Origin of the Induced Pluripotent Stem Cells Affects Their Differentiation into Dopaminergic Neurons


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
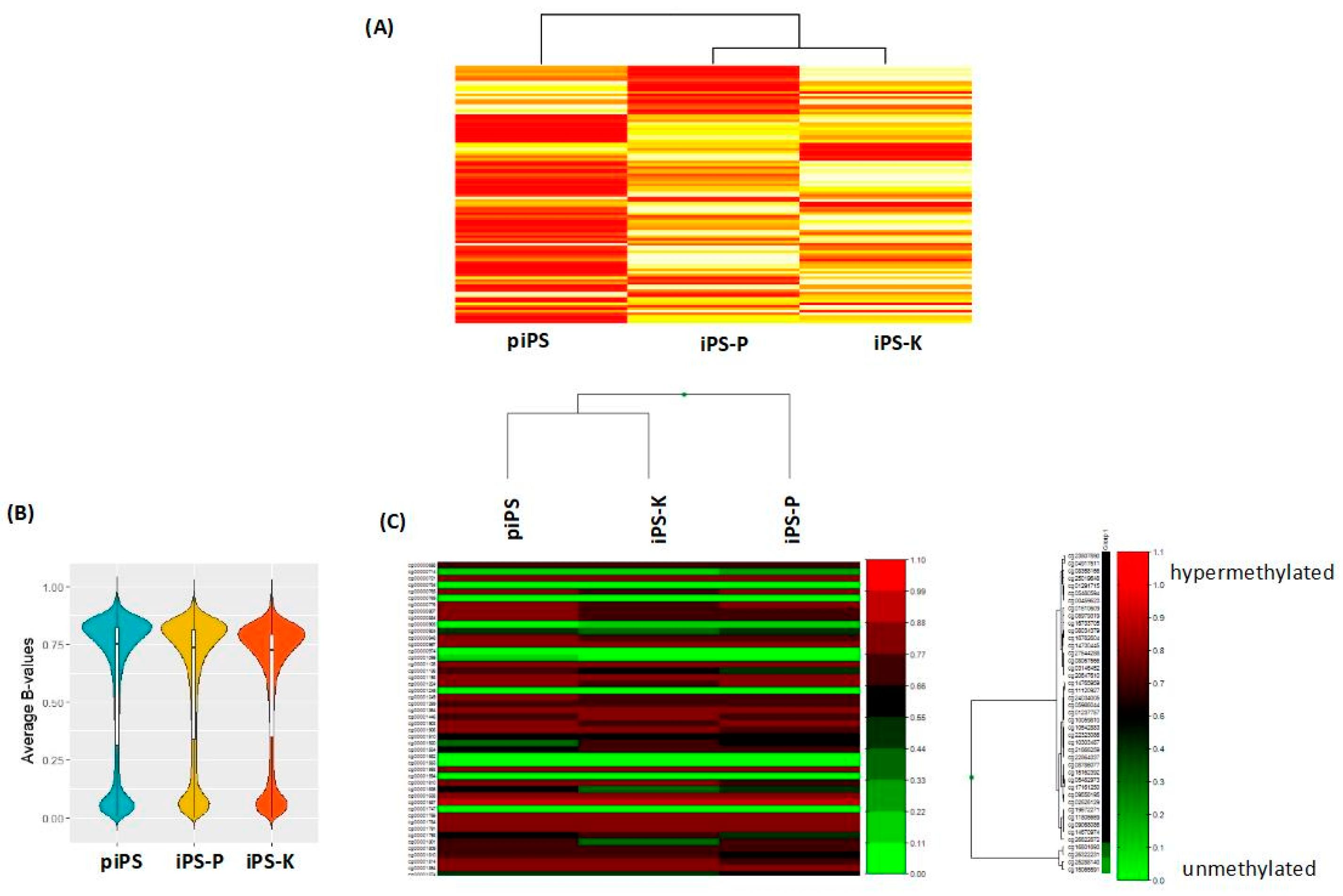
2.1. Keratinocytes and Peripheral Blood Mononuclear Cells (PBMCs) Isolated from Healthy Donors Are Reprogrammed Similarly to Induced Pluripotent Stem (iPS) Cells with the Sendai Viral Vector, Whereas the Generated iPS Cells Display Different Gene Expression Profiles
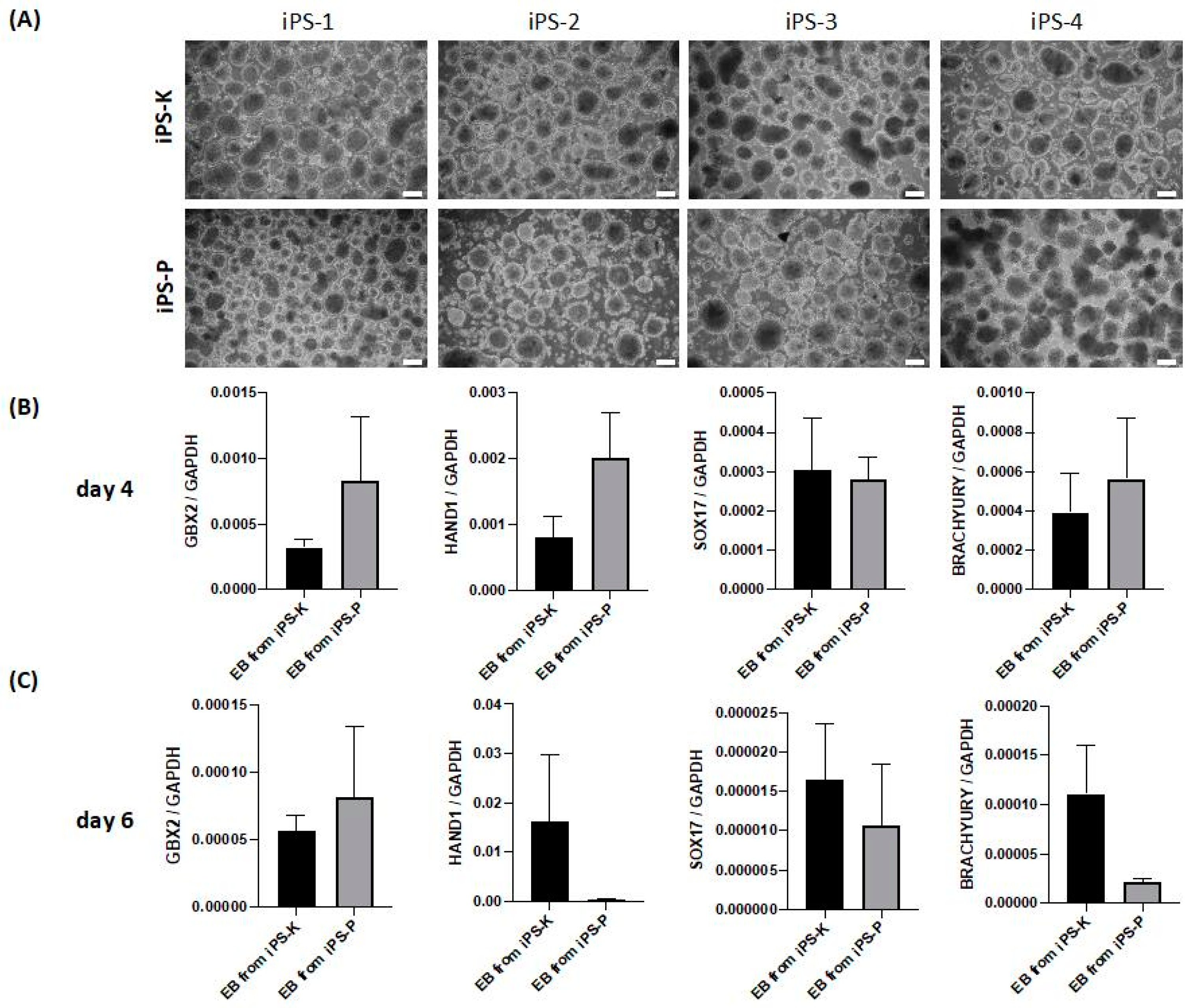
2.2. iPS Cells of Different Origin Form Embryoid Bodies and Three Germ Layers with Similar Effectiveness
2.3. Differentiation of iPS Cells in Teratomas Is Dependent on Origin of iPS Cells
2.4. Origin of iPS Cells Affects Early Stages of Differentiation into 3D Midbrain Organoids
2.5. Origin of iPS Cells Affects Their Capability to Differentiate into 2D Dopaminergic Neurons
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Generation of iPS Cells from Keratinocytes and PBMC
4.3. RNA Isolation and Reverse Transcription
4.4. Polymerase Chain Reaction (PCR) Analysis
4.5. Quantitative Real-Time PCR
4.6. Alkaline Phosphatase (AP) Activity
4.7. Gene Expression and Methylation Quantification
4.8. Formation of Embryoid Bodies
4.9. Differentiation to Three Germ Layers
4.10. Staining and Flow Cytometry
4.11. Formation of Teratomas
4.12. Formation of Midbrain Organoids
4.13. Differentiation of iPS Cells to Dopaminergic Neurons
4.14. Immunofluorescent Staining
4.15. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| iPSCs | Induced pluripotent stem cells |
| iPS-K | Induced pluripotent stem—keratinocytes |
| iPS-P | Induced pluripotent stem—PBMCs |
| PBMCs | Peripheral blood mononuclear cells |
| EB | Embryoid bodies |
| MEFs | Mouse embryonic fibroblasts |
| SeV | Sendai viral vector |
References
- Chlebanowska, P.; Tejchman, A.; Sułkowski, M.; Skrzypek, K.; Majka, M. Use of 3D organoids as a model to study idiopathic form of parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 694. [Google Scholar] [CrossRef] [PubMed]
- Sułkowski, M.; Konieczny, P.; Chlebanowska, P.; Majka, M. Introduction of exogenous HSV-TK suicide gene increases safety of keratinocyte-derived induced pluripotent stem cells by providing genetic “emergency exit” switch. Int. J. Mol. Sci. 2018, 19, 197. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Marton, R.M.; Ioannidis, J.P.A. A Comprehensive Analysis of Protocols for Deriving Dopaminergic Neurons from Human Pluripotent Stem Cells Significance Statement. Stem Cells. Transl. Med. 2018. [Google Scholar] [CrossRef]
- Kriks, S.; Shim, J.W.; Piao, J.; Ganat, Y.M.; Wakeman, D.R.; Xie, Z.; Carrillo-Reid, L.; Auyeung, G.; Antonacci, C.; Buch, A.; et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011, 480, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.-D.; Göke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.-P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem Cell 2016, 19, 248–257. [Google Scholar] [CrossRef]
- Ho, B.X.; Pek, N.M.Q.; Soh, B.S. Disease modeling using 3D organoids derived from human induced pluripotent stem cells. Int. J. Mol. Sci. 2018, 19, 936. [Google Scholar] [CrossRef]
- Jones, V.C.; Atkinson-Dell, R.; Verkhratsky, A.; Mohamet, L. Aberrant astrocytes in human induced stem cell model of Alzheimer’s disease. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders HHS Public Access. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef]
- Juopperi, T.A.; Ryoung Kim, W.; Chiang, C.-H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012. [Google Scholar] [CrossRef]
- Ye, F.; Kang, E.; Yu, C.; Qian, X.; Jacob, F.; Yu, C.; Mao, M.; Poon, R.Y.C.; Kim, J.; Song, H.; et al. DISC1 regulates neurogenesis via modulating kinetochore attachment of Ndel1/Nde1 during mitosis HHS Public Access. Neuron 2017, 96, 1041–1054. [Google Scholar] [CrossRef]
- Gunnar, H.; Marc, E.; Araúzo-Bravo, M.J.; Hemmer, K.; Hallmann, A.-L.; Reinhardt, P.; Kim, K.P.; Adachi, K.; Santourlidis, S.; Ghanjati, F.; et al. Origin-dependent neural cell identities in differentiated human iPSCs in vitro and after transplantation into the mouse brain. Cell Rep. 2014, 8. [Google Scholar] [CrossRef]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.R.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, H.; Liu, J.; Qi, J.; Wei, B.; Yang, J.; Liang, H.; Chen, Y.; Chen, J.; Wu, Y.; et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet. 2013, 45, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Kida, Y.S.; Hawkins, R.D.; Nery, J.R.; Hon, G.; Antosiewicz-Bourget, J.; Ogmalley, R.; Castanon, R.; Klugman, S.; et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 2011, 471, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Krijger, P.H.L.; Di Stefano, B.; De Wit, E.; Limone, F.; Van Oevelen, C.; De Laat, W.; Graf, T. Cell-of-origin-specific 3D genome structure acquired during somatic cell reprogramming. Cell Stem Cell 2016, 18, 597–610. [Google Scholar] [CrossRef]
- Polo, J.M.; Liu, S.; Eugenia Figueroa, M.; Kulalert, W.; Eminli, S.; Yong Tan, K.; Apostolou, E.; Stadtfeld, M.; Li, Y.; Shioda, T.; et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells NIH Public Access Author Manuscript. Nat. Biotechnol. 2010, 28, 848–855. [Google Scholar] [CrossRef]
- Liu, L.; Luo, G.Z.; Yang, W.; Zhao, X.; Zheng, Q.; Lv, Z.; Li, W.; Wu, H.J.; Wang, L.; Wang, X.J.; et al. Activation of the imprinted Dlk1-Dio3 region correlates with pluripotency levels of mouse stem cells. J. Biol. Chem. 2010, 285, 19483–19490. [Google Scholar] [CrossRef]
- Stadtfeld, M.; Apostolou, E.; Akutsu, H.; Fukuda, A.; Follett, P.; Natesan, S.; Kono, T.; Shioda, T.; Hochedlinger, K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 2010, 465, 175–181. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, B. The multi-differentiation potential of peripheral blood mononuclear cells. Stem Cell Res. Ther. 2012, 3, 48. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-O.; Zhu, Y.-F.; Asakawa, M.; Kuma, H.; Hirata, T.; Ueda, Y.; Lee, Y.-S.; Fukumura, M.; Iida, A.; Kato, A.; et al. A Cytoplasmic RNA Vector Derived from Nontransmissible Sendai Virus with Efficient Gene Transfer and Expression. J. Virol. 2000, 74, 6564–6569. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of defective and persistent Sendai virus vector: A unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, H. Methods for inducing embryoid body formation: In vitro differentiation system of embryonic stem cells. J. Biosci. Bioeng. 2007, 103, 389–398. [Google Scholar] [CrossRef]
- Mummery, C.L.; Zhang, J.; Ng, E.S.; Elliott, D.A.; Elefanty, A.G.; Kamp, T.J. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: A methods overview. Circ. Res. 2012, 111, 344–358. [Google Scholar] [CrossRef]
- Faial, T.; Bernardo, A.S.; Mendjan, S.; Diamanti, E.; Ortmann, D.; Gentsch, G.E.; Mascetti, V.L.; Trotter, M.W.B.; Smith, J.C.; Pedersen, R.A. Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development 2015, 142, 2121–2135. [Google Scholar] [CrossRef]
- Barnes, R.M.; Firulli, B.A.; Conway, S.J.; Vincentz, J.W.; Firulli, A.B. Analysis of the Hand1 Cell Lineage Reveals Novel Contributions to Cardiovascular, Neural Crest, Extra-Embryonic, and Lateral Mesoderm Derivatives. Dev. Dyn. 2010, 239, 3086–3097. [Google Scholar] [CrossRef]
- Cserjesi, P.; Brown, D.; Lyons, G.E.; Olson, E.N. Expression of the Novel Basic Helix-Loop-Helix Gene eHAND in Neural Crest Derivatives and Extraembryonic Membranes during Mouse Development. Dev. Biol. 1995, 170, 664–678. [Google Scholar] [CrossRef]
- Séguin, C.A.; Draper, J.S.; Nagy, A.; Rossant, J. Establishment of Endoderm Progenitors by SOX Transcription Factor Expression in Human Embryonic Stem Cells. Cell Stem Cell 2008, 3, 182–195. [Google Scholar] [CrossRef]
- Semrau, S.; Goldmann, J.E.; Soumillon, M.; Mikkelsen, T.S.; Jaenisch, R.; Van Oudenaarden, A. Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Liu, A.; Joyner, A.L. Essential roles of EN and GBX2 in FGF8 signaling. Development 2001, 128, 181–191. [Google Scholar] [PubMed]
- Ferreira, A.; Caceres, A. Expression of the Class III β-tubulin isotype in developing neurons in culture. J. Neurosci. Res. 1992, 32, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Q.; Oblinger, M.M. Differential regulation of beta III and other tubulin genes during peripheral and central neuron development. J. Cell Sci. 1992, 103, 643–651. [Google Scholar] [PubMed]
- Hartfield, E.M.; Yamasaki-Mann, M.; Ribeiro Fernandes, H.J.; Vowles, J.; James, W.S.; Cowley, S.A.; Wade-Martins, R. Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Daubner, S.C.; Le, T.; Wang, S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011, 508, 1–12. [Google Scholar] [CrossRef]
- Kyttälä, A.; Moraghebi, R.; Valensisi, C.; Kettunen, J.; Andrus, C.; Pasumarthy, K.K.; Nakanishi, M.; Nishimura, K.; Ohtaka, M.; Weltner, J.; et al. Stem Cell Reports Ar ticle Genetic Variability Overrides the Impact of Parental Cell Type and Determines iPSC Differentiation Potential. Stem Cell Rep. 2016, 6, 200–212. [Google Scholar] [CrossRef]
- Godini, R.; Lafta, H.Y.; Fallahi, H. Epigenetic modifications in the embryonic and induced pluripotent stem cells. Gene Expr. Patterns 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Rouhani, F.; Kumasaka, N.; De Brito, C.; Bradley, M.; Vallier, A. Genetic Background Drives Transcriptional Variation in Human Induced Pluripotent Stem Cells. PLoS Genet. 2014, 10, 1004432. [Google Scholar] [CrossRef]
- Fukusumi, H.; Shofuda, T.; Bamba, Y.; Yamamoto, A.; Kanematsu, D.; Handa, Y.; Okita, K.; Nakamura, M.; Yamanaka, S.; Okano, H.; et al. Establishment of Human Neural Progenitor Cells from Human Induced Pluripotent Stem Cells with Diverse Tissue Origins. Stem Cells. Int. 2016. [Google Scholar] [CrossRef]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilić, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef]
- Vlahos, K.; Sourris, K.; Mayberry, R.; McDonald, P.; Bruveris, F.F.; Schiesser, J.V.; Bozaoglu, K.; Lockhart, P.J.; Stanley, E.G.; Elefanty, A.G. Generation of iPSC lines from peripheral blood mononuclear cells from 5 healthy adults. Stem Cell Res. 2019, 34, 101380. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Chen, X.; Wei, A.; Song, X.; Wang, W.; Liang, L.; Zhao, Q.; Han, Z.; Han, Z.; Wang, X.; et al. Comparison of Teratoma Formation between Embryonic Stem Cells and Parthenogenetic Embryonic Stem Cells by Molecular Imaging. Stem Cells. Int. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yasudaid, S.; Kusakawa, S.; Kuroda, T.; Miura, T.; Tano, K.; Takada, N.; Matsuyama, S.; Matsuyama, A.; Nasu, M.; Umezawa, A.; et al. Tumorigenicity-associated characteristics of human iPS cell lines. PLoS ONE 2018, 13, e0205022. [Google Scholar] [CrossRef] [PubMed]
- Nasu, A.; Ikeya, M.; Yamamoto, T.; Watanabe, A.; Jin, Y.; Matsumoto, Y.; Hayakawa, K.; Amano, N.; Sato, S.; Osafune, K.; et al. Genetically Matched Human iPS Cells Reveal that Propensity for Cartilage and Bone Differentiation Differs with Clones, not Cell Type of Origin. PLoS ONE 2013, 8, e53771. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sheng, C.; Liu, Z.; Jia, W.; Wang, B.; Li, M.; Fu, L.; Ren, Z.; An, J.; Sang, L.; et al. Lmx1a enhances the effect of iNSCs in a PD model. Stem Cell Res. 2015, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Doucet-Beaupré, H.; Gilbert, C.; Profes, M.S.; Chabrat, A.; Pacelli, C.; Giguère, N.; Rioux, V.; Charest, J.; Deng, Q.; Laguna, A.; et al. Lmx1a and Lmx1b regulate mitochondrial functions and survival of adult midbrain dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2016, 113, 4387–4396. [Google Scholar] [CrossRef] [PubMed]
- Arenas, E. Foxa2: The Rise and Fall of Dopamine Neurons. Cell Stem Cell 2008, 2, 110–112. [Google Scholar] [CrossRef]
- Zetterstrom, R.H.; Solomin, L.; Jansson, L.; Hoffer, B.J.; Olson, L.; Perlmann, T. Dopamine Neuron Agenesis in Nurr1-Deficient Mice. Science 1997, 276, 248–250. [Google Scholar] [CrossRef]
- Oh, S.-M.; Chang, M.-Y.; Song, J.-J.; Rhee, Y.-H.; Joe, E.-H.; Lee, H.-S.; Yi, S.-H.; Lee, S.-H. Combined Nurr1 and Foxa2 roles in the therapy of Parkinson’s disease. EMBO Mol. Med. 2015, 7, 510–525. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Avci, H.X.; Ochalek, A.; Rösingh, L.N.; Molnár, K.; László, L.; Bellák, T.; Téglási, A.; Pesti, K.; Mike, A.; et al. Comparison of 2D and 3D neural induction methods for the generation of neural progenitor cells from human induced pluripotent stem cells. Stem Cell Res. 2017, 25, 139–151. [Google Scholar] [CrossRef]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener 2018, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Zhao, M.-T.; Jahanbani, F.; Shao, N.-Y.; Lee, H.; Chen, H.; Snyder, M.P.; Wu, J.C. Effects of cellular origin on differentiation of human induced pluripotent stem cell-derived endothelial cells. Ref. Inf. JCI Insight 2016, 1, 85558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chlebanowska, P.; Sułkowski, M.; Skrzypek, K.; Tejchman, A.; Muszyńska, A.; Noroozi, R.; Majka, M. Origin of the Induced Pluripotent Stem Cells Affects Their Differentiation into Dopaminergic Neurons. Int. J. Mol. Sci. 2020, 21, 5705. https://doi.org/10.3390/ijms21165705
Chlebanowska P, Sułkowski M, Skrzypek K, Tejchman A, Muszyńska A, Noroozi R, Majka M. Origin of the Induced Pluripotent Stem Cells Affects Their Differentiation into Dopaminergic Neurons. International Journal of Molecular Sciences. 2020; 21(16):5705. https://doi.org/10.3390/ijms21165705
Chicago/Turabian StyleChlebanowska, Paula, Maciej Sułkowski, Klaudia Skrzypek, Anna Tejchman, Agata Muszyńska, Rezvan Noroozi, and Marcin Majka. 2020. "Origin of the Induced Pluripotent Stem Cells Affects Their Differentiation into Dopaminergic Neurons" International Journal of Molecular Sciences 21, no. 16: 5705. https://doi.org/10.3390/ijms21165705
APA StyleChlebanowska, P., Sułkowski, M., Skrzypek, K., Tejchman, A., Muszyńska, A., Noroozi, R., & Majka, M. (2020). Origin of the Induced Pluripotent Stem Cells Affects Their Differentiation into Dopaminergic Neurons. International Journal of Molecular Sciences, 21(16), 5705. https://doi.org/10.3390/ijms21165705