The Remedial Potential of Lycopene in Pancreatitis through Regulation of Autophagy



Abstract
1. Introduction
2. Autophagy Impairment and Pancreatitis
2.1. Steps in the Autophagy Process and Related Cellular Machinery
2.2. Cell Signaling Pathways Regulating Autophagy
2.3. Autophagy and Pancreatitis
2.4. Factors Related to Impaired Autophagy in Pancreatitis
2.4.1. Oxidative Stress
2.4.2. Ca2+ Overload
2.4.3. Mitochondrial Dysfunction
2.4.4. Inflammation
2.4.5. Lysosomal Dysfunction
3. Lycopene and Autophagy
3.1. Inhibition of Cell Death by Suppressing Autophagy
3.2. Inhibition of Cell Death by Activating Autophagy
4. Lycopene and Pancreatitis
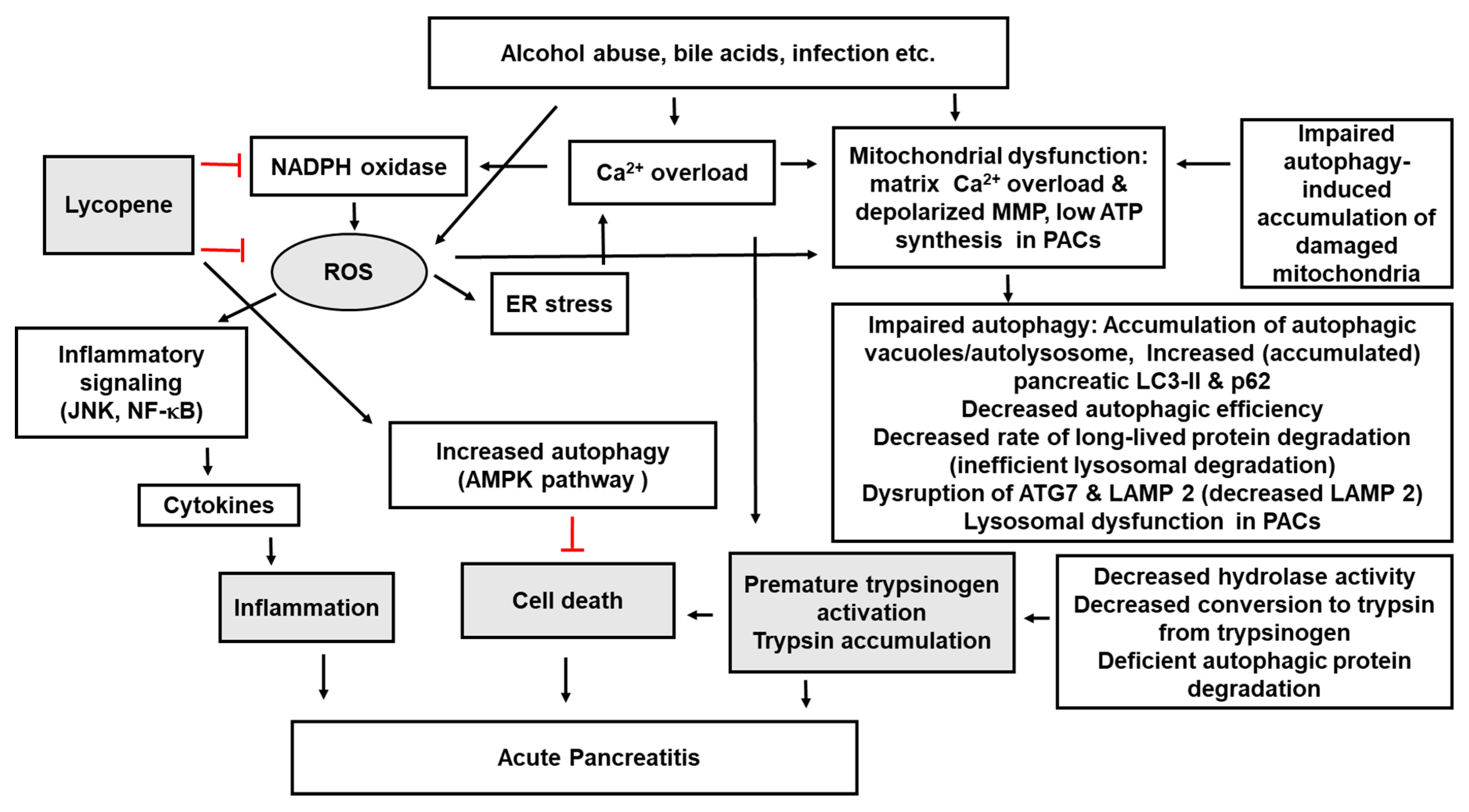
5. Lycopene, Autophagy, and Pancreatitis
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGE | Advanced glycation end product |
| ATG | Autophagy-related gene |
| Bcl-2 | B-cell lymphoma 2 |
| BCO1 | β-Carotene oxygenase 1; β -Carotene 15,15′-oxygenase |
| BCO2 | β-Carotene oxygenase 2; β-Carotene 9′,10′-oxygenase |
| CaMKKβ | Calmodulin-dependent kinase kinase-β |
| CatB | Cathepsin B |
| CAT | Catalase |
| CCK | Cholecystokinin |
| CD36 | Cluster determinant 36 |
| COX | Cyclooxygenase |
| DAMP | Damage-associated molecular patterns |
| DM | Diabetes mellitus |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| FoxO | Forkhead box O |
| GSH | Glutathione |
| GSH-Px | Glutathione peroxidase |
| GSK3β | Glycogen synthase kinase 3β |
| IL | Interleukin |
| iNOS | Inducible nitrogen oxide syntase |
| IKK | IκB kinase |
| IRS | Insulin receptor substrate |
| IκB | Inhibitor of κB |
| JNK | c-Jun NH2-terminal kinase |
| LAMP | Lysosomal-associated membrane protein |
| LC3 | Microtubule-associated 1A/1B-light chain 3 |
| LKB1 | Liver kinase B1 |
| MAPK | Mitogen-activated protein kinase |
| MAPKK/MEK | Mitogen-activated protein kinase kinase |
| MCP | Monocyte chemoattractant protein |
| MDA | Malondialdehyde |
| MIP | Macrophage inflammatory protein |
| MPO | Myeloperoxidase |
| MPTP | Mitochondrial permeability transition pore |
| mTORC | Mammalian target of rapamycin complex |
| NADPH | Nicotinamide adenine dinucleotide phosphate hydrogen |
| NF-κB | Nuclear factor κ B |
| NO | Nitric oxide |
| PACs | Pancreatic acinar cells |
| PAF | Platelet-activating factor |
| PDK | Phosphoinositide-dependent protein kinase 1 |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PKB | Protein kinase B |
| PKC | Protein kinase C |
| PtdIns3KC3 | Class III phosphatidylinositol 3-kinase |
| Raf | Rapidly accelerated fibrosarcoma |
| Rag | Ras-related GTP binding protein |
| Ras | Retrovirus-associated DNA sequences |
| RB1CC1/FIP200 | RB1-inducible coiled-coil 1/Focal adhesion kinase family-interacting protein of 200 kDa |
| Rheb | Ras homolog enriched in brain |
| ROS | Reactive oxygen species |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment protein receptors |
| SOD | Superoxide dismutase |
| SRB | Scavenger receptor class B |
| SR-B1 | Scavenger receptor class B type-1 |
| TNF | Tumor necrosis factor |
| TSC | Tuberous sclerosis |
| ULK | Unc-51-like kinases |
| V-ATPase | Vacuolar ATPase |
| Vps34 | Vacuolar protein sorting 34 |
References
- Mayerle, J.; Sendler, M.; Hegyi, E.; Beyer, G.; Lerch, M.M.; Sahin-Toth, M. Genetics, cell biology, and pathophysiology of pancreatitis. Gastroenterology 2019, 156, 1951–1968. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Papachristou, G.I. New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 479–496. [Google Scholar] [CrossRef]
- Tsai, K.; Wang, S.S.; Chen, T.S.; Kong, C.W.; Chang, F.Y.; Lee, S.D.; Lu, F.J. Oxidative stress: An important phenomenon with pathogenetic significance in the progression of acute pancreatitis. Gut 1998, 42, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Sevillano, S.; de la Mano, A.M.; Manso, M.A.; Orfao, A.; de Dios, I. N-acetylcysteine prevents intra-acinar oxygen free radical production in pancreatic duct obstruction-induced acute pancreatitis. Biochim. Biophys. Acta 2003, 1639, 177–184. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H. Regulation of Autophagy by mTOR Signaling Pathway. Adv. Exp. Med. Biol. 2019, 206, 67–83. [Google Scholar]
- Corona Velazquez, A.F.; Jackson, W.T. So many roads: The multifaceted regulation of autophagy induction. Mol. Cell Biol. 2018, 38, e00303–e00318. [Google Scholar] [CrossRef]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2020, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef] [PubMed]
- Gukovskaya, A.S.; Gukovsky, I. Autophagy and pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G993–G1003. [Google Scholar] [CrossRef]
- Mareninova, O.A.; Hermann, K.; French, S.W.; O’Konski, M.S.; Pandol, S.J.; Webster, P.; Erickson, A.H.; Katunuma, N.; Gorelick, F.S.; Gukovsky, I.; et al. Impaired autophagic flux mediates acinar cell vacuole formation and trypsinogen activation in rodent models of acute pancreatitis. J. Clin. Investig. 2009, 119, 3340–3355. [Google Scholar] [CrossRef]
- Mareninova, O.A.; Sendler, M.; Malla, S.R.; Yakubov, I.; French, S.W.; Tokhtaeva, E.; Vagin, O.; Oorschot, V.; Lüllmann-Rauch, R.; Blanz, J.; et al. Lysosome associated membrane proteins maintain pancreatic acinar cell homeostasis: LAMP-2 deficient mice develop pancreatitis. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 678–694. [Google Scholar] [CrossRef]
- Fortunato, F.; Burgers, H.; Bergmann, F.; Rieger, P.; Büchler, M.W.; Kroemer, G.; Werner, J. Impaired autolysosome formation correlates with Lamp-2 depletion: Role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology 2009, 137, 350–360. [Google Scholar] [CrossRef]
- Helin, H.; Mero, M.; Markkula, H.M.; Helin, M. Pancreatic acinar ultrastructure in human acute pancreatitis. Virchows Arch. A Pathol. Anat. Histol. 1980, 387, 259–270. [Google Scholar] [CrossRef]
- Aho, H.J.; Nevalainen, T.J.; Havia, V.T.; Heinonen, R.J.; Aho, A.J. Human acute pancreatitis: A light and electron microscopic study. Acta Pathol. Microbiol. Immunol. Scand. A 1982, 90, 367–373. [Google Scholar] [CrossRef]
- Gukovsky, I.; Pandol, S.J.; Mareninova, O.A.; Shalbueva, N.; Jia, W.; Gukovskaya, A.S. Impaired autophagy and organellar dysfunction in pancreatitis. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. 2), 27–32. [Google Scholar] [CrossRef]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef] [PubMed]
- Diakopoulos, K.N.; Lesina, M.; Wormann, S.; Song, L.; Aichler, M.; Schild, L.; Anna Artati, A.; Römisch-Margl, W.R.; Wartmann, T.; Fischer, R.; et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology 2015, 148, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D.; et al. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Kim, H. Cerulein pancreatitis: Oxidative stress, inflammation, and apoptosis. Gut Liver 2008, 2, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D.; et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879–44905. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders —A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Wilson, J.S.; Pirola, R.C.; Apte, M.V. Role of alcohol metabolism in chronic pancreatitis. Alcohol Res. Health 2007, 30, 48–54. [Google Scholar]
- Pereda, J.; Sabater, L.; Aparisi, L.; Escobar, J.; Sandoval, J.; Viña, J.; López-Rodas, G.; Sastre, S. Interaction between cytokines and oxidative stress in acute pancreatitis. Curr. Med. Chem. 2006, 13, 2775–2787. [Google Scholar] [CrossRef]
- Escobar, J.; Pereda, J.; Arduini, A.; Sandoval, J.; Sabater, L.; Aparisi, L.; López-Rodas, G.; Sastre, J. Cross-talk between oxidative stress and pro-inflammatory cytokines in acute pancreatitis: A key role for protein phosphatases. Curr. Pharm. Des. 2009, 15, 3027–3042. [Google Scholar] [CrossRef]
- Gukovsky, I.; Li, N.; Todoric, J.; Gukovskaya, A.; Karin, M. Inflammation, autophagy, and obesity: Common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1199–1209. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Gukovsky, I.; Algul, H.; Habtezion, A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology 2017, 153, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Unlu, N.Z.; Bohn, T.; Francis, D.M.; Nagaraja, H.N.; Clinton, S.K.; Schwartz, S.J. Lycopene from heat-induced cis-isomer-rich tomato sauce is more bioavailable than from all-trans-rich tomato sauce in human subjects. Br. J. Nutr. 2007, 98, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Mascio, P.M.; Devasagayam, T.P.A.; Kaiser, S.; Sies, H. Carotenoids, tocopherils and thios as biological signlet molecular oxygen quenchers. Biochem. Soc. Trans. 1990, 18, 1054–1056. [Google Scholar] [CrossRef] [PubMed]
- Milani, A.; Basirnejad, M.; Shahbazi, S.; Bolhassani, A. Carotenoids: Biochemistry, pharmacology and treatment. Br. J. Pharmacol. 2017, 174, 1290–1324. [Google Scholar] [CrossRef]
- Buyuklu, M.; Kandemir, F.M.; Ozkaraca, M.; Set, T.; Bakirci, E.M.; Topal, E.; Ileriturk, M.; Turkmen, K. Benefical effects of lycopene against contrast medium-induced oxidative stress, inflammation, autophagy, and apoptosis in rat kidney. Hum. Exp. Toxicol. 2015, 34, 487–496. [Google Scholar] [CrossRef]
- Reboul, E. Mechanisms of carotenoid intestinal absorption: Where do we stand? Nutrients 2019, 11, 838. [Google Scholar] [CrossRef]
- Reboul, E.; Abou, L.; Mikail, C.; Ghiringhelli, O.; André, M.; Portugal, H.; Jourdheuil-Rahmani, D.; Amiot, M.J.; Lairon, D.; Borel, P. Lutein transport by Caco-2 TC-7 cells occurs partly by a facilitated process involving the scavenger receptor class B type I (SR-BI). Biochem. J. 2005, 387, 455–461. [Google Scholar] [CrossRef]
- During, A.; Doraiswamy, S.; Harrison, E.H. Xanthophylls are preferentially taken up compared with β-carotene by retinal cells via a SRBI-dependent mechanism. J. Lipid Res. 2008, 49, 1715–1724. [Google Scholar] [CrossRef]
- Shyam, R.; Vachali, P.; Gorusupudi, A.; Nelson, K.; Bernstein, P.S. All three human scavenger receptor class B proteins can bind and transport all three macular xanthophyll carotenoids. Arch. Biochem. Biophys. 2017, 634, 21–28. [Google Scholar] [CrossRef]
- Moussa, M.; Landrier, J.F.; Reboul, E.; Ghiringhelli, O.; Comera, C.; Collet, X.; Frohlich, K.; Bohm, V.; Borel, P. Lycopene absorption in human intestinal cells and in mice involves scavenger receptor class b type i but not niemann-pick c1-like 1. J. Nutr. 2008, 138, 1432–1436. [Google Scholar] [CrossRef]
- Moussa, M.; Gouranton, E.; Gleize, B.; El Yazidi, C.; Niot, I.; Besnard, P.; Borel, P.; Landrier, J.F. Cd36 is involved in lycopene and lutein uptake by adipocytes and adipose tissue cultures. Mol. Nutr. Food Res. 2011, 55, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.L.; Swendseid, M.E.; Jacob, R.A.; McKee, R.W. Plasma carotenoid levels in human subjects fed a low carotenoid diet. J. Nutr. 1992, 122, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Redmond, T.M.; Gentleman, S.; Duncan, T.; Yu, S.; Wiggert, B.; Gantt, E.; Cunningham, F.X., Jr. Identification, expression, and substrate specificity of a mammalian beta-carotene 15,15′-dioxygenase. J. Biol. Chem. 2001, 276, 6560–6565. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, A.; Andersson, S. Biochemical properties of purified recombinant human beta-carotene 15,15′-monooxygenase. J. Biol. Chem. 2002, 277, 23942–23948. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D. Lycopene metabolism and its biological significance. Am. J. Clin. Nutr. 2012, 96, 1214S–1222S. [Google Scholar] [CrossRef] [PubMed]
- von Lintig, J.; Vogt, K. Filling the gap in vitamin A research. Molecular identification of an enzyme cleaving beta-carotene to retinal. J. Biol. Chem. 2000, 275, 11915–11920. [Google Scholar] [CrossRef]
- Wyss, A.; Wirtz, G.; Woggon, W.D.; Brugger, R.; Wyss, W.; Friedlein, A.; Bachmann, H.; Hunziker, W. Cloning and expression of beta,beta-carotene 15,15′-dioxygenase. Biochem. Biophys. Res. Commun. 2000, 271, 334–336. [Google Scholar] [CrossRef]
- Kiefer, C.; Hessel, S.; Lampert, J.M.; Vogt, K.; Lederer, M.O.; Breithaupt, D.E.; von Lintig, J. Identification and characterization of a mammalian enzyme catalyzing the asymmetric oxidative cleavage of provitamin A. J. Biol. Chem. 2001, 276, 14110–14116. [Google Scholar] [CrossRef]
- Hessel, S.; Eichinger, A.; Isken, A.; Amengual, J.; Hunzelmann, S.; Hoeller, U.; Elste, V.; Hunziker, W.; Goralczyk, R.; Oberhauser, V.; et al. CMO1 deficiency abolishes vitamin A production from beta-carotene and alters lipid metabolism in mice. J. Biol. Chem. 2007, 282, 33553–33561. [Google Scholar]
- Amengual, J.; Lobo, G.P.; Golczak, M.; Li, H.N.M.; Klimova, T.; Hoppel, C.L.; Wyss, A.; Palczewski, K.; von Lintig, J. A mitochondrial enzyme degrades carotenoids and protects against oxidative stress. FASEB J. 2011, 25, 948–959. [Google Scholar] [CrossRef]
- Coronel, J.; Pinos, I.; Amengual, J. β-carotene in obesity research: Technical considerations and current status of the field. Nutrients 2019, 11, 842. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Moran, N.E.; Cichon, M.J.; Riedl, K.M.; Schwartz, S.J.; Erdman, J.W., Jr.; Pearl, D.K.; Thomas-Ahner, J.M.; Clinton, S.K. β-Carotene-9′,10′-oxygenase status modulates the impact of dietary tomato and lycopene on hepatic nuclear receptor-, stress-, and metabolism-related gene expression in mice. J. Nutr. 2014, 144, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Ford, N.A.; Elsen, A.C.; Erdman, J.W., Jr. Genetic ablation of carotene oxygenases and consumption of lycopene or tomato powder diets modulate carotenoid and lipid metabolism in mice. Nutr. Res. 2013, 33, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Sun, Z.W.; Ye, L.F.; Fu, G.S.; Mou, Y.; Hu, S.J. Lycopene protects against apoptosis in hypoxia/reoxygenation-induced H9C2 myocardioblast cells through increased autophagy. Mol. Med. Rep. 2015, 11, 1358–1365. [Google Scholar] [CrossRef]
- Kim, H. Inhibitory mechanism of lycopene on cytokine expression in experimental pancreatitis. Ann. N. Y. Acad. Sci. 2011, 1229, 99–102. [Google Scholar] [CrossRef]
- Kim, J.; Kim, E. Rag GTPase in amino acid signaling. Amino Acids 2016, 48, 915–928. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef]
- Yoon, M.S. Vps34 and PLD1 take center stage in nutrient signaling: Their dual roles in regulating autophagy. Cell Commun. Signal. 2015, 13, 44. [Google Scholar] [CrossRef]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK pathway and drug response in cancer: A therapeutic perspective. Oxid. Med. Cell Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef]
- Rehman, G.; Shehzad, A.; Khan, A.L.; Hamayun, M. Role of AMP-activated protein kinase in cancer therapy. Arch. Pharm. Weinh. 2014, 347, 457–468. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Furuta, S.; Hidaka, E.; Ogata, A.; Yokota, S.; Kamata, T. Ras is involved in the negative control of autophagy through the class I PI3-kinase. Oncogene 2004, 23, 3898–3904. [Google Scholar] [CrossRef]
- Kim, J.H.; Hong, S.K.; Wu, P.K.; Richards, A.L.; Jackson, W.T.; Park, J.I. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp. Cell Res. 2014, 327, 340–352. [Google Scholar] [CrossRef]
- Yang, J.; Yao, S. JNK-Bcl-2/Bcl-xL-Bax/Bak Pathway Mediates the Crosstalk between Matrine-Induced Autophagy and Apoptosis via Interplay with Beclin 1. Int. J. Mol. Sci. 2015, 16, 25744–25758. [Google Scholar] [CrossRef]
- Park, K.J.; Lee, S.H.; Lee, C.H.; Jang, J.Y.; Chung, J.; Kwon, M.H.; Kim, Y.S. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem. Biophys. Res. Commun. 2009, 382, 726–729. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Lee, B.H.; Ahn, S.G.; Oh, S.H. Proteasome inhibition-induced p38 MAPK/ERK signaling regulates autophagy and apoptosis through the dual phosphorylation of glycogen synthase kinase 3beta. Biochem. Biophys. Res. Commun. 2012, 418, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Jin, Y.; Bai, Y.; Ni, H.; Qiang, L.; Ye, L.; Shan, Y.; Zhou, M. Activation of autophagy through calcium-dependent AMPK/mTOR and PKCθ pathway causes activation of rat hepatic stellate cells under hypoxic stress. FEBS Lett. 2016, 590, 672–682. [Google Scholar] [CrossRef]
- Saluja, A.; Saluja, M.; Villa, A.; Leli, U.; Rutledge, P.; Meldolesi, J.; Steer, M. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J. Clin. Investig. 1989, 84, 1260–1266. [Google Scholar] [CrossRef]
- Chvanov, M.; De Faveri, F.; Moore, D.; Sherwood, M.W.; Awais, M.; Voronina, S.; Sutton, R.; Criddle, D.N.; Haynes, L.; Tepikin, A.V. Intracellular rupture, exocytosis and actin interaction of endocytic vacuoles in pancreatic acinar cells: Initiating events in acute pancreatitis. J. Physiol. 2018, 596, 2547–2564. [Google Scholar] [CrossRef]
- Mareninova, O.A.; Jia, W.; Gretler, S.R.; Holthaus, C.L.; Thomas, D.D.H.; Pimienta, M.; Dillon, D.L.; Gukovskaya, A.S.; Gukovsky, I.; Groblewski, G.E. Transgenic expression of GFP-LC3 perturbs autophagy in exocrine pancreas and acute pancreatitis responses in mice. Autophagy 2020, 16, 1–14. [Google Scholar] [CrossRef]
- Van Acker, G.J.; Weiss, E.; Steer, M.L.; Perides, G. Cause-effect relationships between zymogen activation and other early events in secretagogue-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1738–G1746. [Google Scholar] [CrossRef]
- Hashimoto, D.; Ohmuraya, M.; Hirota, M.; Yamamoto, A.; Suyama, K.; Ida, S.; Okumura, Y.; Takahashi, E.; Kido, H.; Araki, K.; et al. Involvement of autophagy in trypsinogen activation within the pancreatic acinar cells. J. Cell Biol. 2008, 181, 1065–1072. [Google Scholar] [CrossRef]
- Gukovsky, I.; Gukovskaya, A.S. Impaired autophagy underlies key pathological responses of acute pancreatitis. Autophagy 2010, 6, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS. Thiol network is the principal suspect for autophagy commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef]
- da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Bodas, M.; Westphal, C.V.; Carpenter-Thompson, R.; Mohanty, D.K.; Vij, N. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Rad. Biol. Med. 2016, 97, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Sharma, P.K.; Tiwari, R.; Rayavarapu, R.G.; Shankar, J.; Chauhan, L.K.S.; Alok Kumar Pandey, A.K. Impaired lysosomal activity mediated autophagic flux disruption by graphite carbon nanofibers induce apoptosis in human lung epithelial cells through oxidative stress and energetic impairment. Part Fibre Toxicol. 2017, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.D.; Lee, M.S.; Marchetti, P.; Pietropaolo, M.; Towns, R.; Vaccaro, M.I.; Watada, H.; Wiley, J.W. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy 2011, 7, 2–11. [Google Scholar] [CrossRef]
- Booth, D.M.; Murphy, J.A.; Mukherjee, R.; Awais, M.; Neoptolemos, J.P.; Gerasimenko, O.V.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Criddle, D.N. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology 2011, 140, 2116–2125. [Google Scholar] [CrossRef]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. The role of Ca2+ in the pathophysiology of pancreatitis. J. Physiol. 2014, 592, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Habtezion, A.; Gukovskaya, A.S.; Pandol, S.J. Acute pancreatitis: A multifaceted set of organelle and cellular interactions. Gastroenterology 2019, 156, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wei, Q.; Hu, Q.; Huang, X.; Zhou, X.; Luo, G.; Deng, M.; Lu, M. Research progress on the relationship between acute pancreatitis and calcium overload in acinar cells. Dig. Dis. Sci. 2019, 64, 25–38. [Google Scholar] [CrossRef]
- Mukherjee, R.; Mareninova, O.A.; Odinokova, I.V.; Huang, W.; Murphy, J.; Chvanov, M.; Javed, M.A.; Wen, L.; Booth, D.M.; Cane, M.C.; et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: Inhibition prevents acute pancreatitis by protecting production of ATP. Gut 2016, 65, 1333–1346. [Google Scholar] [CrossRef]
- Jakkampudi, A.; Jangala, R.; Reddy, B.R.; Mitnala, S.; Reddy, D.N.; Talukdar, R. NF-κB in acute pancreatitis: Mechanisms and therapeutic potential. Pancreatology 2016, 16, 477–488. [Google Scholar] [CrossRef]
- Netea-Maier, R.T.; Plantinga, T.S.; van de Veerdonk, F.L.; Smit, J.W.; Netea, M.G. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy 2016, 12, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Piplani, H.; Marek-Iannucci, S.; Sin, J.; Hou, J.; Takahashi, T.; Sharma, A.; de Freitas Germano, J.; Waldron, R.T.; Saadaeijahromi, H.; Song, Y.; et al. Simvastatin induces autophagic flux to restore cerulein-impaired phagosome-lysosome fusion in acute pancreatitis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xing, S.; Li, Z. Antagonistic effects of lycopene on cadmium-induced hippocampal dysfunctions in autophagy, calcium homeostatis and redox. Oncotarget 2017, 8, 44720–44731. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.C.; Peng, L.S.; Zou, L.; Huang, S.F.; Xie, Y.; Mu, G.P.; Zeng, X.H.; Zhou, X.L.; Zeng, Y.C. Protective effect and mechanism of lycopene on endothelial progenitor cells (EPCs) from type 2 diabetes mellitus rats. Biomed. Pharmacother. 2017, 92, 86–94. [Google Scholar] [CrossRef]
- Bayomy, N.A.; Elbakary, R.H.; Ibrahim, M.A.A.; Abdelaziz, E.Z. Effect of lycopene and rosmarinic acid on gentamicin-induced renal cortical oxidative stress, apoptosis, and autophagy in adult male albino rat. Anat. Rec. Hoboken 2017, 300, 1137–1149. [Google Scholar] [CrossRef]
- Liu, C.; Wang, R.; Zhang, B.; Hu, C.; Zhang, H. Protective effects of lycopene on oxidative stress, proliferation and autophagy in iron supplementation rats. Biol. Res. 2013, 46, 189–200. [Google Scholar] [CrossRef]
- Zhan, X.; Wang, F.; Bi, Y.; Ji, B. Animal models of gastrointestinal and liver diseases. Animal models of acute and chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G343–G355. [Google Scholar] [CrossRef]
- Muili, K.A.; Wang, D.; Orabi, A.I.; Sarwar, S.; Luo, Y.; Javed, T.A.; Eisses, J.F.; Mahmood, S.M.; Jin, S.; Singh, V.P.; et al. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J. Biol. Chem. 2013, 288, 570–580. [Google Scholar] [CrossRef]
- Kang, M.; Park, K.S.; Seo, J.Y.; Kim, H. Lycopene inhibits IL-6 expression in cerulein-stimulated pancreatic acinar cells. Genes Nutr. 2011, 6, 117–123. [Google Scholar] [CrossRef]
- El-Ashmawy, N.E.; Khedr, N.F.; El-Bahrawy, H.A.; Hamada, O.B. Suppression of inducible nitric oxide synthase and tumor necrosis factor-alpha level by lycopene is comparable to methylprednisolone in acute pancreatitis. Dig. Liver Dis. 2018, 50, 601–607. [Google Scholar] [CrossRef]
- Lv, J.C.; Wang, G.; Pan, S.H.; Bai, X.W.; Sun, B. Lycopene protects pancreatic acinar cells against severe acute pancreatitis by abating the oxidative stress through JNK pathway. Free Radic. Res. 2015, 49, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, E.; Akyuz, C.; Dulundu, E.; Topaloglu, U.; Sehirli, A.O.; Ercan, F.; Sener, G. Protective effects of lycopene on cerulein-induced experimental acute pancreatitis in rats. J. Surg. Res. 2012, 176, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Perez, S.; Pereda, J.; Sabater, L.; Sastre, J. Redox signaling in acute pancreatitis. Redox Biol. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Experimental Model | Dose | Signaling Mediators | Notable Results | Ref. |
|---|---|---|---|---|
| Cadmium-induced hippocampal dysfunction in mice and TH22 cell line | 5 mg/kg for mice, 10 μM for cells | Reduced Akt1, MAPK1, signaling | Reduced Cd-induced autophagy (ATG expression) and cell death | [103] |
| Endothelial progenitor cells isolated from diabetes mellitus rats | 10–50 μg/mL | Reduced mitochondrial dysfunction | Reduced apoptosis and oxidative autophagy | [104] |
| Contrast-induced nephropathy in rats | 4 mg/kg | Enhanced antioxidant enzymes | Reduced cell death | [35] |
| Gentamicin-induced nephrotoxicity in rats | 4 mg/kg | Enhanced SOD, GSH | Reduced oxidative stress-induced apoptosis and autophagy | [105] |
| Iron-induced oxidative damage in rats | 10 mg/kg, 15 mg/kg, 20 mg/kg | Reduced formation of autophagic vesicles | Inhibited oxidative stress, and pathologic autophagy | [106] |
| Hypoxia/reoxygenation-induced H9C2 myocardioblast cells | 2.5 μM, 5 μM | Increased AMPK activity | Reduced apoptotic cell death through increased autophagy | [54] |
| Experimental Model | Dose | Signaling Mediators | Notable Results | Ref. |
|---|---|---|---|---|
| Cerulein-induced rats pancreatic acinar cells | 2 μmol/L, 5 μmol/L | Reduced NF-κB activity | Decreased cytotoxicity | [109] |
| L-arginine-induced acute pancreatitis in rats | 50 mg/kg | Reduced TNF-β and increased GSH | Pancreatitis amelioration | [110] |
| Sodium taurocholate-induced severe acute pancreatitis in rats | 10 mg/kg | Reduced NF-κB p65 activity | Pancreatitis amelioration | [111] |
| Cerulein-stimulated rats pancreatic acinar cells | 2 μmol/L, 10 μmol/L | Blocked JNK–caspase-3 axis | Decreased cytotoxicity | [112] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.; Kim, H. The Remedial Potential of Lycopene in Pancreatitis through Regulation of Autophagy. Int. J. Mol. Sci. 2020, 21, 5775. https://doi.org/10.3390/ijms21165775
Choi S, Kim H. The Remedial Potential of Lycopene in Pancreatitis through Regulation of Autophagy. International Journal of Molecular Sciences. 2020; 21(16):5775. https://doi.org/10.3390/ijms21165775
Chicago/Turabian StyleChoi, Suyun, and Hyeyoung Kim. 2020. "The Remedial Potential of Lycopene in Pancreatitis through Regulation of Autophagy" International Journal of Molecular Sciences 21, no. 16: 5775. https://doi.org/10.3390/ijms21165775
APA StyleChoi, S., & Kim, H. (2020). The Remedial Potential of Lycopene in Pancreatitis through Regulation of Autophagy. International Journal of Molecular Sciences, 21(16), 5775. https://doi.org/10.3390/ijms21165775