Transmission
2.3. Tropism and Associated Clinical Manifestations
2.3.1. Nasal Cavity
2.3.2. Olfactory Epithelium
2.3.3. Lung
2.3.4. Gastrointestinal Tract
2.3.5. Liver
2.3.6. Cardiovascular System
2.3.7. Brain
2.3.8. Kidney
2.3.9. Secondary Lymphoid Organs
2.3.10. Adipose Tissue
2.3.11. Musculoskeletal Symptoms
3. Immunopathology of SARS-CoV-2
3.1. Cellular Response
3.1.1. Innate Immune Response
Pattern Recognition Receptor
Dendritic Cells (DCs)
Macrophages
Polymorphonuclear Neutrophils (PMN)
Complement
Natural Killer (NK) Cells
3.1.2. Adaptive Immune Response
T Cells
B Cells
3.2. Molecular Response
3.3. Immune Evasion
4. Regards to Discrepancies in Diverse Immune Conditions
4.1. Elderly
4.2. Children
4.3. Pregnancy
4.4. Men/Women Susceptibility
4.5. Genetic Susceptibility
5. Immunotherapy
5.1. Antibody-Based Therapy
5.2. Immunomodulators
5.3. Stem Cell-Based Therapy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the origin and continuing evolution of SARS-CoV-2. Nat. Sci. Rev. 2020. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Knipe, D.M.; Howley, P.M. Fields Virology; Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013. [Google Scholar]
- Bos, E.C.W.; Luytjes, W.; Meulen, H.V.D.; Koerten, H.K.; Spaan, W.J.M. The Production of Recombinant Infectious DI-Particles of a Murine Coronavirus in the Absence of Helper Virus. Virology 1996, 218, 52–60. [Google Scholar] [CrossRef]
- Vennema, H.; Godeke, G.J.; Rossen, J.W.; Voorhout, W.F.; Horzinek, M.C.; Opstelten, D.J.; Rottier, P.J. Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 1996, 15, 2020–2028. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Ion Channel Activity Promotes Virus Fitness and Pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor-binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef]
- Peng, Q.; Peng, R.; Yuan, B.; Zhao, J.; Wang, M.; Wang, X.; Wang, Q.; Sun, Y.; Fan, Z.; Qi, J.; et al. Structural and biochemical characterization of nsp12-nsp7-nsp8 core polymerase complex from COVID-19 virus. Microbiology 2020, 31, 107774. [Google Scholar]
- Kim, J.Y.; Ko, J.-H.; Kim, Y.; Kim, Y.-J.; Kim, J.-M.; Chung, Y.-S.; Kim, H.M.; Han, M.-G.; Kim, S.Y.; Chin, B.S. Viral Load Kinetics of SARS-CoV-2 Infection in First Two Patients in Korea. J. Korean Med. Sci. 2020, 35, e86. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. Microbiology 2020. [Google Scholar] [CrossRef]
- Baranov, P.V.; Henderson, C.M.; Anderson, C.B.; Gesteland, R.F.; Atkins, J.F.; Howard, M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology 2005, 332, 498–510. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLOS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.W.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Bárcena, M.; et al. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. Microbiology 2020. [Google Scholar] [CrossRef]
- Sethna, P.B.; Hung, S.L.; Brian, D.A. Coronavirus subgenomic minus-strand RNAs and the potential for mRNA replicons. Nat. Acad. Sci. 1989, 86, 5626–5630. [Google Scholar] [CrossRef]
- Masters, P.S. The Molecular Biology of Coronaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2006; Volume 66, pp. 193–292. [Google Scholar]
- Fischer, F.; Stegen, C.F.; Masters, P.S.; Samsonoff, W.A. Analysis of Constructed E Gene Mutants of Mouse Hepatitis Virus Confirms a Pivotal Role for E Protein in Coronavirus Assembly. J. Virol. 1998, 72, 7885–7894. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.-W.; Yuen, T.T.-T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.-Y.; Tsang, J.O.-L.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Schifanella, L.; Anderson, J.L.; Galli, M.; Corbellino, M.; Lai, A.; Wieking, G.; Grzywacz, B.; Klatt, N.R.; Haase, A.T.; Schacker, T.W. Massive viral replication and cytopathic effects in early COVID-19 pneumonia. arXiv 2020, arXiv:2005.00004. [Google Scholar]
- Ghinai, I.; McPherson, T.D.; Hunter, J.C.; Kirking, H.L.; Christiansen, D.; Joshi, K.; Rubin, R.; Morales-Estrada, S.; Black, S.R.; Pacilli, M.; et al. First known person-to-person transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the USA. Lancet 2020, 395, 1137–1144. [Google Scholar] [CrossRef]
- Han, Y.; Yang, H. The transmission and diagnosis of 2019 novel coronavirus infection disease (COVID-19): A Chinese perspective. J. Med. Virol. 2020, 92, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.-W.; Zhang, A.J.; Yuan, S.; Poon, V.K.-M.; Chan, C.C.-S.; Lee, A.C.-Y.; Chan, W.-M.; Fan, Z.; Tsoi, H.-W.; Wen, L.; et al. Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: Implications for disease pathogenesis and transmissibility. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Yeo, C.; Kaushal, S.; Yeo, D. Enteric involvement of coronaviruses: Is faecal–oral transmission of SARS-CoV-2 possible? Lancet Gastroenterol. Hepatol. 2020, 5, 335–337. [Google Scholar] [CrossRef]
- Gu, J.; Han, B.; Wang, J. COVID-19: Gastrointestinal Manifestations and Potential Fecal–Oral Transmission. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Napoli, P.E.; Nioi, M.; d’Aloja, E.; Fossarello, M. The Ocular Surface and the Coronavirus Disease 2019: Does a Dual ‘Ocular Route’ Exist? J. Clin. Med. 2020, 9, 1269. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020. [Google Scholar] [CrossRef]
- Muus, C.; Luecken, M.D.; Eraslan, G.; Waghray, A.; Heimberg, G.; Sikkema, L.; Kobayashi, Y.; Vaishnav, E.D.; Subramanian, A.; Smilie, C.; et al. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor Necrosis Factor-α Convertase (ADAM17) Mediates Regulated Ectodomain Shedding of the Severe-acute Respiratory Syndrome-Coronavirus (SARS-CoV) Receptor, Angiotensin-converting Enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF- -converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF- production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential Downregulation of ACE2 by the Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lorenzo, Ó.; Rupérez, M.; Blanco, J.; Egido, J. Systemic Infusion of Angiotensin II into Normal Rats Activates Nuclear Factor-κB and AP-1 in the Kidney: Role of AT1 and AT2 Receptors. Am. J. Pathol. 2001, 158, 1743–1756. [Google Scholar] [CrossRef]
- Wolf, G.; Wenzel, U.; Burns, K.D.; Harris, R.C.; Stahl, R.A.K.; Thaiss, F. Angiotensin II activates nuclear transcription factor-κB through AT1 and AT2 receptors11See Editorial by Luft, p. 2272. Kidney Int. 2002, 61, 1986–1995. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Ruperez, M.; Esteban, V.; Egido, J. Molecular mechanisms of angiotensin II-induced vascular injury. Curr. Sci. 2003, 5, 73–79. [Google Scholar] [CrossRef]
- Xu, J.; Sriramula, S.; Xia, H.; Moreno-Walton, L.; Culicchia, F.; Domenig, O.; Poglitsch, M.; Lazartigues, E. Clinical Relevance and Role of Neuronal AT1 Receptors in ADAM17-Mediated ACE2 Shedding in Neurogenic Hypertension. Circ. Res. 2017, 121, 43–55. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guo, F.; Liu, K.; Wang, H.; Rao, S.; Yang, P.; Jiang, C. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008, 136, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Han, D.P.; Lohani, M.; Cho, M.W. Specific Asparagine-Linked Glycosylation Sites Are Critical for DC-SIGN- and L-SIGN-Mediated Severe Acute Respiratory Syndrome Coronavirus Entry. J. Virol. 2007, 81, 12029–12039. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Y.; Huang, Y.; Ganesh, L.; Leung, K.; Kong, W.-P.; Schwartz, O.; Subbarao, K.; Nabel, G.J. pH-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus Is Mediated by the Spike Glycoprotein and Enhanced by Dendritic Cell Transfer through DC-SIGN. J. Virol. 2004, 78, 5642–5650. [Google Scholar] [CrossRef]
- Chen, Z.; Mi, L.; Xu, J.; Yu, J.; Wang, X.; Jiang, J.; Xing, J.; Shang, P.; Qian, A.; Li, Y.; et al. Function of HAb18G/CD147 in Invasion of Host Cells by Severe Acute Respiratory Syndrome Coronavirus. J. Infect. Dis. 2005, 191, 755–760. [Google Scholar] [CrossRef]
- Pushkarsky, T.; Zybarth, G.; Dubrovsky, L.; Yurchenko, V.; Tang, H.; Guo, H.; Toole, B.; Sherry, B.; Bukrinsky, M. CD147 facilitates HIV-1 infection by interacting with virus-associated cyclophilin A. Proc. Natl. Acad. Sci. USA 2001, 98, 6360–6365. [Google Scholar] [CrossRef]
- Watanabe, A.; Yoneda, M.; Ikeda, F.; Terao-Muto, Y.; Sato, H.; Kai, C. CD147/EMMPRIN Acts as a Functional Entry Receptor for Measles Virus on Epithelial Cells. J. Virol. 2010, 84, 4183–4193. [Google Scholar] [CrossRef]
- Xiong, L.; Edwards, C.K.; Zhou, L. The Biological Function and Clinical Utilization of CD147 in Human Diseases: A Review of the Current Scientific Literature. Int. J. Mol. Sci. 2014, 15, 17411–17441. [Google Scholar] [CrossRef]
- Yurchenko, V.; Constant, S.; Eisenmesser, E.; Bukrinsky, M. Cyclophilin–CD147 interactions: A new target for anti-inflammatory therapeutics. Clin. Exp. Immunol. 2010, 160, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Shilts, J.; Wright, G.J. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. bioRxiv 2020. [Google Scholar] [CrossRef]
- Softic, L.; Brillet, R.; Berry, F.; Ahnou, N.; Nevers, Q.; Morin-Dewaele, M.; Hamadat, S.; Bruscella, P.; Fourati, S.; Pawlotsky, J.-M.; et al. Inhibition of SARS-CoV-2 Infection by the Cyclophilin Inhibitor Alisporivir (Debio 025). Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.T.-C.; Chien, S.-C.; Chen, I.-Y.; Lai, C.-T.; Tsay, Y.-G.; Chang, S.C.; Chang, M.-F. Surface vimentin is critical for the cell entry of SARS-CoV. J. Biomed. Sci. 2016, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Antón-Plágaro, C.; Williamson, M.K.; Shoemark, D.K.; Simón-Gracia, L.; Klein, K.; Bauer, M.; Hollandi, R.; Greber, U.F.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; Kallio, K.; Kaya, T.; Anastasina, M.; Smura, T.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and provides a possible pathway into the central nervous system. bioRxiv 2020. [Google Scholar] [CrossRef]
- García, S.R.; Deprez, M.; Lebrigand, K.; Cavard, A.; Paquet, A.; Arguel, M.-J.; Magnone, V.; Truchi, M.; Caballero, I.; Leroy, S.; et al. Novel dynamics of human mucociliary differentiation revealed by single-cell RNA sequencing of nasal epithelial cultures. Development 2019, 146. [Google Scholar] [CrossRef]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- Lechien, J.R.; Chiesa-Estomba, C.M.; De Siati, D.R.; Horoi, M.; Le Bon, S.D.; Rodriguez, A.; Dequanter, D.; Blecic, S.; El Afia, F.; Distinguin, L.; et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study. Eur. Arch. Otorhinolaryngol. 2020. [Google Scholar] [CrossRef]
- Di Carlo, D.T.; Montemurro, N.; Petrella, G.; Siciliano, G.; Ceravolo, R.; Perrini, P. Exploring the clinical association between neurological symptoms and COVID-19 pandemic outbreak: A systematic review of current literature. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Rocke, J.; Hopkins, C.; Philpott, C.; Kumar, N. Is loss of sense of smell a diagnostic marker in COVID-19: A Systematic Review and Meta-analysis. Clin. Otolaryngol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Butowt, R.; Bilinska, K. SARS-CoV-2: Olfaction, Brain Infection, and the Urgent Need for Clinical Samples Allowing Earlier Virus Detection. ACS Chem. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; den Berge, K.V.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-C.; Liu, Y.H.; Wang, C.-Y.; Wang, Y.-H.; Hsueh, S.-C.; Yen, M.-Y.; Ko, W.-C.; Hsueh, P.-R. Asymptomatic carrier state, acute respiratory disease, and pneumonia due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2): Facts and myths. J. Microbiol. Immunol. Infect. 2020. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients with 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1299–1300. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Tian, S.; Xiong, Y.; Liu, H.; Niu, L.; Guo, J.; Liao, M.; Xiao, S.-Y. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod. Pathol. 2020, 1–8. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Copin, M.-C.; Parmentier, E.; Duburcq, T.; Poissy, J.; Mathieu, D.; Caplan, M.; Cousin, N.; Durand, A.; Goutay, J.; Kalioubie, A.E.; et al. Time to consider histologic pattern of lung injury to treat critically ill patients with COVID-19 infection. Intensive Care Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.H.; Li, T.Y.; He, Z.C.; Ping, Y.F.; Liu, H.W.; Yu, S.C.; Mou, H.M.; Wang, L.H.; Zhang, H.R.; Fu, W.J.; et al. A pathological report of three COVID-19 cases by minimally invasive autopsies. Chin. J. Pathol. 2020, 49, E009. [Google Scholar] [CrossRef]
- Ingraham, N.E.; Barakat, A.G.; Reilkoff, R.; Bezdicek, T.; Schacker, T.; Chipman, J.G.; Tignanelli, C.J.; Puskarich, M.A. Understanding the Renin-Angiotensin-Aldosterone-SARS-CoV-Axis: A Comprehensive Review. Eur. Respir. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.; Lui, R.N.; Sung, J.J. Covid-19 and the digestive system. J. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Tang, M.; Zheng, X.; Liu, Y.; Li, X.; Shan, H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, Y.; Jiang, N.; Zhou, Q.; Ma, W.-L. Persistence of intestinal SARS-CoV-2 infection in patients with COVID-19 leads to re-admission after pneumonia resolved. Int. J. Infect. Dis. 2020, 95, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Alsaad, K.O.; Hajeer, A.H.; Al Balwi, M.; Al Moaiqel, M.; Al Oudah, N.; Al Ajlan, A.; AlJohani, S.; Alsolamy, S.; Gmati, G.E.; Balkhy, H.; et al. Histopathology of Middle East respiratory syndrome coronovirus (MERS-CoV) infection—Clinicopathological and ultrastructural study. Histopathology 2018, 72, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ni, C.; Gao, R.; Wang, Y.; Yang, L.; Wei, J.; Lv, T.; Liang, J.; Zhang, Q.; Xu, W.; et al. Recapitulation of SARS-CoV-2 Infection and Cholangiocyte Damage with Human Liver Organoids. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 1–16. [Google Scholar] [CrossRef]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac Involvement in a Patient with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 819–824. [Google Scholar] [CrossRef]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Alhogbani, T. Acute myocarditis associated with novel Middle East respiratory syndrome coronavirus. Annal. Saudi Med. 2016, 36, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Fitzek, A.; Bräuninger, H.; Aleshcheva, G.; Edler, C.; Meissner, K.; Scherschel, K.; Kirchhof, P.; Escher, F.; Schultheiss, H.-P.; et al. Association of Cardiac Infection with SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol. 2020. [Google Scholar] [CrossRef]
- Bojkova, D.; Wagner, J.U.G.; Shumliakivska, M.; Aslan, G.S.; Saleem, U.; Hansen, A.; Luxán, G.; Günther, S.; Pham, M.D.; Krishnan, J.; et al. SARS-CoV-2 infects and induces cytotoxic effects in human cardiomyocytes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Glass, W.G.; Subbarao, K.; Murphy, B.; Murphy, P.M. Mechanisms of Host Defense following Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) Pulmonary Infection of Mice. J. Immunol. 2004, 173, 4030–4039. [Google Scholar] [CrossRef]
- Li, K.; Wohlford-Lenane, C.; Perlman, S.; Zhao, J.; Jewell, A.K.; Reznikov, L.R.; Gibson-Corley, K.N.; Meyerholz, D.K.; McCray, P.B. Middle East Respiratory Syndrome Coronavirus Causes Multiple Organ Damage and Lethal Disease in Mice Transgenic for Human Dipeptidyl Peptidase 4. J. Infect. Dis. 2016, 213, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, L.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. 2004, 203, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhong, S.; Liu, J.; Li, L.; Li, Y.; Wu, X.; Li, Z.; Deng, P.; Zhang, J.; Zhong, N.; et al. Detection of Severe Acute Respiratory Syndrome Coronavirus in the Brain: Potential Role of the Chemokine Mig in Pathogenesis. Clin. Infect. Dis. 2005, 41, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bai, W.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef]
- Yachou, Y.; El Idrissi, A.; Belapasov, V.; Ait Benali, S. Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: Understanding the neurological manifestations in COVID-19 patients. J. Neurol. Sci. 2020. [Google Scholar] [CrossRef]
- Asadi-Pooya, A.A.; Simani, L. Central nervous system manifestations of COVID-19: A systematic review. J. Neurol. Sci. 2020, 413, 116832. [Google Scholar] [CrossRef]
- Sedaghat, Z.; Karimi, N. Guillain Barre syndrome associated with COVID-19 infection: A case report. J. Clin. Neurosci. 2020, 76, 233–235. [Google Scholar] [CrossRef]
- Zhao, H.; Shen, D.; Zhou, H.; Liu, J.; Chen, S. Guillain-Barré syndrome associated with SARS-CoV-2 infection: Causality or coincidence? Lancet Neurol. 2020, 19, 383–384. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; Liu, Y.; Liu, Y.; et al. Human Kidney is a Target for Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Fanelli, V.; Fiorentino, M.; Cantaluppi, V.; Gesualdo, L.; Stallone, G.; Ronco, C.; Castellano, G. Acute kidney injury in SARS-CoV-2 infected patients. Crit. Care 2020, 24, 155. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.S.; Ng, J.H.; Ross, D.W.; Sharma, P.; Shah, H.H.; Barnett, R.L.; Hazzan, A.D.; Fishbane, S.; Jhaveri, K.D.; Abate, M.; et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020, 98, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.-Y.; Wang, B.; Liu, B.-C. Acute Kidney Injury in the 2019 Novel Coronavirus Disease. Kidney Dis. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cummings, M.J.; Baldwin, M.R.; Abrams, D.; Jacobson, S.D.; Meyer, B.J.; Balough, E.M.; Aaron, J.G.; Claassen, J.; Rabbani, L.E.; Hastie, J.; et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: A prospective cohort study. Lancet 2020, 395, 1763–1770. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, Z.; Diao, B.; Wang, R.; Wang, G.; Wang, C.; Tan, Y.; Liu, L.; Wang, C.; Liu, Y.; et al. The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Fathi, N.; Rezaei, N. Lymphopenia in COVID-19: Therapeutic opportunities. Cell Biol. Int. 2020. [Google Scholar] [CrossRef]
- Dierckx, T.; Khouri, R.; Menezes, S.M.; Decanine, D.; Farre, L.; Bittencourt, A.; Vandamme, A.M.; Van Weyenbergh, J. IFN-β induces greater antiproliferative and proapoptotic effects and increased p53 signaling compared with IFN-α in PBMCs of Adult T-cell Leukemia/Lymphoma patients. Blood Cancer J. 2017, 7, e519. [Google Scholar] [CrossRef]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity 2020. [Google Scholar] [CrossRef]
- Venkata, C.; Sampathkumar, P.; Afessa, B. Hospitalized Patients with 2009 H1N1 Influenza Infection: The Mayo Clinic Experience. Mayo Clin. Proc. 2010, 85, 798–805. [Google Scholar] [CrossRef]
- Maier, H.E.; Lopez, R.; Sanchez, N.; Ng, S.; Gresh, L.; Ojeda, S.; Burger-Calderon, R.; Kuan, G.; Harris, E.; Balmaseda, A.; et al. Obesity Increases the Duration of Influenza A Virus Shedding in Adults. Infect. Dis. 2018, 218, 1378–1382. [Google Scholar] [CrossRef]
- Kruglikov, I.L.; Scherer, P.E. The role of adipocytes and adipocyte-like cells in the severity of COVID-19 infections. Obesity 2020. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Skiba, D.S.; Touyz, R.M.; Harrison, D.G. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc. Res. 2017, 113, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.M.; Caplice, N.M. Is Adipose Tissue a Reservoir for Viral Spread, Immune Activation and Cytokine Amplification in COVID-19. Obesity 2020. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Lechien, J.R.; Chiesa-Estomba, C.M.; Place, S.; Laethem, Y.V.; Cabaraux, P.; Mat, Q.; Huet, K.; Plzak, J.; Horoi, M.; Hans, S.; et al. Clinical and Epidemiological Characteristics of 1,420 European Patients with mild-to-moderate Coronavirus Disease 2019. J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X.-S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Severin, R.; Arena, R.; Lavie, C.J.; Bond, S.; Phillips, S.A. Respiratory Muscle Performance Screening for Infectious Disease Management Following COVID-19: A Highly Pressurized Situation. Am. J. Med. 2020. [Google Scholar] [CrossRef]
- Prompetchara, E.; Chutitorn, K.; Tanapat, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Dandekar, A.A.; Perlman, S. Immunopathogenesis of coronavirus infections: Implications for SARS. Nat. Rev. Immunol. 2005, 5, 917–927. [Google Scholar] [CrossRef]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; Breillat, P.; et al. Impaired type I interferon activity and exacerbated inflammatory responses in severe Covid-19 patients. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Sa Ribero, M.; Jouvenet, N.; Dreux, M.; Nisole, S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020, 16, e1008737. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Zhou, J.; Ho-Yin Wong, B.; Li, C.; Cheng, Z.-S.; Lin, X.; Kwok-Man Poon, V.; Sun, T.; Choi-Yi Lau, C.; Fuk-Woo Chan, J.; et al. Productive replication of Middle East respiratory syndrome coronavirus in monocyte-derived dendritic cells modulates innate immune response. Virology 2014, 454–455, 197–205. [Google Scholar] [CrossRef]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef]
- Wang, F.; Hou, H.; Luo, Y.; Tang, G.; Wu, S.; Huang, M.; Liu, W.; Zhu, Y.; Lin, Q.; Mao, L.; et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight 2020. [Google Scholar] [CrossRef]
- Sanchez-Cerrillo, I.; Landete, P.; Aldave, B.; Sanchez-Alonso, S.; Sanchez-Azofra, A.; Marcos-Jimenez, A.; Avalos, E.; Alcaraz-Serna, A.; de los Santos, I.; Mateu-Albero, T.; et al. Differential Redistribution of Activated Monocyte and Dendritic Cell Subsets to the Lung Associates with Severity of COVID-19. MedRxiv 2020. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol. Sin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020. [Google Scholar] [CrossRef] [PubMed]
- Potey, P.M.; Rossi, A.G.; Lucas, C.D.; Dorward, D.A. Neutrophils in the initiation and resolution of acute pulmonary inflammation: Understanding biological function and therapeutic potential. J. Pathol. 2019, 247, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Twaddell, S.H.; Baines, K.J.; Grainge, C.; Gibson, P.G. The Emerging Role of Neutrophil Extracellular Traps in Respiratory Disease. CHEST 2019, 156, 774–782. [Google Scholar] [CrossRef]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. RES 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Ward, P.A.; Fattahi, F. New strategies for treatment of infectious sepsis. J. Leukoc. Biol. 2019, 106, 187–192. [Google Scholar] [CrossRef]
- Haick, A.K.; Rzepka, J.P.; Brandon, E.; Balemba, O.B.; Miura, T.A. Neutrophils are needed for an effective immune response against pulmonary rat coronavirus infection, but also contribute to pathology. J. Gen. Virol. 2014, 95, 578–590. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martínez-Colón, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps (NETs) as markers of disease severity in COVID-19. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020. [Google Scholar] [CrossRef]
- Li, C.K.; Xu, X. Host Immune Responses to SARS Coronavirus in Humans. In Molecular Biology of the SARS-Coronavirus; Lal, S.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 259–278. [Google Scholar]
- He, R.; Lu, Z.; Zhang, L.; Fan, T.; Xiong, R.; Shen, X.; Feng, H.; Meng, H.; Lin, W.; Jiang, W.; et al. The clinical course and its correlated immune status in COVID-19 pneumonia. J. Clin. Virol. 2020, 127, 104361. [Google Scholar] [CrossRef]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Wang, F.; Nie, J.; Wang, H.; Zhao, Q.; Xiong, Y.; Deng, L.; Song, S.; Ma, Z.; Mo, P.; Zhang, Y. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Rokni, M.; Ghasemi, V.; Tavakoli, Z. Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: Comparison with SARS and MERS. Rev. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Oh, H.-L.J.; Chia, A.; Chang, C.X.L.; Leong, H.N.; Ling, K.L.; Grotenbreg, G.M.; Gehring, A.J.; Tan, Y.J.; Bertoletti, A. Engineering T Cells Specific for a Dominant Severe Acute Respiratory Syndrome Coronavirus CD8 T Cell Epitope. J. Virol. 2011, 85, 10464–10471. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Zhao, J.; Perlman, S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res. 2014, 59, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-Y.; Huang, Z.-T.; Li, L.; Wu, M.-H.; Yu, T.; Koup, R.A.; Bailer, R.T.; Wu, C.-Y. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch. Virol. 2009, 154, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Yang, L.; Wang, L.; Li, J.; Huang, J.; Lu, Z.; Koup, R.A.; Bailer, R.T.; Wu, C. Long-lived memory T lymphocyte responses against SARS coronavirus nucleocapsid protein in SARS-recovered patients. Virology 2006, 351, 466–475. [Google Scholar] [CrossRef]
- Yang, L.-T.; Peng, H.; Zhu, Z.-L.; Li, G.; Huang, Z.-T.; Zhao, Z.-X.; Koup, R.A.; Bailer, R.T.; Wu, C.-Y. Long-lived effector/central memory T-cell responses to severe acute respiratory syndrome coronavirus (SARS-CoV) S antigen in recovered SARS patients. Clin. Immunol. 2006, 120, 171–178. [Google Scholar] [CrossRef]
- Yang, L.; Peng, H.; Zhu, Z.; Li, G.; Huang, Z.; Zhao, Z.; Koup, R.A.; Bailer, R.T.; Wu, C. Persistent memory CD4+ and CD8+ T-cell responses in recovered severe acute respiratory syndrome (SARS) patients to SARS coronavirus M antigen. J. Gen. Virol. 2007, 88, 2740–2748. [Google Scholar] [CrossRef]
- Yang, K.; Sun, K.; Srinivasan, K.N.; Salmon, J.; Marques, E.T.; Xu, J.; August, J.T. Immune responses to T-cell epitopes of SARS CoV-N protein are enhanced by N immunization with a chimera of lysosome-associated membrane protein. Gene Ther. 2009, 16, 1353–1362. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Pre-existing immunity to SARS-CoV-2: The knowns and unknowns. Nat. Rev. Immunol. 2020, 20, 457–458. [Google Scholar] [CrossRef]
- Yaqinuddin, A. Cross-immunity between respiratory coronaviruses may limit COVID-19 fatalities. Med. Hypotheses 2020, 144, 110049. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Schmitz, K.S.; Raadsen, M.P.; Grifoni, A.; Okba, N.M.A.; Endeman, H.; van den Akker, J.P.C.; Molenkamp, R.; Koopmans, M.P.G.; van Gorp, E.C.M.; et al. Phenotype of SARS-CoV-2-specific T-cells in COVID-19 patients with acute respiratory distress syndrome. medRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. Presence of SARS-CoV-2 reactive T cells in COVID-19 patients and healthy donors. medRxiv 2020. [Google Scholar] [CrossRef]
- Meckiff, B.J.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; Sette, A.; et al. Single-cell transcriptomic analysis of SARS-CoV-2 reactive CD4+ T cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bert, N.L.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.; Lin, M.; Tan, N.; Linster, M.; et al. Different pattern of pre-existing SARS-COV-2 specific T cell immunity in SARS-recovered and uninfected individuals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hsueh, P.-R.; Huang, L.-M.; Chen, P.-J.; Kao, C.-L.; Yang, P.-C. Chronological evolution of IgM, IgA, IgG and neutralisation antibodies after infection with SARS-associated coronavirus. Clin. Microbiol. Infect. 2004, 10, 1062–1066. [Google Scholar] [CrossRef]
- Gorse, G.J.; Donovan, M.M.; Patel, G.B. Antibodies to coronaviruses are higher in older compared with younger adults and binding antibodies are more sensitive than neutralizing antibodies in identifying coronavirus-associated illnesses. J. Med. Virol. 2020, 92, 512–517. [Google Scholar] [CrossRef]
- Wu, L.-P.; Wang, N.-C.; Chang, Y.-H.; Tian, X.-Y.; Na, D.-Y.; Zhang, L.-Y.; Zheng, L.; Lan, T.; Wang, L.-F.; Liang, G.-D. Duration of Antibody Responses after Severe Acute Respiratory Syndrome. Emerg. Infect. Dis. 2007, 13, 1562–1564. [Google Scholar] [CrossRef]
- Tang, F.; Quan, Y.; Xin, Z.-T.; Wrammert, J.; Ma, M.-J.; Lv, H.; Wang, T.-B.; Yang, H.; Richardus, J.H.; Liu, W.; et al. Lack of Peripheral Memory B Cell Responses in Recovered Patients with Severe Acute Respiratory Syndrome: A Six-Year Follow-Up Study. J. Immunol. 2011, 186, 7264–7268. [Google Scholar] [CrossRef]
- Cao, W.-C.; Liu, W.; Zhang, P.-H.; Zhang, F.; Richardus, J.H. Disappearance of Antibodies to SARS-Associated Coronavirus after Recovery. N. Engl. J. Med. 2007, 357, 1162–1163. [Google Scholar] [CrossRef]
- Wang, B.; Wang, L.; Kong, X.; Geng, J.; Xiao, D.; Ma, C.; Jiang, X.-M.; Wang, P.-H. Long-term coexistence of SARS-CoV-2 with antibody response in COVID-19 patients. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, X.; Zhu, C.; Song, Y.; Feng, F.; Qiu, Y.; Feng, J.; Jia, Q.; Song, Q.; Zhu, B.; et al. Immune Phenotyping Based on the Neutrophil-to-Lymphocyte Ratio and IgG Level Predicts Disease Severity and Outcome for Patients with COVID-19. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Dobi, A.; Frumence, E.; Rakoto, M.L.; Lebeau, G.; Vagner, D.; Seteyen, A.-L.S.; Giry, C.; Septembre-Malaterre, A.; Jaffar-Bandjee, M.-C.; Raffray, L.; et al. Serological surveys in Reunion Island of the first hospitalized patients revealed that long-lived immunoglobulin G antibodies specific against SARS-CoV2 virus are rapidly vanishing in severe cases. medRxiv 2020. [Google Scholar] [CrossRef]
- To, K.K.-W.; Tsang, O.T.-Y.; Leung, W.-S.; Tam, A.R.; Wu, T.-C.; Lung, D.C.; Yip, C.C.-Y.; Cai, J.-P.; Chan, J.M.-C.; Chik, T.S.-H.; et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infect. Dis. 2020, 20, 565–574. [Google Scholar] [CrossRef]
- Long, Q.-X.; Tang, X.-J.; Shi, Q.-L.; Li, Q.; Deng, H.-J.; Yuan, J.; Hu, J.-L.; Xu, W.; Zhang, Y.; Lv, F.-J.; et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020, 26, 1–5. [Google Scholar] [CrossRef]
- Sun, B.; Feng, Y.; Mo, X.; Zheng, P.; Wang, Q.; Li, P.; Peng, P.; Liu, X.; Chen, Z.; Huang, H.; et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 940–948. [Google Scholar] [CrossRef]
- Huang, W.; Berube, J.; McNamara, M.; Saksena, S.; Hartman, M.; Arshad, T.; Bornheimer, S.J.; O’Gorman, M. Lymphocyte Subset Counts in COVID-19 Patients: A Meta-Analysis. Cytom. Part A 2020. [Google Scholar] [CrossRef]
- Crotty, S.; Ahmed, R. Immunological memory in humans. Semin. Immunol. 2004, 16, 197–203. [Google Scholar] [CrossRef]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral Immunity Due to Long-Lived Plasma Cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef]
- Elsner, R.A.; Hastey, C.J.; Olsen, K.J.; Baumgarth, N. Suppression of Long-Lived Humoral Immunity Following Borrelia burgdorferi Infection. PLoS Pathog. 2015, 11, e1004976. [Google Scholar] [CrossRef]
- Burton, D.R. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2002, 2, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Kucharski, A.J.; Lessler, J.; Cummings, D.A.T.; Riley, S. Timescales of influenza A/H3N2 antibody dynamics. PLoS Biol. 2018, 16, e2004974. [Google Scholar] [CrossRef] [PubMed]
- Pasmans, H.; Schurink-van’t Klooster, T.M.; Bogaard, M.J.M.; van Rooijen, D.M.; de Melker, H.E.; Welters, M.J.P.; van der Burg, S.H.; van der Klis, F.R.M.; Buisman, A.-M. Long-term HPV-specific immune response after one versus two and three doses of bivalent HPV vaccination in Dutch girls. Vaccine 2019, 37, 7280–7288. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.H.L.; Skehel, J.J.; Kassiotis, G.; Langhorne, J. Recovery of an Antiviral Antibody Response following Attrition Caused by Unrelated Infection. PLoS Pathog. 2014, 10, e1003843. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.; Oehme, L.; Zehentmeier, S.; Zhang, Y.; Niesner, R.; Hauser, A.E. Tracking plasma cell differentiation and survival. Cytom. Part A 2014, 85, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-F.; Tseng, S.-P.; Yen, C.-H.; Yang, J.-Y.; Tsao, C.-H.; Shen, C.-W.; Chen, K.-H.; Liu, F.-T.; Liu, W.-T.; Chen, Y.-M.A.; et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochem. Biophys. Res. Commun. 2014, 451, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Yip, M.S.; Leung, N.H.L.; Cheung, C.Y.; Li, P.H.; Lee, H.H.Y.; Daëron, M.; Peiris, J.S.M.; Bruzzone, R.; Jaume, M. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virology 2014, 11, 82. [Google Scholar] [CrossRef]
- Tetro, J.A. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020, 22, 72–73. [Google Scholar] [CrossRef]
- Sharma, A. It is too soon to attribute ADE to COVID-19. Microbes Infect. 2020. [Google Scholar] [CrossRef]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. Covid-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar] [CrossRef]
- Galeotti, C.; Bayry, J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020, 16, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Zhang, Y.; Zhang, S.; Qin, X.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; et al. Brief Report: Anti-phospholipid antibodies in critically ill patients with Coronavirus Disease 2019 (COVID-19). Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bomhof, G.; Mutsaers, P.G.N.J.; Leebeek, F.W.G.; te Boekhorst, P.A.W.; Hofland, J.; Croles, F.N.; Jansen, A.J.G. COVID-19-associated immune thrombocytopenia. Br. J. Haematol. 2020, 190, e61–e64. [Google Scholar] [CrossRef] [PubMed]
- Zulfiqar, A.-A.; Lorenzo-Villalba, N.; Hassler, P.; Andrès, E. Immune Thrombocytopenic Purpura in a Patient with Covid-19. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef]
- Lazarian, G.; Quinquenel, A.; Bellal, M.; Siavellis, J.; Jacquy, C.; Re, D.; Merabet, F.; Mekinian, A.; Braun, T.; Damaj, G.; et al. Autoimmune haemolytic anaemia associated with COVID-19 infection. Br. J. Haematol. 2020, 190, 29–31. [Google Scholar] [CrossRef]
- Lopez, C.; Kim, J.; Pandey, A.; Huang, T.; DeLoughery, T.G. Simultaneous onset of COVID-19 and autoimmune haemolytic anaemia. Br. Haematol. 2020, 190, 31–32. [Google Scholar] [CrossRef]
- Berger, J.R. COVID-19 and the nervous system. J. Neurovirol. 2020, 26, 143–148. [Google Scholar] [CrossRef]
- Dalakas, M.C. Guillain-Barré syndrome: The first documented COVID-19–triggered autoimmune neurologic disease: More to come with myositis in the offing. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e781. [Google Scholar] [CrossRef]
- Pouletty, M.; Borocco, C.; Ouldali, N.; Caseris, M.; Basmaci, R.; Lachaume, N.; Bensaid, P.; Pichard, S.; Kouider, H.; Morelle, G.; et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 mimicking Kawasaki disease (Kawa-COVID-19): A multicentre cohort. Annal. Rheum. Dis. 2020, 79, 999–1006. [Google Scholar] [CrossRef]
- Wang, H.; Ma, S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am. J. Emerg. Med. 2008, 26, 711–715. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 102537. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.V.; Jonsson, C.B. A Role for Neutrophils in Viral Respiratory Disease. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Cassatella, M.A. Neutrophil-derived chemokines on the road to immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A.; Chávez-Valencia, V. High IL-6/IFN-γ ratio could be associated with severe disease in COVID-19 patients. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, C.; Li, J.; Yuan, J.; Yang, M.; Wang, F.; Li, G.; Li, Y.; Xing, L.; Peng, L.; et al. Exuberant elevation of IP-10, MCP-3 and IL-1ra during SARS-CoV-2 infection is associated with disease severity and fatal outcome. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Kumar, Y.; Liang, C.; Bo, Z.; Rajapakse, J.C.; Ooi, E.E.; Tannenbaum, S.R. Serum Proteome and Cytokine Analysis in a Longitudinal Cohort of Adults with Primary Dengue Infection Reveals Predictive Markers of DHF. PLoS Negl. Trop. Dis. 2012, 6, e1887. [Google Scholar] [CrossRef]
- Liu, K.-T.; Liu, Y.-H.; Lin, C.-Y.; Kuo, P.-L.; Yen, M.-C. Inflammatory molecules expression pattern for identifying pathogen species in febrile patient serum. Exp. Ther. Med. 2016, 12, 312–318. [Google Scholar] [CrossRef]
- Reis, E.A.G.; Hagan, J.E.; Ribeiro, G.S.; Teixeira-Carvalho, A.; Martins-Filho, O.A.; Montgomery, R.R.; Shaw, A.C.; Ko, A.I.; Reis, M.G. Cytokine Response Signatures in Disease Progression and Development of Severe Clinical Outcomes for Leptospirosis. PLoS Negl. Trop. Dis. 2013, 7, e2457. [Google Scholar] [CrossRef]
- Reddy, V.; Mani, R.S.; Desai, A.; Ravi, V. Correlation of plasma viral loads and presence of Chikungunya IgM antibodies with cytokine/chemokine levels during acute Chikungunya virus infection. J. Med. Virol. 2014, 86, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Brenner, E.J.; Ungaro, R.C.; Gearry, R.B.; Kaplan, G.G.; Kissous-Hunt, M.; Lewis, J.D.; Ng, S.C.; Rahier, J.-F.; Reinisch, W.; Ruemmele, F.M.; et al. Corticosteroids, But Not TNF Antagonists, Are Associated with Adverse COVID-19 Outcomes in Patients with Inflammatory Bowel Diseases: Results From an International Registry. Gastroenterology 2020. [Google Scholar] [CrossRef] [PubMed]
- Dolinger, M.T.; Person, H.; Smith, R.; Jarchin, L.; Pittman, N.; Dubinsky, M.C.; Lai, J. Pediatric Crohn Disease and Multisystem Inflammatory Syndrome in Children (MIS-C) and COVID-19 Treated with Infliximab. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the alert for cytokine storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef]
- Cron, R.Q.; Behrens, E.M. Cytokine Storm Syndrome; Springer International Publishing: Cham, Switzerland, 2019. [Google Scholar]
- Ichikawa, A.; Kuba, K.; Morita, M.; Chida, S.; Tezuka, H.; Hara, H.; Sasaki, T.; Ohteki, T.; Ranieri, V.M.; dos Santos, C.C.; et al. CXCL10-CXCR3 Enhances the Development of Neutrophil-mediated Fulminant Lung Injury of Viral and Nonviral Origin. Am. J. Respir. Crit. Care Med. 2013, 187, 65–77. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, J.-E.; Hyun, M.C. Cytokine response in pediatric patients with pandemic influenza H1N1 2009 virus infection and pneumonia: Comparison with pediatric pneumonia without H1N1 2009 infection. Pediatr. Pulmonol. 2011, 46, 1233–1239. [Google Scholar] [CrossRef]
- Tang, X.; Du, R.; Wang, R.; Cao, T.; Guan, L.; Yang, C.; Zhu, Q.; Hu, M.; Li, X.; Li, Y.; et al. Comparison of Hospitalized Patients with ARDS Caused by COVID-19 and H1N1. Chest 2020. [Google Scholar] [CrossRef]
- Kindler, E.; Thiel, V.; Weber, F. Chapter Seven—Interaction of SARS and MERS Coronaviruses with the Antiviral Interferon Response. In Advances in Virus Research; Ziebuhr, J., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 96, pp. 219–243. [Google Scholar]
- Astuti, I. Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. Clin. Res. Rev. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Wong, L.-Y.R.; Lui, P.-Y.; Jin, D.-Y. A molecular arms race between host innate antiviral response and emerging human coronaviruses. Virol. Sin. 2016, 31, 12–23. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Inhibits Type I Interferon Production by Interfering with TRIM25-Mediated RIG-I Ubiquitination. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Chen, J.; Ly, H. Immunosuppression by viral N proteins. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Shokri, S.; Mahmoudvand, S.; Taherkhani, R.; Farshadpour, F. Modulation of the immune response by Middle East respiratory syndrome coronavirus. J. Cell. Physiol. 2019, 234, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Comar, C.E.; Goldstein, S.A.; Li, Y.; Yount, B.; Baric, R.S.; Weiss, S.R. Antagonism of dsRNA-Induced Innate Immune Pathways by NS4a and NS4b Accessory Proteins during MERS Coronavirus Infection. mBio 2019, 10. [Google Scholar] [CrossRef]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.M.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; de Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef]
- Thornbrough, J.M.; Jha, B.K.; Yount, B.; Goldstein, S.A.; Li, Y.; Elliott, R.; Sims, A.C.; Baric, R.S.; Silverman, R.H.; Weiss, S.R. Middle East Respiratory Syndrome Coronavirus NS4b Protein Inhibits Host RNase L Activation. mBio 2016, 7. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, G.; Yang, Y.; Shi, J.; Fang, L.; Li, F.; Xiao, S.; Fu, Z.F.; Peng, G. A conserved region of nonstructural protein 1 from alphacoronaviruses inhibits host gene expression and is critical for viral virulence. J. Biol. Chem. 2019, 294, 13606–13618. [Google Scholar] [CrossRef]
- Jauregui, A.R.; Savalia, D.; Lowry, V.K.; Farrell, C.M.; Wathelet, M.G. Identification of Residues of SARS-CoV nsp1 That Differentially Affect Inhibition of Gene Expression and Antiviral Signaling. PLoS ONE 2013, 8, e62416. [Google Scholar] [CrossRef]
- Menachery, V.D.; Debbink, K.; Baric, R.S. Coronavirus non-structural protein 16: Evasion, attenuation, and possible treatments. Virus Res. 2014, 194, 191–199. [Google Scholar] [CrossRef]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef]
- Yang, J.-R.; Deng, D.-T.; Wu, N.; Yang, B.; Li, H.-J.; Pan, X.-B. Persistent viral RNA positivity during recovery period of a patient with SARS-CoV-2 infection. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, W.; Yu, X.; Hu, D.; Bao, M.; Liu, H.; Zhou, J.; Jiang, H. Coronavirus disease 2019 in elderly patients: Characteristics and prognostic factors based on 4-week follow-up. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, A.N. Age-dependent alterations of the T cell repertoire and functional diversity of T cells of the aged. Immunol. Res. 2006, 36, 221–228. [Google Scholar] [CrossRef]
- Lega, S.; Naviglio, S.; Volpi, S.; Tommasini, A. Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19. Vaccines 2020, 8, 224. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Curtis, N. Coronavirus Infections in Children Including COVID-19: An Overview of the Epidemiology, Clinical Features, Diagnosis, Treatment and Prevention Options in Children. Pediatr. Infect. Dis. J. 2020, 39, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Kevat, A. Children may be less affected than adults by novel coronavirus (COVID-19). Paediatr. Child Health 2020, 56, 657. [Google Scholar] [CrossRef]
- Prendergast, A.J.; Klenerman, P.; Goulder, P.J.R. The impact of differential antiviral immunity in children and adults. Nat. Rev. Immunol. 2012, 12, 636–648. [Google Scholar] [CrossRef]
- Belot, A.; Antona, D.; Renolleau, S.; Javouhey, E.; Hentgen, V.; Angoulvant, F.; Delacourt, C.; Iriart, X.; Ovaert, C.; Bader-Meunier, B.; et al. SARS-CoV-2-related paediatric inflammatory multisystem syndrome, an epidemiological study, France, 1 March to 17 May 2020. Eurosurveillance 2020, 25, 2001010. [Google Scholar] [CrossRef]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Dashraath, P.; Wong, J.L.J.; Lim, M.X.K.; Lim, L.M.; Li, S.; Biswas, A.; Choolani, M.; Mattar, C.; Su, L.L. Coronavirus disease 2019 (COVID-19) pandemic and pregnancy. Am. J. Obstet. Gynecol. 2020. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.-L.; Zhao, S.-J.; Kwak-Kim, J.; Mor, G.; Liao, A.-H. Why are pregnant women susceptible to COVID-19? An immunological viewpoint. J. Reprod. Immunol. 2020, 139, 103122. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, M.J.; Haddadi, S.; Tahvildari, A.; Farsi, Y.; Arbabi, M.; Hasanzadeh, S.; Jamshidi, P.; Murthi, M.; Mirsaeidi, M. COVID-19 clinical characteristics, and sex-specific risk of mortality: Systematic Review and Meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fett, C.; Mack, M.; Eyck, P.P.T.; Meyerholz, D.K.; Perlman, S. Sex-Based Differences in Susceptibility to Severe Acute Respiratory Syndrome Coronavirus Infection. J. Immunol. 2017, 198, 4046–4053. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Kadel, S.; Kovats, S. Sex Hormones Regulate Innate Immune Cells and Promote Sex Differences in Respiratory Virus Infection. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Kachuri, L.; Francis, S.S.; Morrison, M.; Bossé, Y.; Cavazos, T.B.; Rashkin, S.R.; Ziv, E.; Witte, J.S. The landscape of host genetic factors involved in infection to common viruses and SARS-CoV-2. medRxiv 2020. [Google Scholar] [CrossRef]
- Scepanovic, P.; Alanio, C.; Hammer, C.; Hodel, F.; Bergstedt, J.; Patin, E.; Thorball, C.W.; Chaturvedi, N.; Charbit, B.; Abel, L.; et al. Human genetic variants and age are the strongest predictors of humoral immune responses to common pathogens and vaccines. Genome Med. 2018, 10, 59. [Google Scholar] [CrossRef]
- Kenney, A.D.; Dowdle, J.A.; Bozzacco, L.; McMichael, T.M.; St. Gelais, C.; Panfil, A.R.; Sun, Y.; Schlesinger, L.S.; Anderson, M.Z.; Green, P.L.; et al. Human Genetic Determinants of Viral Diseases. Annu. Rev. Genet. 2017, 51, 241–263. [Google Scholar] [CrossRef]
- Lin, M.; Tseng, H.-K.; Trejaut, J.A.; Lee, H.-L.; Loo, J.-H.; Chu, C.-C.; Chen, P.-J.; Su, Y.-W.; Lim, K.H.; Tsai, Z.-U.; et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med. Genet. 2003, 4, 9. [Google Scholar] [CrossRef]
- Cheng, Y.; Cheng, G.; Chui, C.H.; Lau, F.Y.; Chan, P.K.S.; Ng, M.H.L.; Sung, J.J.Y.; Wong, R.S.M. ABO Blood Group and Susceptibility to Severe Acute Respiratory Syndrome. JAMA 2005, 293, 1447–1451. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO Blood Group and the COVID-19 Susceptibility. medRxiv 2020. [Google Scholar] [CrossRef]
- Zietz, M.; Tatonetti, N.P. Testing the association between blood type and COVID-19 infection, intubation, and death. medRxiv 2020. [Google Scholar] [CrossRef]
- Alzoughool, F.; Alanagreh, L. Coronavirus drugs: Using plasma from recovered patients as a treatment for COVID-19. JRS 2020, 1–5. [Google Scholar] [CrossRef]
- Jawhara, S. Could Intravenous Immunoglobulin Collected from Recovered Coronavirus Patients Protect against COVID-19 and Strengthen the Immune System of New Patients? IJMS 2020, 21, 2272. [Google Scholar] [CrossRef]
- Keith, P.; Day, M.; Perkins, L.; Moyer, L.; Hewitt, K.; Wells, A. A novel treatment approach to the novel coronavirus: An argument for the use of therapeutic plasma exchange for fulminant COVID-19. Crit. Care 2020, 24, 128. [Google Scholar] [CrossRef]
- Casadevall, A.; Scharff, M.D. Serum therapy revisited: Animal models of infection and development of passive antibody therapy. Antimicrob. Agents Chemother. 1994, 38, 1695–1702. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L. Antibody-mediated regulation of cellular immunity and the inflammatory response. Trends Immunol. 2003, 24, 474–478. [Google Scholar] [CrossRef]
- Rojas, M.; Rodríguez, Y.; Monsalve, D.M.; Acosta-Ampudia, Y.; Camacho, B.; Gallo, J.E.; Rojas-Villarraga, A.; Ramírez-Santana, C.; Díaz-Coronado, J.C.; Manrique, R.; et al. Convalescent plasma in Covid-19: Possible mechanisms of action. Autoimmun. Rev. 2020, 19, 102554. [Google Scholar] [CrossRef]
- Saghazadeh, A.; Rezaei, N. Towards treatment planning of COVID-19: Rationale and hypothesis for the use of multiple immunosuppressive agents: Anti-antibodies, immunoglobulins, and corticosteroids. Int. Immunopharm. 2020, 84, 106560. [Google Scholar] [CrossRef]
- Soo, Y.O.Y.; Cheng, Y.; Wong, R.; Hui, D.S.; Lee, C.K.; Tsang, K.K.S.; Ng, M.H.L.; Chan, P.; Cheng, G.; Sung, J.J.Y. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin. Microbiol. Infect. 2004, 10, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.-M.; Chiueh, T.-S.; Siu, L.K.; Lin, J.-C.; Chan, P.K.S.; Peng, M.-Y.; Wan, H.-L.; Chen, J.-H.; Hu, B.-S.; Perng, C.-L.; et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. J. Antimicrob. Chemother. 2005, 56, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Bloch, E.M.; Shoham, S.; Casadevall, A.; Sachais, B.S.; Shaz, B.; Winters, J.L.; van Buskirk, C.; Grossman, B.J.; Joyner, M.; Henderson, J.P.; et al. Deployment of convalescent plasma for the prevention and treatment of COVID-19. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Perotti, C.; Baldanti, F.; Bruno, R.; Fante, C.D.; Seminari, E.; Casari, S.; Percivalle, E.; Glingani, C.; Musella, V.; Belliato, M.; et al. Mortality reduction in 46 severe Covid-19 patients treated with hyperimmune plasma. A proof of concept single arm multicenter trial. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; Yang, N.; Ma, Y.; Zhou, Q.; Li, W.; Wang, X.; Huang, L.; Luo, X.; Fukuoka, T.; et al. Effectiveness of intravenous immunoglobulin for children with severe COVID-19: A rapid review. Annal. Transl. Med. 2020, 8, 625. [Google Scholar] [CrossRef]
- Cao, W.; Liu, X.; Bai, T.; Fan, H.; Hong, K.; Song, H.; Han, Y.; Lin, L.; Ruan, L.; Li, T. High-Dose Intravenous Immunoglobulin as a Therapeutic Option for Deteriorating Patients with Coronavirus Disease 2019. Open Forum Infect. Dis. 2020, 7. [Google Scholar] [CrossRef]
- Mohtadi, N.; Ghaysouri, A.; Shirazi, S.; Shafiee, E.; Bastani, E.; Kokhazadeh, T.; Tavan, H. Recovery of severely ill COVID-19 patients by intravenous immunoglobulin (IVIG) treatment: A case series. Virology 2020, 548, 1–5. [Google Scholar] [CrossRef]
- Feldstein, L.R.; Rose, E.B.; Horwitz, S.M.; Collins, J.P.; Newhams, M.M.; Son, M.B.F.; Newburger, J.W.; Kleinman, L.C.; Heidemann, S.M.; Martin, A.A.; et al. Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef]
- Sokolovsky, S.; Soni, P.; Hoffman, T.; Kahn, P.; Scheers-Masters, J. COVID-19 associated Kawasaki-like multisystem inflammatory disease in an adult. Am. J. Emerg. Med. 2020. [Google Scholar] [CrossRef]
- Yiğenoğlu, T.N.; Ulas, T.; Dal, M.S.; Korkmaz, S.; Erkurt, M.A.; Altuntaş, F. Extracorporeal blood purification treatment options for COVID-19: The role of immunoadsorption. Transfus. Apher. Sci. 2020, 102855. [Google Scholar] [CrossRef]
- Daoud, A.M.; Soliman, K.M.; Ali, H.K. Potential Limitations of Plasmapheresis in Treatment of COVID-19 Patients and How to Overcome Them? Ther. Apher. Dial. 2020. [Google Scholar] [CrossRef] [PubMed]
- Honore, P.M.; Mugisha, A.; Kugener, L.; Redant, S.; Attou, R.; Gallerani, A.; De Bels, D. Therapeutic plasma exchange as a routine therapy in septic shock and as an experimental treatment for COVID-19: We are not sure. Critic. Care 2020, 24, 226. [Google Scholar] [CrossRef] [PubMed]
- Khamis, F.; Al-Zakwani, I.; Al Hashmi, S.; Al Dowaiki, S.; Al Bahrani, M.; Pandak, N.; Al Khalili, H.; Memish, Z. Therapeutic Plasma Exchange in Adults with Severe COVID-19 Infection. Int. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kesici, S.; Yavuz, S.; Bayrakci, B. Get rid of the bad first: Therapeutic plasma exchange with convalescent plasma for severe COVID-19. Proc. Natl. Acad. Sci. USA 2020, 117, 12526–12527. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.D.M.E.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Microbiology 2020. [CrossRef]
- Pan, X.; Zhou, P.; Fan, T.; Wu, Y.; Zhang, J.; Shi, X.; Shang, W.; Fang, L.; Jiang, X.; Shi, J.; et al. Immunoglobulin fragment F(ab’)2 against RBD potently neutralizes SARS-CoV-2 in vitro. Pharmacol. Toxicol. 2020. [Google Scholar] [CrossRef]
- Alsoussi, W.B.; Turner, J.S.; Case, J.B.; Zhao, H.; Schmitz, A.J.; Zhou, J.Q.; Chen, R.E.; Lei, T.; Rizk, A.A.; McIntire, K.M.; et al. A Potently Neutralizing Antibody Protects Mice against SARS-CoV-2 Infection. J. Immunol. 2020. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef]
- Díez, J.-M.; Romero, C.; Gajardo, R. Currently available intravenous immunoglobulin (Gamunex®-C and Flebogamma® DIF) contains antibodies reacting against SARS-CoV-2 antigens. Immunology 2020. [Google Scholar] [CrossRef]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020. [Google Scholar] [CrossRef]
- Tu, Y.-F.; Chien, C.-S.; Yarmishyn, A.A.; Lin, Y.-Y.; Luo, Y.-H.; Lin, Y.-T.; Lai, W.-Y.; Yang, D.-M.; Chou, S.-J.; Yang, Y.-P.; et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Moroni, G. Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin. Drug Saf. 2017, 16, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.S.; Rathod, J.; Gernsheimer, J. A Rapid Systematic Review of Clinical Trials Utilizing Chloroquine and Hydroxychloroquine as a Treatment for COVID-19. Acad. Emerg. Med. 2020, 27, 493–504. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef]
- Picot, S.; Marty, A.; Bienvenu, A.-L.; Blumberg, L.H.; Dupouy-Camet, J.; Carnevale, P.; Kano, S.; Jones, M.K.; Daniel-Ribeiro, C.T.; Mas-Coma, S. Coalition: Advocacy for prospective clinical trials to test the post-exposure potential of hydroxychloroquine against COVID-19. One Health 2020, 9, 100131. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Million, M.; Gautret, P.; Colson, P.; Cortaredona, S.; Giraud-Gatineau, A.; Honoré, S.; Gaubert, J.-Y.; Fournier, P.-E.; Tissot-Dupont, H.; et al. Outcomes of 3737 COVID-19 patients treated with hydroxychloroquine/azithromycin and other regimens in Marseille, France: A retrospective analysis. Travel Med. Infect. Dis. 2020, 36, 101791. [Google Scholar] [CrossRef]
- Molina, J.M.; Delaugerre, C.; Le Goff, J.; Mela-Lima, B.; Ponscarme, D.; Goldwirt, L.; de Castro, N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Méd. Mal. Infect. 2020, 50, 384. [Google Scholar] [CrossRef]
- Putman, M.; Chock, Y.P.E.; Tam, H.; Kim, A.H.J.; Sattui, S.E.; Berenbaum, F.; Danila, M.I.; Korsten, P.; Sanchez-Alvarez, C.; Sparks, J.A.; et al. Antirheumatic Disease Therapies for the Treatment of COVID-19: A Systematic Review and Meta-analysis. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef]
- Bauman, J.L.; Tisdale, J.E. Chloroquine and Hydroxychloroquine in the Era of SARS—CoV2: Caution on Their Cardiac Toxicity. Pharmacotherapy 2020, 40, 387–388. [Google Scholar] [CrossRef]
- Lofgren, S.M.M.; Nicol, M.R.; Bangdiwala, A.S.; Pastick, K.A.; Okafor, E.C.; Skipper, C.P.; Pullen, M.F.; Engen, N.W.; Abassi, M.; Williams, D.A.; et al. Safety of Hydroxychloroquine among Outpatient Clinical Trial Participants for COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Nanni, O.; Viale, P.; Vertogen, B.; Lilli, C.; Zingaretti, C.; Donati, C.; Masini, C.; Monti, M.; Serra, P.; Vespignani, R.; et al. PROTECT Trial: A cluster-randomized study with hydroxychloroquine versus observational support for prevention or early-phase treatment of Coronavirus disease (COVID-19): A structured summary of a study protocol for a randomized controlled trial. Trials 2020, 21, 689. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Dai, L.; Zhang, X.; Zhang, Z.; Zhang, Z. Hydroxychloroquine and chloroquine: A potential and controversial treatment for COVID-19. Arch. Pharm. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.D.; Millar, J.E.; Baillie, J.K. Clinical evidence does not support corticosteroid treatment for 2019-nCoV lung injury. Lancet 2020, 395, 473–475. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, J.; Zhou, Y.; Zhao, X.; Zhao, Q.; Liu, J. The effect of corticosteroid treatment on patients with coronavirus infection: A systematic review and meta-analysis. J. Infect. 2020. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Jiang, Q.; Yang, J.; Li, J.; Gao, C.; Jiang, H.; Ge, L.; Liu, Y. No Clear Benefit to the Use of Corticosteroid as Treatment in Adult Patients with Coronavirus Disease 2019: A Retrospective Cohort Study. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Hu, F.; Yin, G.; Chen, Y.; Song, J.; Ye, M.; Liu, J.; Chen, C.; Song, Y.; Tang, X.; Zhang, Y. Corticosteroid, oseltamivir, and delayed admission are independent risk factors for prolonged viral shedding in patients with Coronavirus Disease 2019. Clin. Respir. J. 2020. [Google Scholar] [CrossRef]
- Lee, K.H.; Yoon, S.; Jeong, G.H.; Kim, J.Y.; Han, Y.J.; Hong, S.H.; Ryu, S.; Kim, J.S.; Lee, J.Y.; Yang, J.W.; et al. Efficacy of Corticosteroids in Patients with SARS, MERS and COVID-19: A Systematic Review and Meta-Analysis. J. Clin. Med. 2020, 9, 2392. [Google Scholar] [CrossRef]
- Hasan, S.S.; Capstick, T.; Ahmed, R.; Kow, C.S.; Mazhar, F.; Merchant, H.A.; Zaidi, S.T.R. Mortality in COVID-19 patients with acute respiratory distress syndrome and corticosteroids use: A systematic review and meta-analysis. Expert Rev. Respir. Med. 2020. [Google Scholar] [CrossRef]
- Group, T.R.C. Dexamethasone in Hospitalized Patients with Covid-19—Preliminary Report. N. Eng. J. Med. 2020. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, Y.; Tian, D.; Wang, C.; Wang, S.; Cheng, J.; Hu, M.; Fang, M.; Gao, Y. Potential benefits of precise corticosteroids therapy for severe 2019-nCoV pneumonia. Sig. Transduct. Target. Ther. 2020, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Ferro, F.; Elefante, E.; Puxeddu, I.; Baldini, C.; Bartoloni, E.; Baratè, C.; Galimberti, S.; Talarico, R.; Mosca, M.; Bombardieri, S. COVID-19: The new challenge for rheumatologists. First update. Clin. Exp. Rheumatol. 2020, 38, 373–382. [Google Scholar] [PubMed]
- Sarzi-Puttini, P.; Giorgi, V.; Sirotti, S.; Marotto, D.; Ardizzone, S.; Rizzardini, G.; Antinori, S.; Galli, M. COVID-19, cytokines and immunosuppression: What can we learn from severe acute respiratory syndrome? Clin. Exp. Rheumatol. 2020, 38, 337–342. [Google Scholar] [PubMed]
- Buonaguro, F.M.; Puzanov, I.; Ascierto, P.A. Anti-IL6R role in treatment of COVID-19-related ARDS. J. Transl. Med. 2020, 18, 165. [Google Scholar] [CrossRef]
- Guo, C.; Li, B.; Ma, H.; Wang, X.; Cai, P.; Yu, Q.; Zhu, L.; Jin, L.; Jiang, C.; Fang, J.; et al. Tocilizumab treatment in severe COVID-19 patients attenuates the inflammatory storm incited by monocyte centric immune interactions revealed by single-cell analysis. Genomics 2020. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Wang, T.S.; Lee, E.; Cremer, P.C.; Carey, B.; Rajendram, P.; Hudock, K.M.; Korbee, L.; Van Tassell, B.W.; et al. Targeting GM-CSF in COVID-19 Pneumonia: Rationale and Strategies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Luca, G.D.; Cavalli, G.; Campochiaro, C.; Della-Torre, E.; Angelillo, P.; Tomelleri, A.; Boffini, N.; Tentori, S.; Mette, F.; Farina, N.; et al. GM-CSF blockade with mavrilimumab in severe COVID-19 pneumonia and systemic hyperinflammation: A single-centre, prospective cohort study. Lancet Rheumatol. 2020, 2, e465–e473. [Google Scholar] [CrossRef]
- Lokugamage, K.G.; Hage, A.; Schindewolf, C.; Rajsbaum, R.; Menachery, V.D. SARS-CoV-2 is sensitive to type I interferon pretreatment. Microbiology 2020. [Google Scholar] [CrossRef]
- Sallard, E.; Lescure, F.-X.; Yazdanpanah, Y.; Mentre, F.; Peiffer-Smadja, N. Type 1 interferons as a potential treatment against COVID-19. Antivir. Res. 2020, 178, 104791. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef]
- Cauchois, R.; Koubi, M.; Delarbre, D.; Manet, C.; Carvelli, J.; Blasco, V.B.; Jean, R.; Fouche, L.; Bornet, C.; Pauly, V.; et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. PNAS 2020. [Google Scholar] [CrossRef] [PubMed]
- Diurno, F.; Numis, F.G.; Porta, G.; Cirillo, F.; Maddaluno, S.; Ragozzino, A.; Negri, P.D.; Gennaro, C.D.; Pagano, A.; Allegorico, E.; et al. Eculizumab treatment in patients with COVID-19: Preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med. Pharmacol. Sci. 2020, 8. [Google Scholar] [CrossRef]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Cantini, F.; Niccoli, L.; Nannini, C.; Matarrese, D.; Natale, M.E.D.; Lotti, P.; Aquilini, D.; Landini, G.; Cimolato, B.; Pietro, M.A.D.; et al. Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, A.; Tummolo, A.M.; Montemurro, G.; Gremese, E.; Larosa, L.; Cipriani, M.C.; Pasciuto, G.; Liperoti, R.; Murri, R.; Pirronti, T.; et al. Baricitinib as rescue therapy in a patient with COVID-19 with no complete response to sarilumab. Infection 2020. [Google Scholar] [CrossRef]
- Titanji, B.K.; Farley, M.M.; Mehta, A.; Connor-Schuler, R.; Moanna, A.; Cribbs, S.K.; O’Shea, J.; DeSilva, K.; Chan, B.; Edwards, A.; et al. Use of Baricitinib in Patients with Moderate and Severe COVID-19. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Bari, E.; Ferrarotti, I.; Saracino, L.; Perteghella, S.; Torre, M.L.; Corsico, A.G. Mesenchymal Stromal Cell Secretome for Severe COVID-19 Infections: Premises for the Therapeutic Use. Cells 2020, 9, 924. [Google Scholar] [CrossRef]
- Leng, Z.; Zhu, R.; Hou, W.; Feng, Y.; Yang, Y.; Han, Q.; Shan, G.; Meng, F.; Du, D.; Wang, S.; et al. Transplantation of ACE2-Mesenchymal Stem Cells Improves the Outcome of Patients with COVID-19 Pneumonia. Aging Dis. 2020, 11, 216. [Google Scholar] [CrossRef]
- Shetty, A.K. Mesenchymal Stem Cell Infusion Shows Promise for Combating Coronavirus (COVID-19)-Induced Pneumonia. Aging Dis. 2020, 11, 462. [Google Scholar] [CrossRef]
- Zhao, R.C. Stem Cell–Based Therapy for Coronavirus Disease 2019. Stem Cells Dev. 2020. [Google Scholar] [CrossRef]
- Atluri, S.; Manchikanti, L.; Hirsch, J.A. Expanded Umbilical Cord Mesenchymal Stem Cells (UC-MSCs) as a Therapeutic Strategy In Managing Critically Ill COVID-19 Patients: The Case for Compassionate Use. Pain Physician 2020, 23, E71–E83. [Google Scholar] [PubMed]






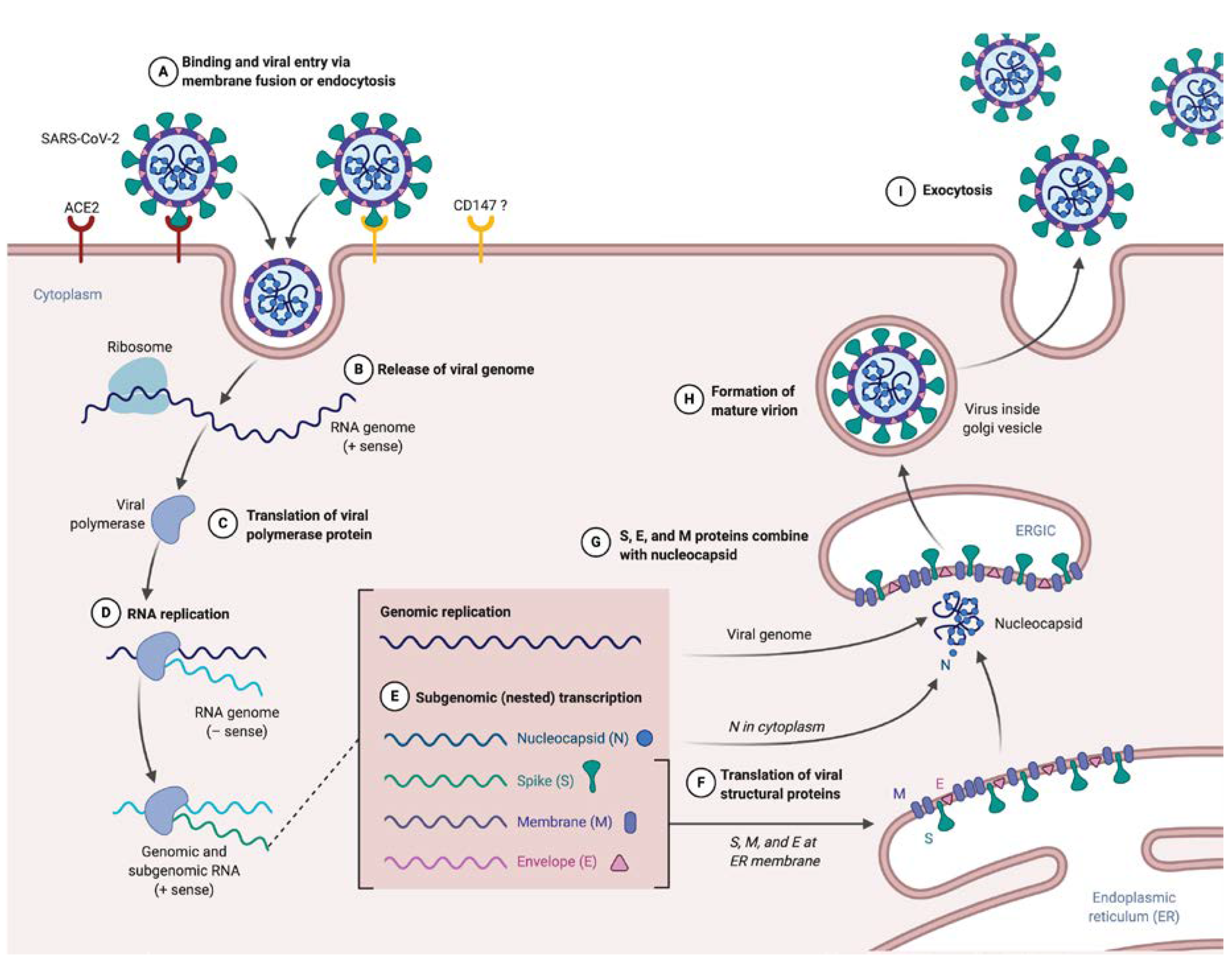{kind=link}
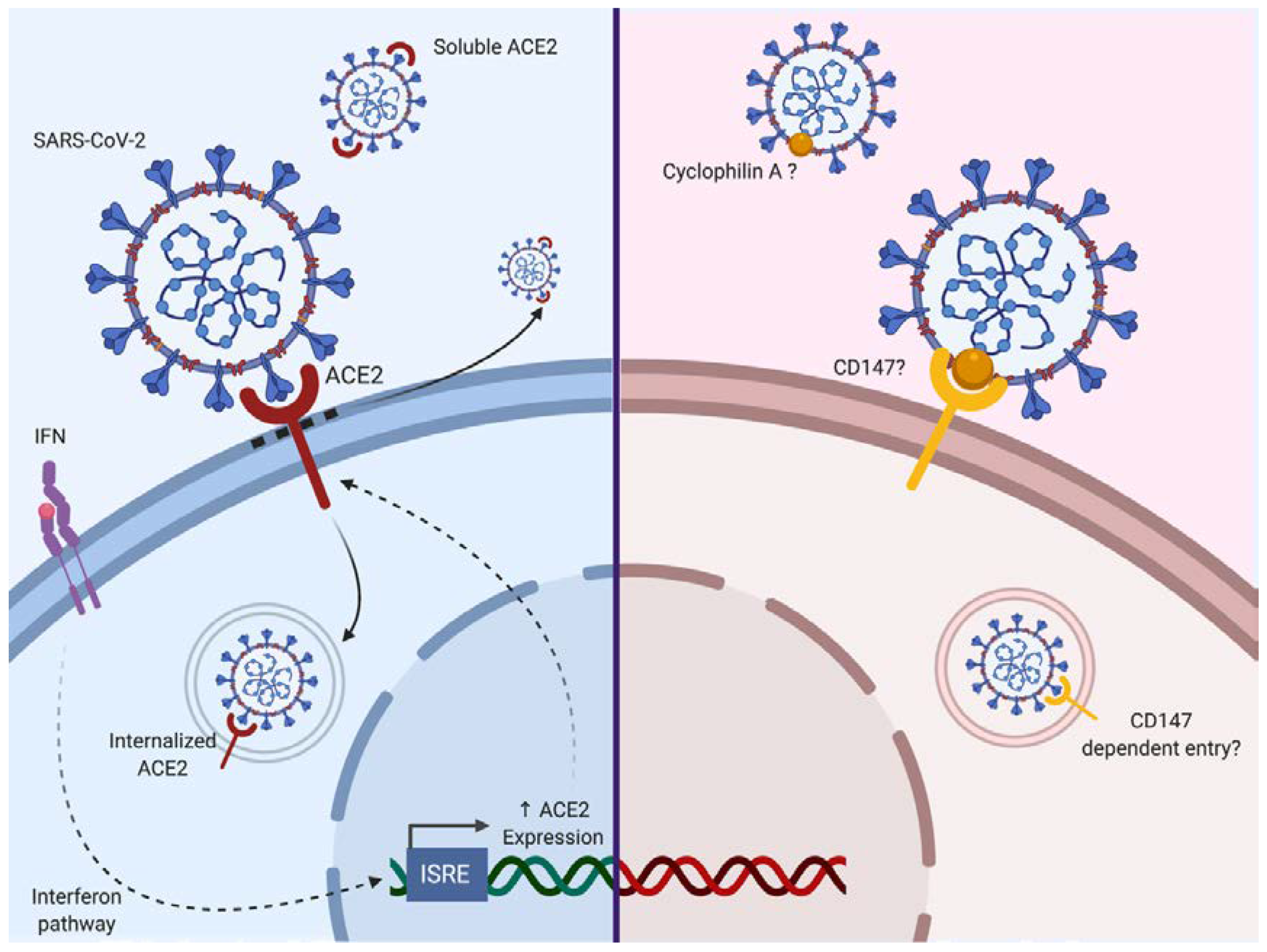{kind=link}
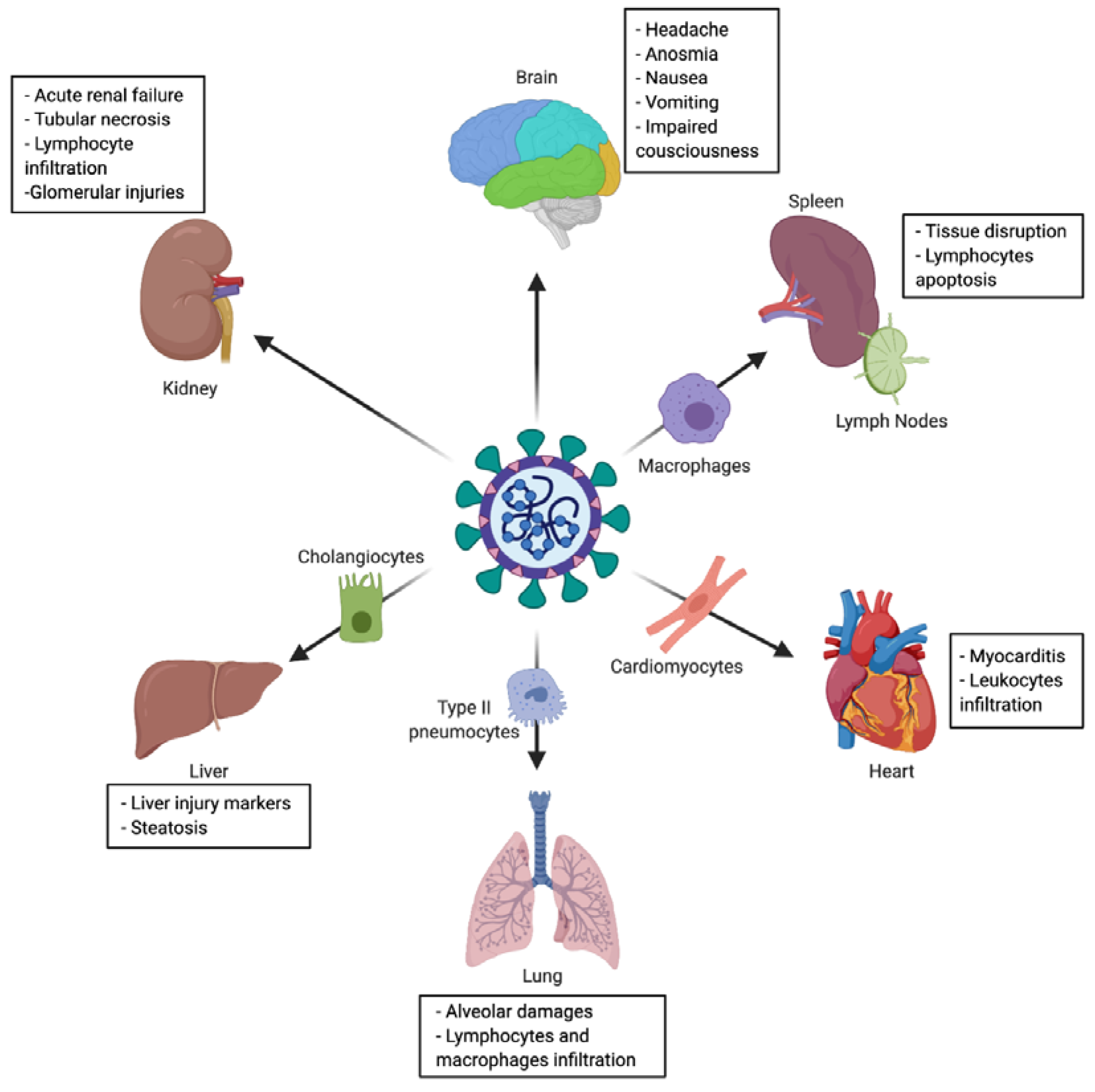{kind=link}
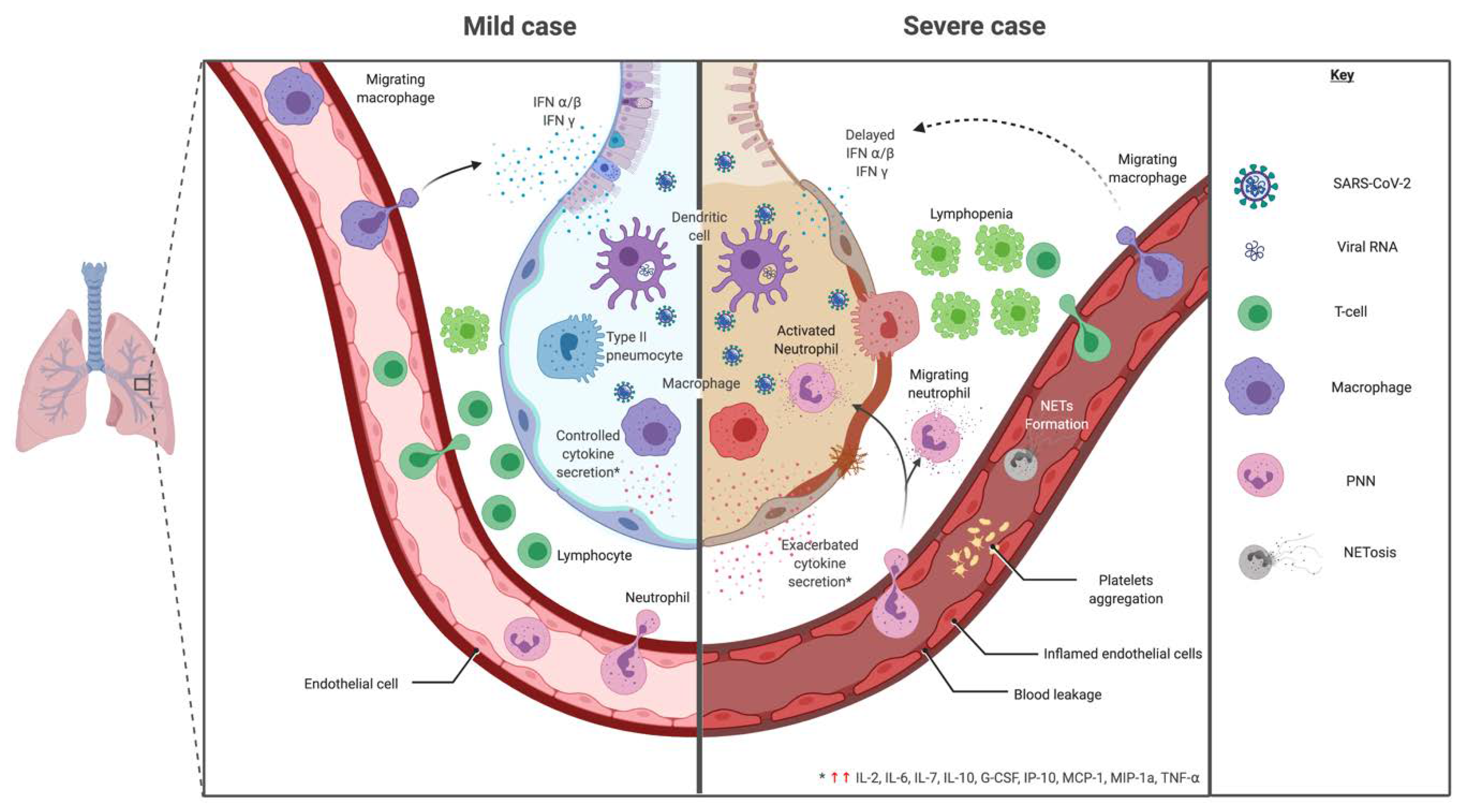{kind=link}
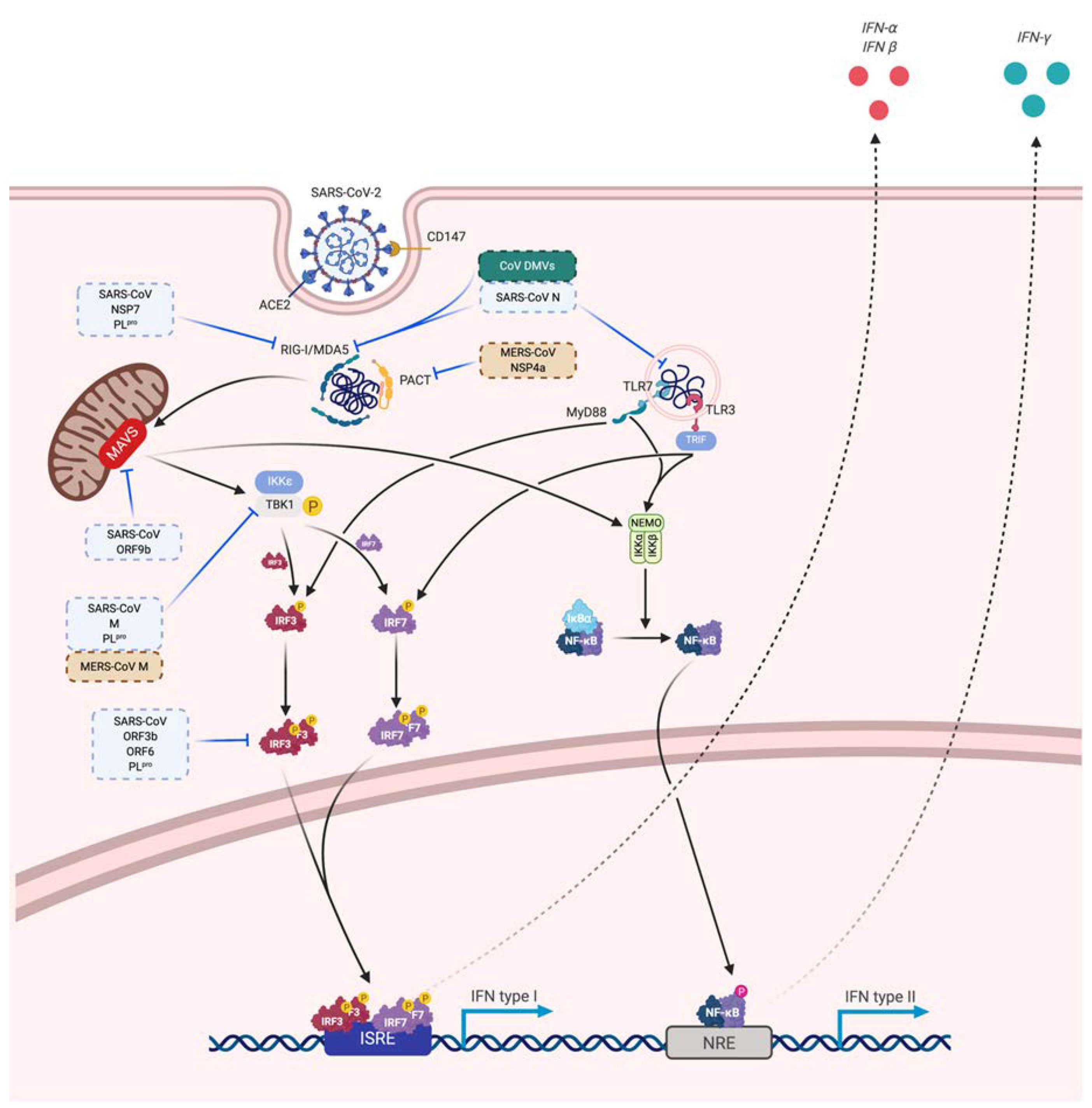{kind=link}
| Organ | Main Reported Pathologic Findings |
|---|---|
| Lungs | Edema |
| Mononuclear inflammatory cells (lymphocytes, plasmocytes) | |
| Cellular or proteinaceous exudate | |
| Type II pneumocyte hyperplasia with cytologic atypia | |
| Vascular congestion | |
| Fibrinoid vascular necrosis | |
| Hyaline membrane formation (hallmark of DAD) | |
| Fibrin deposition with early organization | |
| Extensive intra-alveolar “fibrin balls” and organizing pneumonia (AFOP) | |
| Desquamative pneumocytes | |
| Gastrointestinal tract | Plasma cell and lymphocyte infiltrate |
| Edema in the lamina propria | |
| Partial epithelial degeneration, necrosis, shedding of gastric and intestinal mucosa | |
| Liver | Mild microvesicular steatosis |
| Mild lobular and portal activity | |
| Centrilobular sinusoidal dilatation | |
| Heart | Cardiomyocyte hypertrophy |
| Degeneration and necrosis of some cardiomyocytes, mild interstitial hyperemia | |
| Edema | |
| Infiltration of a small number of lymphocytes, monocytes and neutrophils | |
| Kidney | Diffuse proximal tubule injuries (loss of brush border, vacuolar degeneration, acute tubular necrosis) |
| Lymphocyte infiltration | |
| Glomerular injuries |
| Cytokine (pg/mL) | COVID-19 [220] * [126] # | Dengue Fever [221] | Severe Chikungunya [224] | Viral Sepsis [222] | Leptospirosis [223] | Bacterial Sepsis [222] |
|---|---|---|---|---|---|---|
| IFNγ | 9 # | 772.4 ± 1762.7 | 12.6 ± 10.8 | 9.54 ± 19.16 | 7.2 | 16.8 ± 39.61 |
| IL-1 | 5 (no detectable) * | 3.3 ± 2.2 | 209.6 ± 936.8 | 9.6 | ||
| IL-2 | 8 # | 2.5 ± 0.8 | 1.46 ± 1.04 | 4 | 3.2 ± 6.67 | |
| IL-6 | 25.2 (9.6–54.5) * | 24 ± 47 | 671.9 ± 1261.3 | 63.23 ± 265 | 74.7 | 2533 ± 6559 |
| IL-8 | 18.4 (11.3–28.4) * | 21.1 ± 10.1 | 734.1 ± 1721.9 | 30.31 ± 38.01 | 251.1 | 1249 ± 6944 |
| IL-10 | 6.6 (5–11.3) * | 32.5 ± 54 | 90.9 ± 417 | 14.27 ± 16.75 | 21 | 205.11 ± 741.85 |
| TNF-⍺ | 8.7 (7.1–11.6) * | 15.1 ± 107.1 | 4 | |||
| IP-10 | >1000 # | 56.6 ± 278.5 | 291.8 ± 460 | 1944 ± 1295 | 666.72 ± 766 | |
| MCP-1 | 50 * | 389.6 ± 929.3 | 545.4 ± 418.9 | 494.31 ± 679 | 4533 ± 1458 | |
| MIG | 791.7 ± 434.9 | 660.71 ± 661 | 1377 ± 1906 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebeau, G.; Vagner, D.; Frumence, É.; Ah-Pine, F.; Guillot, X.; Nobécourt, E.; Raffray, L.; Gasque, P. Deciphering SARS-CoV-2 Virologic and Immunologic Features. Int. J. Mol. Sci. 2020, 21, 5932. https://doi.org/10.3390/ijms21165932
Lebeau G, Vagner D, Frumence É, Ah-Pine F, Guillot X, Nobécourt E, Raffray L, Gasque P. Deciphering SARS-CoV-2 Virologic and Immunologic Features. International Journal of Molecular Sciences. 2020; 21(16):5932. https://doi.org/10.3390/ijms21165932
Chicago/Turabian StyleLebeau, Grégorie, Damien Vagner, Étienne Frumence, Franck Ah-Pine, Xavier Guillot, Estelle Nobécourt, Loïc Raffray, and Philippe Gasque. 2020. "Deciphering SARS-CoV-2 Virologic and Immunologic Features" International Journal of Molecular Sciences 21, no. 16: 5932. https://doi.org/10.3390/ijms21165932
APA StyleLebeau, G., Vagner, D., Frumence, É., Ah-Pine, F., Guillot, X., Nobécourt, E., Raffray, L., & Gasque, P. (2020). Deciphering SARS-CoV-2 Virologic and Immunologic Features. International Journal of Molecular Sciences, 21(16), 5932. https://doi.org/10.3390/ijms21165932