Abstract
Inward rectifier potassium ion channels (IK1-channels) of the Kir2.x family are responsible for maintaining a stable negative resting membrane potential in excitable cells, but also play a role in processes of non-excitable tissues, such as bone development. IK1-channel loss-of-function, either congenital or acquired, has been associated with cardiac disease. Currently, basic research and specific treatment are hindered by the absence of specific and efficient Kir2.x channel activators. However, twelve different compounds, including approved drugs, show off-target IK1 activation. Therefore, these compounds contain valuable information towards the development of agonists of Kir channels, AgoKirs. We reviewed the mechanism of IK1 channel activation of these compounds, which can be classified as direct or indirect activators. Subsequently, we examined the most viable starting points for rationalized drug development and possible safety concerns with emphasis on cardiac and skeletal muscle adverse effects of AgoKirs. Finally, the potential value of AgoKirs is discussed in view of the current clinical applications of potentiators and activators in cystic fibrosis therapy.
1. Kir2.x Expression, Structure and Rectification
Kir2.x potassium channels are part of the inward rectifier K+ (IK1) channels family, which are of major importance for stabilizing the resting membrane potential of excitable cells and contribute to final action potential repolarization in cardiomyocytes [1,2]. Additionally, IK1 has an important role in non-excitable cells, for example, in osteoblasts, in which IK1 channels are involved in chondrogenesis and osteoblastogenesis [3].
Kir2.x channels are expressed in several excitable tissues, such as skeletal muscle (Kir2.1, Kir2.2, and Kir2.6), brain (Kir2.1, Kir2.2, and Kir2.3), and heart (Kir2.1, Kir2.2, and Kir2.3) [4]. Within tissues, isoform expression can vary. For example, Kir2.3 is dominantly expressed in the atria, whereas Kir2.1 is mainly present in the ventricles and has a higher channel density [5]. An intermediate level of expression of Kir2.x channels is present in smooth muscle tissue and the retina [4].
IK1 channels are homo- or heterotetrameric assemblies of Kir2.x monomeric subunits [6,7]. An individual subunit consists of a transmembrane and a cytoplasmic region. The transmembrane region of the channel controls ion selectivity and channel gating, and the cytoplasmic domain functions as a gating regulator [1].
Depending on the membrane potential (Vm) relative to the potassium equilibrium potential (Ek), IK1 channels conduct either inward or outward current. The outward currents are smaller due to rectification properties. Inward rectification is based on binding of polyamines and Mg2+ in the conducting pore region, at Vm more positive than Ek and inhibiting outward current [8]. Polyamine and Mg2+ dependent blocks are absent at more negative Vm than Ek, which results in inward current [9]. Each Kir2.x isoform displays its own characteristic rectification profile [5].
Whereas much knowledge on Kir2.x function has been derived from experimental in vitro studies and transgenic mice, insights on the physiological contributions of IK1 in larger organisms can only be deduced from a few experimental large animal studies using specific inhibitors [10,11] and patients with gain- or loss-of-function mutations [5]. For example, specific IK1 inhibition in awake dogs induced adverse effects like premature ventricular contractions, respiratory distress, and mild generalized muscle weakness [11]. On the contrary, the effects of specific IK1 activation have not been investigated in large animals. Therefore, as argued before [12], specific IK1 modifying compounds, i.e., inhibitors and activators, would benefit experimental studies on the role of IK1 in muscle contraction, cardiac function, neuronal excitation, and bone development.
2. Kir2.x Disease Relationships
IK1 function and disease have a bidirectional relationship. On the one hand, mutations can affect Kir2.x function, while, on the other hand, diseased states influence Kir2.x function. Both congenital and acquired loss-of-function may result from impaired functional Kir2.x expression at the plasma membrane, for example, due to aberrant transcription, trafficking, or gating kinetics.
Gain-of-function mutations in the KCNJ2 gene encoding Kir2.1 are associated with ventricular arrhythmia like short QT syndrome type 3 [13] or atrial arrhythmia such as congenital atrial fibrillation (AF) [14,15]. Loss-of-function mutations in KCNJ2 are associated with autosomal dominant Andersen syndrome (AS) [16]. This disease is characterized by symptoms like ventricular arrhythmias, periodic muscle paralysis, and dysmorphologies such as a broad forehead, a cleft palate, and small hands and feet [17].
AF results in increased IK1 densities in atrial tissue [18,19,20], which thereafter causes a shortening of the atrial effective refractory period that ultimately promotes and stabilizes atrial re-entry. Chronic heart failure (HF) induces loss-of-function of IK1. In experimental models of HF, IK1 is reduced in canine and rabbit ventricles [21,22,23]. More recently, heart failure following myocardial infarction (MI) in a porcine model shows significantly decreased IK1 density [24]. Also, declined density of whole-cell IK1 was also found in terminal HF patients [25].
Inward rectifier current inhibition has been suggested as a potential avenue in AF therapy [26]. Indeed, in preclinical and clinical settings, pharmacological IK1 inhibition is an effective method of AF reduction [10,27]. Moreover, pharmacological activators have been developed for several ion channels and successfully implemented in clinical therapy to treat cystic fibrosis, hypertension, and alopecia, to name a few applications [28,29,30]. Currently, no drugs are available that specifically target impaired IK1, whereas such a treatment may be effective in AS and HF. However, twelve drugs and compounds exert IK1 activating capacity (Figure 1, Table 1). We will review these drugs for indirect and direct properties to activate the different Kir2.x channels, focusing on Kir2.1, Kir2.2, and Kir2.3.
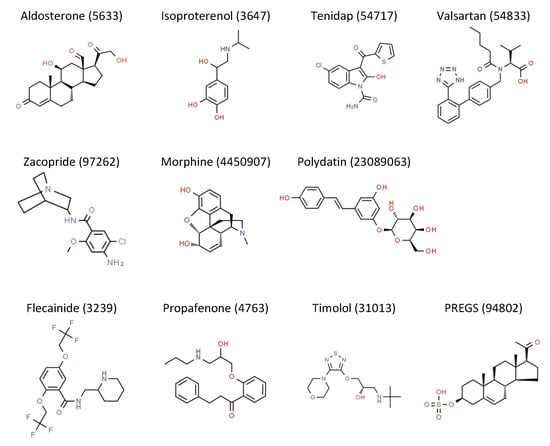
Figure 1.
Chemical structures of IK1 activating compounds, obtained from the Royal Society of Chemistry-owned ChemSpider website (accessed on 12 June 2020). Structure name and ChemSpider ID is given. Full records can be retrieved at http://www.chemspider.com/Chemical-Structure.ID.html, in which ID should be substituted by the ID number provided above the structure.

Table 1.
Compounds with IK1 activating properties.
3. AgoKirs, Agonists of Kir2.x Function
3.1. Indirect Activators
3.1.1. Aldosterone
Aldosterone is part of the renin-angiotensin-aldosterone-system (RAAS) and is essential in Na+ homeostasis. Activation of aldosterone and the mineralocorticoïdreceptor (MR) play an important role in the pathophysiology of cardiovascular disease.
In vitro studies using isolated rabbit ventricular cardiomyocytes demonstrated that 10 nmol/L of aldosterone caused a rapid activation (within minutes to days) of a 30 pS K+-selective current.
This current had pharmacological and biophysical properties that are consistent with those of the IK1-current. By use of RU28318, a specific MR-antagonist, and potassium canrenoate, a non-specific MR-antagonist, the underlying mechanism was determined as independent of the MR pathway [34].
Furthermore, cardiac tissue of rats implanted with osmotic minipumps showed a dose-dependent aldosterone effect on Kir2.1 expression after four weeks. At a dose of 0.5 µg/h, a moderate increase of Kir2.1 and Kir2.3 was observed [35], whereas, in another study, decreased expression levels of Kir2.1 surprisingly was shown at 1 µg/h aldosterone [45].
3.1.2. Isoproterenol
Isoproterenol, or isoprenaline (ISO), is a synthetic β-adrenoceptor (β-AR) agonist that has a cardiac muscle stimulating and bronchodilating effect. β3-AR stimulation leads to intracellular signal transduction pathways, including protein kinase A and C (PKA and PKC) [46]. In Xenopus oocytes, 10 µM ISO activated Kir2.2 currents via a PKA-dependent pathway [36], consistent with findings on the role of PKA in Kir2.2 activation [47]. Furthermore, ISO also activated Kir2.1 currents via a PKC-dependent pathway [36].
3.1.3. Tenidap
Tenidap is a non-steroidal cyclooxygenase and lipoxygenase inhibitor [48] but also a potent Kir2.3 opener. By use of 86Rb+ cell efflux and patch clamp electrophysiology, it was found that tenidap increased Kir2.3 carried inward and outward current in Chinese hamster ovary (CHO) cells [44]. Tenidap showed some channel specificity as it did not enhance Kir2.1 and Kv1.5 channel activity [44]; however, it did activate IKATP channels [49].
3.1.4. Valsartan
Valsartan is a highly selective angiotensin type 1 receptor antagonist and is widely used in the treatment of mild to moderate essential hypertension [50]. In rats with MI, induced by coronary artery ligation, Kir2.1 mRNA, protein, and the resulting IK1 became downregulated, which was associated with ventricular arrhythmias [51]. Such downregulation of Kir2.1/IK1 was prevented by Valsartan [37,38,39], possibly involving multiple mechanisms.
Firstly, casein kinase 2 dependent Kir2.1 downregulation after MI was prevented by valsartan. Secondly, valsartan treatment after MI decreased T-helper cell levels and thereby ameliorated IK1/Kir2.1 downregulation. Thirdly, valsartan reduced miRNA-16 levels by the prevention of NF-κB upregulation and thereby prevented downregulation of Kir2.1/KCNJ2/IK1 in infarcted hearts. Currently, it is unknown to what extent the working mechanism of valsartan on each individual signaling pathway contributed to restoring normal Kir2.1 expression in MI rat hearts. Nevertheless, the authors stated that a direct interaction of valsartan with the Kir2.1 ion channel, resulting in its activation, appeared improbable.
3.1.5. Zacopride
Zacopride (ZAC) is an antiemetic, gastroprokinetic, and anxiolytic drug. It is a selective antagonist of the 5-hydroxytryptamine (5-HT)3 receptor and agonist of 5-HT4 receptor. In the adrenal glands, ZAC is known to stimulate the secretion of aldosterone [52].
Rats treated with ZAC showed elevated levels of Kir2.1 protein in left ventricular tissue [53]. Furthermore, ZAC treatment prevented ischemia-mediated downregulation of left ventricular Kir2.1 protein [54] (note [55]). Additionally, ZAC enhanced both the inward and outward IK1 current in rat ventricular myocytes [56,57] but not in atrial cardiac myocytes [40]. ZAC-induced Kir2.1 channel activation appeared to be mediated by PKA-dependent phosphorylation of Ser425 in the Kir2.1 C-terminus [40]. In human embryonic kidney 293 cells and CHO cells, ZAC increased IK1 carried by ectopic homotetrameric Kir2.1 channels but not current carried by homotetrameric Kir2.2 or Kir2.3 channels or heterotetrameric channels containing Kir2.1, Kir2.2, or Kir2.3 [40,57].
3.2. Direct Activators
Within the class of direct activators, interactions with the extracellular domain and cytoplasmic domain have been indicated. The direct activators currently described display isoform specificity and may open the way towards tissue-specific activation of IK1.
3.2.1. Flecainide
Flecainide, a class Ic antiarrhythmic drug, is known to block Na+ channels and voltage-dependent K+ (Kv) channels. Thereby, flecainide effectively prolongs action potential duration (APD) in the atria but not in the ventricles [58]. On the other hand, flecainide increases IK1 selectively in the ventricles, offering a possible explanation for a difference in effect on atrial and ventricular APD [31].
Flecainide specifically activated Kir2.1 channels by a mechanism involving Cysteine311 (Cys311) and had no effect on Kir2.2 or Kir2.3 channels that contain an alanine instead of a cysteine residue on their equivalent positions (312 and 303, respectively) [31].
Flecainide’s mode of action likely involved antagonizing spermine-mediated rectification, resulting in increased outward current. Spermine was shown to inhibit IK1 in a concentration-dependent manner. The presence of flecainide decreased spermine’s inhibiting effect, as a rightward shift in the concentration-effect curve was observed [31]. The fact that the Emax of spermine was saturated at 82.1 ± 5.5% in the presence of flecainide suggests that spermine block was decreased in a noncompetitive manner by flecainide through allosteric changes to the binding site for polyamines [31].
3.2.2. Propafenone
Propafenone, just like flecainide, is a class Ic antiarrhythmic drug, mainly used in the treatment of ventricular tachycardias. Supra-therapeutic concentrations (>1 µM) of propafenone inhibited Kir2.x current [59]. Propafenone interacted with the cytoplasmic domain of the channel, which decreased the negative charge of the pore and the channel affinity for PIP2, a lipid critical for channel activation [59].
Given its structural similarities with flecainide, the effect of propafenone on Kir2.1 carried current was tested at lower concentrations. At 0.5 μM, propafenone enlarged inward and outward Kir2.1 current [33]. Propafenone significantly decreased spermine-induced block and thus relieved inward rectification, similarly as observed for flecainide [33]. However, propafenone failed to enhance both inward and outward Kir2.2 and Kir2.3 carried current [33]. In cells expressing Kir2.1/2.2, Kir2.1/2.3, and Kir2.2/2.3 heterotetrameric channels, propafenone also failed to increase inward and outward current [33]. Propafenone also did not modify IK1 recorded in human right atrial myocytes [33].
The molecular structure of propafenone can be described as an L-like shape, in which a long and short arm are joined by an aromatic ring [33]. Molecular dynamics simulations predicted interaction with Kir2.1, in which the hydrophobic long arm of propafenone embeds in a hydrophobic pocket formed by subunit A of the channel. The short arm, on the other hand, contains a group that formed a hydrogen bond with cysteine residue Cys311, which is part of the G-loop [33,60]. This critical hydrogen bond between Cys311 and propafenone was promoted due to the binding orientation of the alkylamino tail, which enabled propafenone’s hydroxyl group to be in close proximity to Cys311 [33].
To determine a potential role of Cys311 and propafenone interaction, mutations creating a cysteine were made in Kir2.2 (A312C) and Kir2.3 (A303C). These mutations made the channels responsive to activation with 0.5 µM propafenone. With respect to channel kinetics, it was found that opening frequency and mean open probability were increased at all tested voltages [33].
3.2.3. Timolol
Structural similarities and the common Cys311 binding site of flecainide and propafenone have led to the development of a pharmacophore model for binding to this particular site. The model predicted that a drug must have the following molecular properties that enable interaction with Cys311: (1) an “L-like” configuration with a short and long arm, joined by an aromatic ring at an angle of approximately 100 degrees; (2) a hydrogen bond acceptor/donor group in the short arm that interacts with Cys311; (3) a hydrophobic group in the long arm that interacts with a hydrophobic pocket in subunit A of Kir2.1; (4) a hydrogen bond between Arg67 or Glu63 and the drug for stabilization. By screening drugs for these properties, timolol was found as a potential activator of Kir2.1 [33]. Timolol, a non-selective β-receptor antagonist, selectively activated Kir2.1 by directly binding to Cys311 and thereby increased IK1 in the ventricles [33].
3.2.4. Pregnenolone Sulfate
Pregnenolone sulfate (PREGS) is an endogenous neurosteroid. PREGS modulates the function of multiple neurotransmitter receptors and channels, among which were voltage-gated K+ channels [61]. PREGS-enhanced Kir2.3 carried current in Xenopus oocytes when applied extracellularly only, whereas no current response in Kir1.1, Kir2.1, Kir2.2, or Kir3.1/Kir3.2 channels was observed [32].
3.3. Unknown Mechanism of Activation
3.3.1. LPS (lipopolysaccharides)
Lipopolysaccharides (LPS) are an important constituent of the cell wall of gram-negative bacteria. LPS can injure pulmonary microvascular walls. In those vessels, Kir2.1 plays a role in vasodilation by modulating the membrane potential and intracellular Ca2+ concentration. Treatment of mouse pulmonary microvascular endothelial cells with LPS enhanced Kir2.1 channel expression and Ba2+ sensitive IK1 current [41].
3.3.2. Morphine
Morphine is the main element of opium and is used as an anesthetic or sedative as agonist of the μ, δ, and κ opioid receptors [62]. In rabbit ventricular myocytes, morphine significantly increased IK1, independent of the opioid-receptor pathway [42]. In human atrial myocytes, morphine was unable to enlarge IK1 [63].
3.3.3. Polydatin
Polydatin (PD), also known as piceid (3,4′,5-trihydroxystilbene-3-β-d-glucoside), is a monocrystalline compound found, for example, in Polygonum cuspidatum Sieb. et Zucc. (Polygonaceae), peanuts, and grapes. PD has a therapeutic effect on hypertension, arrhythmia, hypertrophy, cardiac ischemia, and heart failure by means of manipulation of Ca2+ mobilization [64]. In rat ventricular myocytes, PD increased IK1 in a concentration-independent manner [43].
4. Lead Compounds and Clinical Perspective
Due to their similar chemical structure, the drugs flecainide, propafenone, and timolol are able to directly activate Kir2.1 channels by means of their interaction with cysteine residue Cys311. This off-target effect can be exploited for rationalized drug development towards specific IK1 activators. This provides a promising perspective in the search for a suitable AgoKir.
As mentioned above, timolol has been found to be an activator of Kir2.1 after being selected based on the criteria proposed by a pharmacophore model developed by [33]. This opens the possibility to develop a new drug with a higher specificity for Kir2.1 channels that meets the requirements of the model. Medicinal chemistry approaches involving modifications to the long and short arm of one of these three existing drugs might be an important step to explore properties that increase specificity for the Kir2.1 channel. For example, flecainide’s principal mechanism of action is the inhibition of cardiac Nav1.5 sodium channels [65]. It might be possible to modify parts of the flecainide molecule that, as per the pharmacophore model, are not essential for binding to Kir2.1, but are important for interaction with Nav1.5 to steer specificity.
Out of all the drugs reviewed in this article, the ones directly activating Kir2.1 by binding to Cys311 provide the most viable basis for the development of AgoKirs. As discussed earlier, IK1 channels are homo- or heterotetrameric assemblies of Kir2.x monomeric subunits. Only the Kir2.1 subunit has the cysteine residue Cys311 as part of its G-loop. Kir2.2 and Kir2.3 subunits contain an alanine instead of a cysteine residue on their equivalent positions (312 and 303, respectively) [31]. Propafenone and flecainide both cannot activate homo- or heterotetramers of Kir2.2 and Kir2.3 channels. These observations indicate the potential for isoform-specific Kir2.x activating compound development by targeting this channel domain but also indicate that other regions of the channel protein should be targeted for the development of multi-isoform activators. Furthermore, high throughput screening methods, such as automated patch-clamp, optical membrane potential detection, and ion-flux measurements, are being developed that will further aid the generation of AgoKirs [66].
The reports on aldosterone and valsartan on Kir2.x/IK1 regulation/activation are contradictory. Aldosterone production is part of RAAS, whereas valsartan is an antagonist of that system. Therefore, one would expect that those compounds have opposite effects on the activation and expression of the Kir2.1 subunits. The findings, however, showed that valsartan was able to ameliorate MI-induced Kir2.1 downregulation. Unfortunately, the results for aldosterone were inconclusive. Two studies demonstrated upregulation, and only one study proved downregulation and therefore matched our expectations. This inconsistency between the outcomes of aldosterone and valsartan might indicate that both compounds use additional, RAAS independent pathways in order to activate IK1.
Specific AgoKirs may come with potential adverse effects. Gain-of-function mutations indicate the potential of developing reentry-based arrhythmias, either atrial, ventricular, or both. The underlying mechanism is likely AP shortening and reduction in the effective refractory period. Furthermore, due to the strong stabilization of the resting membrane potential, muscle and neuronal cells harboring IK1 may become less or even unexcitable. Therefore, it would be beneficial to develop (1) Kir2 isoform-specific AgoKirs and (2) AgoKirs with a wide therapeutic range.
Ion channel activators have been pursued in several other research fields, and, for some, they entered clinical practice. The research field advanced furthest with respect to channel activation is undoubtedly that of cystic fibrosis. This disease results mainly from insufficient cystic fibrosis transmembrane conductance regulator (CFTR) channel activity at the plasma membrane of glandular epithelial cells of, for example, the lungs, sweat glands, and gastrointestinal tract [29]. The CFTR channel research field developed both potentiators and correctors. Potentiators act on plasma membrane-localized channels and increase their open probability, whereas correctors address the impaired trafficking deficiencies and enhance forward trafficking of the channels by approaching several different steps of the trafficking machinery [29]. Clinically, a combination of potentiators and correctors appeared most effective (e.g., [67,68]). With respect to cardiac potassium channel activators, progress has been made for Kv11.1/IKr. Delayed rectifier IKr loss-of-function is associated with long QT syndrome type 2 and cardiac arrhythmia [69]. A number of compounds have been developed and tested in animal models for antiarrhythmic properties [70,71,72,73]. Currently, these activators mainly function to enhance channel kinetics resulting in increased potassium current. However, many of the forms of IKr loss-of-function are due to defective forward trafficking of the Kv11.1 proteins [74,75,76], and the development of compounds directly addressing this issue may be favorable. Unfortunately, existing enhancers of forward trafficking are also strong Kv11.1 inhibitors [77]. However, molecular insights in the rescue mechanism, including drug-channel interactions, may result in new rescuers of trafficking that do not display inhibition effects. In addition, activators alone might be sufficient in several conditions to increase current to sufficient levels to counteract arrhythmia [78]. With respect to inward rectifier currents, advances have been made in the application of drugs targeting IKATP channels. For example, minoxidil, an IKATP channel opener, is used as a vasodilator in the treatment of resistant hypertension [28] and to promote hair growth in androgenetic alopecia patients [30]. To our knowledge, specific AgoKirs have not been developed, but giving the new insights from the direct channel activators (flecainide, propafenone, timolol, PREGS) and the successes in other research fields, this development will be a valid way to counteract disease-associated loss-of-function of IK1 channels.
Author Contributions
Conceptualization, M.B. and M.A.G.v.d.H.; writing—original draft preparation, all authors; writing—review and editing, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AF | Atrial Fibrillation |
| AgoKir | Agonist of Kir channel |
| AS | Andersen Syndrome |
| CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
| CHO | Chinese Hamster Ovary |
| EK | Potassium equilibrium potential |
| HEK | Human Embryonal Kidney |
| HF | Heart Failure |
| IK1 | Inward rectifier current |
| IKATP | ATP regulated inward rectifier current |
| ISO | Isoproterenol/isoprenaline |
| Kir2.x | Isoform x of the inward rectifier protein Kir2 family |
| LPS | Lipopolysacharides |
| MI | Myocardial Infarction |
| MR | Mineral corticoid Receptor |
| PD | Polydatin |
| PKA | Protein Kinase A |
| PKC | Protein Kinase C |
| PREGS | Pregnenolone Sulfate |
| RAAS | Renin-Angiotensin-Aldosterone-System |
| Vm | Membrane potential |
| ZAC | Zacopride |
References
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef]
- Van der Heyden, M.A.G.; Jespersen, T. Pharmacological exploration of the resting membrane potential reserve: Impact on atrial fibrillation. Eur. J. Pharmacol. 2016, 771, 56–64. [Google Scholar] [CrossRef]
- Pini, J.; Rouleau, M.; Desnuelle, C.; Sacconi, S.; Bendahhou, S. Modeling Andersen’s Syndrome in Human Induced Pluripotent Stem Cells. Stem Cells Dev. 2016, 25, 151–159. [Google Scholar] [CrossRef]
- De Boer, T.P.; Houtman, M.J.C.; Compier, M.; Van der Heyden, M.A.G. The mammalian KIR2.x inward rectifier ion channel family: Expression pattern and pathophysiology. Acta Physiol. 2010, 199, 243–256. [Google Scholar] [CrossRef]
- Anumonwo, J.M.B.; Lopatin, A.N. Cardiac strong inward rectifier potassium channels. J. Mol. Cell. Cardiol. 2010, 48, 45–54. [Google Scholar] [CrossRef]
- Glowatzki, E.; Fakler, G.; Brändle, U.; Rexhausen, U.; Zenner, H.P.; Ruppersberg, J.P.; Fakler, B. Subunit-dependent assembly of inward-rectifier K+ channels. Proc. R. Soc. B Biol. Sci. 1995, 261, 251–261. [Google Scholar] [CrossRef]
- Preisig-Müller, R.; Schlichthörl, G.; Goerge, T.; Heinen, S.; Brüggemann, A.; Rajan, S.; Derst, C.; Veh, R.W.; Daut, J. Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen’s syndrome. Proc. Natl. Acad. Sci. USA 2002, 99, 7774–7779. [Google Scholar] [CrossRef]
- Nichols, C.G.; Lee, S.J. Polyamines and potassium channels: A 25-year romance. J. Biol. Chem. 2018, 293, 18779–18788. [Google Scholar] [CrossRef]
- Bichet, D.; Haass, F.A.; Jan, L.Y. Merging functional studies with structures of inward-rectifier K+ channels. Nat. Rev. Neurosci. 2003, 4, 957–967. [Google Scholar] [CrossRef]
- Ji, Y.; Varkevisser, R.; Opacic, D.; Bossu, A.; Kuiper, M.; Beekman, J.D.M.; Yang, S.; Khan, A.P.; Dobrev, D.; Voigt, N.; et al. The inward rectifier current inhibitor PA-6 terminates atrial fibrillation and does not cause ventricular arrhythmias in goat and dog models. Br. J. Pharmacol. 2017, 174, 2576–2590. [Google Scholar] [CrossRef]
- Szatmári, V.; Ji, Y.; Herwijnen, B.V.; Feng, M.; Wang, M.Z.; Bossu, A.; Van der Heyden, M.A.G. Efficacy of pentamidine analogue 6 in dogs with chronic atrial fibrillation. J. Vet. Intern. Med. 2018, 32, 1549–1554. [Google Scholar] [CrossRef]
- Van der Heyden, M.A.G.; Sánchez-Chapula, J.A. Toward specific cardiac I(K1) modulators for in vivo application: Old drugs point the way. Heart Rhythm 2011, 8, 1076–1080. [Google Scholar] [CrossRef]
- Priori, S.G.; Pandit, S.V.; Rivolta, I.; Berenfeld, O.; Ronchetti, E.; Dhamoon, A.; Napolitano, C.; Anumonwo, J.; di Barletta, M.R.; Gudapakkam, S.; et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 2005, 96, 800–807. [Google Scholar] [CrossRef]
- Xia, M.; Jin, Q.; Bendahhou, S.; He, Y.; Larroque, M.M.; Chen, Y.; Zhou, Q.; Yang, Y.; Liu, Y.; Liu, B.; et al. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem. Biophys. Res. Commun. 2005, 332, 1012–1019. [Google Scholar] [CrossRef]
- Veldhuis, M.G.; Ji, Y.; van der Heyden, M.A.G. A Little Too Much: Cardiac Electrophysiological Effects of Elevated Inward Rectifying Current Carried by the KIR2.1 Ion Channel Protein. Adapt. Med. 2015, 7, 1–8. [Google Scholar] [CrossRef]
- Tristani-Firouzi, M.; Jensen, J.L.; Donaldson, M.R.; Sansone, V.; Meola, G.; Hahn, A.; Bendahhou, S.; Kwiecinski, H.; Fidzianska, A.; Plaster, N.; et al. Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome). J. Clin. Investig. 2002, 110, 381–388. [Google Scholar] [CrossRef]
- Andersen, E.D.; Krasilnikoff, P.A.; Overvad, H. Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies: A new syndrome? Acta Paediatr. 1971, 60, 559–564. [Google Scholar] [CrossRef]
- Bosch, R.F.; Zeng, X.; Grammer, J.B.; Popovic, K.; Mewis, C.; Kühlkamp, V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc. Res. 1999, 44, 121–131. [Google Scholar] [CrossRef]
- Dobrev, D.; Graf, E.; Wettwer, E.; Himmel, H.M.; Hála, O.; Doerfel, C.; Christ, T.; Schüler, S.; Ravens, U. Molecular basis of downregulation of G-protein-coupled inward rectifying K(+) current (I(K,ACh) in chronic human atrial fibrillation: Decrease in GIRK4 mRNA correlates with reduced I(K,ACh) and muscarinic receptor-mediated shortening of action potentials. Circulation 2001, 104, 2551–2557. [Google Scholar] [CrossRef]
- Dobrev, D.; Wettwer, E.; Kortner, A.; Knaut, M.; Schüler, S.; Ravens, U. Human inward rectifier potassium channels in chronic and postoperative atrial fibrillation. Cardiovasc. Res. 2002, 54, 397–404. [Google Scholar] [CrossRef]
- Kääb, S.; Nuss, H.B.; Chiamvimonvat, N.; O’Rourke, B.; Pak, P.H.; Kass, D.A.; Marban, E.; Tomaselli, G.F. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ. Res. 1996, 78, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Rose, J.; Armoundas, A.A.; Tian, Y.; DiSilvestre, D.; Burysek, M.; Halperin, V.; O’Rourke, B.; Kass, D.A.; Marbán, E.; Tomaselli, G.F. Molecular correlates of altered expression of potassium currents in failing rabbit myocardium. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2077–H2087. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, A.; Nishijima, Y.; Terentyev, D.; Khan, M.; Terentyeva, R.; Hamlin, R.L.; Nakayama, T.; Gyorke, S.; Cardounel, A.J.; Carnes, C.A. Chronic heart failure and the substrate for atrial fibrillation. Cardiovasc. Res. 2009, 84, 227–236. [Google Scholar] [CrossRef]
- Hegyi, B.; Bossuyt, J.; Griffiths, L.G.; Shimkunas, R.; Coulibaly, Z.; Jian, Z.; Grimsrud, K.N.; Sondergaard, S.S.; Ginsburg, K.S.; Chiamvimonvat, N.; et al. Complex electrophysiological remodeling in postinfarction ischemic heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, E3036–E3044. [Google Scholar] [CrossRef]
- Beuckelmann, D.J.; Näbauer, M.; Erdmann, E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ. Res. 1993, 73, 379–385. [Google Scholar] [CrossRef]
- Ehrlich, J.R. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J. Cardiovasc. Pharmacol. 2008, 52, 129–135. [Google Scholar] [CrossRef]
- Tobón, C.; Palacio, L.C.; Chidipi, B.; Slough, D.P.; Tran, T.; Tran, N.; Reiser, M.; Lin, Y.S.; Herweg, B.; Sayad, D.; et al. The Antimalarial Chloroquine Reduces the Burden of Persistent Atrial Fibrillation. Front. Pharmacol. 2019, 10, 1392. [Google Scholar] [CrossRef]
- Mundt, H.M.; Matenaer, M.; Lammert, A.; Göttmann, U.; Krämer, B.K.; Birck, R.; Benck, U. Minoxidil for Treatment of Resistant Hypertension in Chronic Kidney Disease--A Retrospective Cohort Analysis. J. Clin. Hypertens. 2016, 18, 1162–1167. [Google Scholar] [CrossRef]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Primers 2015, 1, 15010. [Google Scholar] [CrossRef]
- Varothai, S.; Bergfeld, W.F. Androgenetic alopecia: An evidence-based treatment update. Am. J. Clin. Dermatol. 2014, 15, 217–230. [Google Scholar] [CrossRef]
- Caballero, R.; Dolz-Gaitón, P.; Gómez, R.; Amorós, I.; Barana, A.; de la Fuente, M.G.; Osuna, L.; Duarte, J.; López-Izquierdo, A.; Moraleda, I.; et al. Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc. Natl. Acad. Sci. USA 2010, 107, 15631–15636. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Wahiyama, K.; Ikeda, K. Pregnenolone Sulfate Potentiates the Inwardly Rectifying K+ Channel Kir2.3. PLoS ONE 2009, 4, e6311. [Google Scholar] [CrossRef] [PubMed]
- Gómez, R.; Caballero, R.; Barana, A.; Amorós, I.; De Palm, S.H.; Matamoros, M.; Núñez, M.; Pérez-Hernández, M.; Iriepa, I.; Tamargo, J.; et al. Structural basis of drugs that increase cardiac inward rectifier Kir2.1 currents. Cardiovasc. Res. 2014, 104, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, J.; Hof, T.; Puddu, P.E.; Rouet, R.; Guinamard, R.; Manrique, A.; Beygui, F.; Sallé, L.; Milliez, P. Rapid and MR-Independent IK1 Activation by Aldosterone during Ischemia-Reperfusion. PLoS ONE 2015, 10, e0132592. [Google Scholar] [CrossRef]
- Lammers, C.; Dartsch, T.; Brandt, M.C.; Rottländer, D.; Halbach, M.; Peinkofer, G.; Ockenpoehler, S.; Weierbraeger, M.; Schneider, T.; Reuter, H.; et al. Spironolactone prevents aldosterone induced increased duration of atrial fibrillation in rat. Cell. Physiol. Biochem. 2012, 29, 833–840. [Google Scholar] [CrossRef]
- Scherer, D.; Kiesecker, C.; Kulzer, M.; Günth, M.; Scholz, E.P.; Kathöfer, S.; Thomas, D.; Maurer, M.; Kreuzer, J.; Bauer, A.; et al. Activation of inwardly rectifying Kir2.x potassium channels by β3-adrenoceptors is mediated via different signaling pathways with a predominant role of PKC for Kir2.1 and of PKA for Kir2.2. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2007, 375, 311–322. [Google Scholar] [CrossRef]
- Li, X.; Hu, H.; Wang, Y.; Xue, M.; Li, X.; Cheng, W.; Xuan, Y.; Yin, J.; Yang, N.; Yan, S. Valsartan Upregulates Kir2.1 in Rats Suffering from Myocardial Infarction via Casein Kinase 2. Cardiovasc. Drugs Ther. 2015, 29, 209–218. [Google Scholar] [CrossRef]
- Li, X.; Hu, H.; Wang, Y.; Xue, M.; Li, X.; Cheng, W.; Xuan, Y.; Yin, J.; Yang, N.; Yan, S. Valsartan Attenuates KIR2.1 by Downregulating the Th1 Immune Response in Rats Following Myocardial Infarction. J. Cardiovasc. Pharmacol. 2016, 67, 252–259. [Google Scholar] [CrossRef]
- Li, X.; Hu, H.; Wang, Y.; Xue, M.; Li, X.; Cheng, W.; Xuan, Y.; Yin, J.; Yang, N.; Yan, S. Valsartan ameliorates KIR2.1 in rats with myocardial infarction via the NF-κB-mir-16 pathway. Gene 2016, 590, 201–209. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Q.H.; Liu, C.F.; Zhai, X.W.; Feng, Q.L.; Xu, R.L.; Cui, X.L.; Zhao, Z.Q.; Cao, J.M.; Wu, B. Zacopride selectively activates the Kir2.1 channel via a PKA signaling pathway in rat cardiomyocytes. Sci. China Life Sci. 2013, 56, 788–796. [Google Scholar] [CrossRef][Green Version]
- Fang, W.; Xia, L.; Sun, X.; Cai, S.; Bai, S.; Sun, Y.; Zhou, P.; Liu, X.; Zhao, R.; Shen, B. Lipopolysaccharides increase Kir2.1 expression in lung endothelial cells. Int. J. Clin. Exp. Pathol. 2018, 11, 2959–2967. [Google Scholar] [PubMed]
- Xiao, G.-S.; Zhou, J.-J.; Wang, G.-Y.; Cao, C.-M.; Li, G.-R.; Wong, T.-M. In vitro electrophysiologic effects of morphine in rabbit ventricular myocytes. Anesthesiology 2005, 103, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhou, J.-J.; Yang, J.; Zhang, J.; Zhang, L.-P.; Zhang, Y. Enhancement of polydatin on inward rectifier potassium channel current in rat ventricular myocytes. Acta Physiol. Sin. 2013, 65, 285–292. [Google Scholar]
- Liu, Y.; Liu, D.; Printzenhoff, D.; Coghlan, M.J.; Harris, R.; Krafte, D.S. Tenidap, a novel anti-inflammatory agent, is an opener of the inwardly rectifying K+ channel hKir2.3. Eur. J. Pharmacol. 2002, 435, 153–160. [Google Scholar] [CrossRef]
- Dartsch, T.; Fischer, R.; Gapelyuk, A.; Weiergraeber, M.; Ladage, D.; Schneider, T.; Schirdewan, A.; Reuter, H.; Müeller-Ehmsen, J.; Zobel, C. Aldosterone induces electrical remodeling independent of hypertension. Int. J. Cardiol. 2013, 164, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Sterin-Borda, L.; Bernabeo, G.; Ganzinelli, S.; Joensen, L.; Borda, E. Role of nitric oxide/cyclic GMP and cyclic AMP in β3 adrenoceptor-chronotropic response. J. Mol. Cell. Cardiol. 2006, 40, 580–588. [Google Scholar] [CrossRef]
- Zitron, E.; Kiesecker, C.; Lück, S.; Kathöfer, S.; Thomas, D.; Kreye, V.A.W.; Kiehn, J.; Katus, H.A.; Schoels, W.; Karle, C.A. Human cardiac inwardly rectifying current IKir2.2 is upregulated by activation of protein kinase A. Cardiovasc. Res. 2004, 63, 520–527. [Google Scholar] [CrossRef]
- Hwang, S.H.; Wecksler, A.T.; Wagner, K.; Hammock, B.D. Rationally designed multitarget agents against inflammation and pain. Curr. Med. Chem. 2013, 20, 1783–1799. [Google Scholar] [CrossRef]
- Best, L.; Brown, P.D.; Sener, A.; Malaisse, W.J. Opposing effects of tenidap on the volume-regulated anion channel and K(ATP) channel activity in rat pancreatic beta-cells. Eur. J. Pharmacol. 2010, 629, 159–163. [Google Scholar] [CrossRef]
- Kızılirmak, P.; Üresin, A.Y. Hypertension and valsartan. Anadolu Kardiyol. Derg. 2014, 14, S20–S24. [Google Scholar] [CrossRef]
- Liao, S.-Y.; Tse, H.-F.; Chan, Y.-C.; Yip, P.M.-C.; Zhang, Y.; Liu, Y.; Li, R.A. Overexpression of Kir2.1 channel in embryonic stem cell-derived cardiomyocytes attenuates posttransplantation proarrhythmic risk in myocardial infarction. Heart Rhythm 2013, 10, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, H.; Contesse, V.; Delarue, C.; Soubrane, C.; Legrand, A.; Kuhn, J.M.; Wolf, L.M.; Vaudry, H. Effect of the serotonin-4 receptor agonist zacopride on aldosterone secretion from the human adrenal cortex: In vivo and in vitro studies. J. Clin. Endocrinol. Metab. 1993, 77, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-H.; Qiao, X.; Zhang, L.-J.; Wang, J.; Zhang, L.; Zhai, X.-W.; Re, X.-Z.; Li, Y.; Cao, X.-N.; Feng, Q.-L.; et al. IK1 Channel Agonist Zacopride Alleviates Cardiac Hypertrophy and Failure via Alterations in Calcium Dyshomeostasis and Electrical Remodeling in Rats. Front. Pharmacol. 2019, 10, 929. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, E.; Luo, T.; Zhang, W.; He, R. Opening of the inward rectifier potassium channel alleviates maladaptive tissue repair following myocardial infarction. Acta Biochim. Biophys. Sin. 2016, 48, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Korte, S.M.; Van der Heyden, M.A.G. Preventing publication of falsified and fabricated data: Roles of scientists, editors, reviewers, and readers. J. Cardiovasc. Pharmacol. 2017, 69, 65–70. [Google Scholar] [CrossRef]
- Liu, Q.-H.; Li, X.-L.; Xu, Y.-W.; Lin, Y.-Y.; Cao, J.-M.; Wu, B.-W. A novel discovery of IK1 channel agonist: Zacopride selectively enhances IK1 current and suppresses triggered arrhythmias in the rat. J. Cardiovasc. Pharmacol. 2012, 59, 37–48. [Google Scholar] [CrossRef]
- Zhai, X.-W.; Zhang, L.; Guo, Y.-F.; Yang, Y.; Wang, D.-M.; Zhang, Y.; Li, P.; Niu, Y.-F.; Feng, Q.-L.; Wu, B.-W.; et al. The IK1/Kir2.1 channel agonist zacopride prevents and cures acute ischemic arrhythmias in the rat. PLoS ONE 2017, 12, e0177600. [Google Scholar] [CrossRef]
- Katritsis, D.; Rowland, E.; O’Nunain, S.; Shakespeare, C.F.; Poloniecki, J.; Camm, A.J. Effect of flecainide on atrial and ventricular refractoriness and conduction in patients with normal left ventricle. Implications for possible antiarrhythmic and proarrhythmic mechanisms. Eur. Heart J. 1995, 16, 1930–1935. [Google Scholar] [CrossRef]
- Amorós, I.; Dolz-Gaitón, P.; Gómez, R.; Matamoros, M.; Barana, A.; de la Fuente, M.G.; Núñez, M.; Pérez-Hernández, M.; Moraleda, I.; Gálvez, E.; et al. Propafenone blocks human cardiac Kir2.x channels by decreasing the negative electrostatic charge in the cytoplasmic pore. Biochem. Pharmacol. 2013, 86, 267–278. [Google Scholar] [CrossRef]
- Pegan, S.; Arrabit, C.; Zhou, W.; Kwiatkowski, W.; Collins, A.; Slesinger, P.A.; Choe, S. Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 2005, 8, 279–287. [Google Scholar] [CrossRef]
- Schumacher, M.; Liere, P.; Akwa, Y.; Rajkowski, K.; Griffiths, W.; Bodin, K.; Sjövall, J.; Baulieu, E.-E. Pregnenolone sulfate in the brain: A controversial neurosteroid. Neurochem. Int. 2008, 52, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W. Pharmacological mechanisms of opioid analgesics. Clin. NeuroPharmacol. 1993, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.F.; Tsai, C.H.; Su, M.J. Opioid receptor independent effects of morphine on membrane currents in single cardiac myocytes. Br. J. Anaesth. 1998, 81, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, P.; Deng, J.; Lv, J.; Liu, J. Resveratrol and polydatin as modulators of Ca2+ mobilization in the cardiovascular system. Ann. N. Y. Acad. Sci. 2017, 1403, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; O’leary, M.E. State-dependent trapping of flecainide in the cardiac sodium channel. J. Physiol. 2004, 560, 37–49. [Google Scholar] [CrossRef]
- Walsh, K.B. Screening Technologies for Inward Rectifier Potassium Channels: Discovery of New Blockers and Activators. SLAS Discov. 2020, 25, 420–433. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K+ channels: Structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef]
- Perry, M.; Sanguinetti, M.; Mitcheson, J. Revealing the structural basis of action of hERG potassium channel activators and blockers. J. Physiol. 2010, 588, 3157–3167. [Google Scholar] [CrossRef]
- Meng, J.; Shi, C.; Li, L.; Du, Y.; Xu, Y. Compound ICA-105574 prevents arrhythmias induced by cardiac delayed repolarization. Eur. J. Pharmacol. 2013, 718, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C. HERG1 channel agonists and cardiac arrhythmia. Curr. Opin. Pharmacol. 2014, 15, 22–27. [Google Scholar] [CrossRef]
- Qile, M.; Beekman, H.D.M.; Sprenkeler, D.J.; Houtman, M.J.C.; van Ham, W.B.; Stary-Weinzinger, A.; Beyl, S.; Hering, S.; van den Berg, D.-J.; de Lange, E.C.M.; et al. LUF7244, an allosteric modulator/activator of Kv11.1 channels, counteracts dofetilide-induced torsades de pointes arrhythmia in the chronic atrioventricular block dog model. Br. J. Pharmacol. 2019, 176, 3871–3885. [Google Scholar] [CrossRef]
- Anderson, C.L.; Delisle, B.P.; Anson, B.D.; Kilby, J.A.; Will, M.L.; Tester, D.J.; Gong, Q.; Zhou, Z.; Ackerman, M.J.; January, C.T. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 2016, 113, 365–373. [Google Scholar] [CrossRef]
- Anderson, C.L.; Kuzmicki, C.E.; Childs, R.R.; Hintz, C.J.; Delisle, B.P.; January, C.T. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 2014, 5, 5535. [Google Scholar] [CrossRef] [PubMed]
- Wible, B.A.; Hawryluk, P.; Ficker, E.; Kuryshev, Y.A.; Kirsch, G.; Brown, A.M. HERG-Lite: A novel comprehensive high-throughput screen for drug-induced hERG risk. J. Pharmacol. Toxicol. Methods 2005, 52, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Varkevisser, R.; Houtman, M.J.C.; Linder, T.; de Git, K.C.G.; Beekman, H.D.M.; Tidwell, R.R.; IJzerman, A.P.; Star-Weinziger, A.; Vos, M.A.; Van der Heyden, M.A.G. Structure-activity relationships of pentamidine-affected ion channel trafficking and dofetilide mediated rescue. Br. J. Pharmacol. 2013, 169, 1322–1334. [Google Scholar] [CrossRef]
- Qile, M.; Ji, Y.; Golden, T.D.; Houtman, M.J.C.; Romunde, F.; Fransen, D.; van Ham, W.B.; IJzerman, A.P.; January, C.T.; Heitman, L.H.; et al. LUF7244 plus dofetilide rescues aberrant Kv11.1 trafficking and produces functional IKv11.1. Mol. Pharmacol. 2020, 97, 355–364. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).