Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis

 ,
, 
 ,
,  , , , and
, , , and 
Abstract
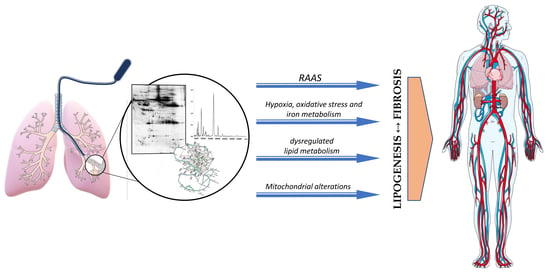
1. Introduction
2. Metabolic Alterations in IPF
2.1. Proteomic Studies Suggesting Metabolic Alterations in IPF Onset
2.2. Metabolic Dysregulation: Renin–Angiotensin–Aldosterone System in Fibrosis
2.3. Hypoxia, Oxidative Stress and Iron Metabolism
3. Onset of Fibrosis: The Contribution of Dysregulated Lipid Metabolism
Mitochondrial Alterations at Onset of Fibrosis
4. Discussion
5. Future Perspectives
6. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Joo, S.; Kim, D.K.; Sim, H.J.; Lee, G.D.; Hwang, S.K.; Choi, S.; Kim, H.R.; Kim, Y.-H.; Park, S.-I. Clinical results of sublobar resection versus lobectomy or more extensive resection for lung cancer patients with idiopathic pulmonary fibrosis. J. Thorac. Dis. 2016, 8, 977–984. [Google Scholar] [CrossRef]
- Ghatol, A.; Ruhl, A.P.; Danoff, S.K. Exacerbations in idiopathic pulmonary fibrosis triggered by pulmonary and nonpulmonary surgery: A case series and comprehensive review of the literature. Lung 2012, 190, 373–380. [Google Scholar] [CrossRef]
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015, 147, 157–164. [Google Scholar] [CrossRef]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Khalil, W.; Xia, H.; Bodempudi, V.; Kahm, J.; Hergert, P.; Smith, K.; Peterson, M.; Parker, M.; Herrera, J.; Bitterman, P.B.; et al. Pathologic Regulation of Collagen I by an Aberrant Protein Phosphatase 2A/Histone Deacetylase C4/MicroRNA-29 Signal Axis in Idiopathic Pulmonary Fibrosis Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2015, 53, 391–399. [Google Scholar] [CrossRef]
- Bargagli, E.; Cameli, P.; Carleo, A.; Refini, R.M.; Bergantini, L.; D’alessandro, M.; Vietri, L.; Perillo, F.; Volterrani, L.; Rottoli, P.; et al. The effect of cigarette smoking on bronchoalveolar lavage protein profiles from patients with different interstitial lung diseases. Panminerva Med. 2019, 62, 109–115. [Google Scholar] [CrossRef]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Richeldi, L.; Kolb, M.; Jouneau, S.; Wuyts, W.A.; Schinzel, B.; Stowasser, S.; Quaresma, M.; Raghu, G. Efficacy and safety of nintedanib in patients with advanced idiopathic pulmonary fibrosis. BMC Pulm. Med. 2020, 20, 3. [Google Scholar] [CrossRef]
- Vietri, L.; Cameli, P.; Perruzza, M.; Cekorja, B.; Bergantini, L.; d’Alessandro, M.; Refini, R.M.; Pieroni, M.; Fossi, A.; Bennett, D.; et al. Pirfenidone in idiopathic pulmonary fibrosis: Real-life experience in the referral centre of Siena. Ther. Adv. Respir. Dis. 2020, 14, 1753466620906326. [Google Scholar] [CrossRef]
- Lanzarone, N.; Gentili, F.; Alonzi, V.; Bergantini, L.; d’Alessandro, M.; Rottoli, P.; Refini, R.M.; Pieroni, M.; Vietri, L.; Bianchi, F.; et al. Bronchoalveolar lavage and serum KL-6 concentrations in chronic hypersensitivity pneumonitis: Correlations with radiological and immunological features. Intern. Emerg. Med. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- Bergantini, L.; d’Alessandro, M.; Cameli, P.; Carleo, A.; Landi, C.; Vietri, L.; Lanzarone, N.; Pieroni, M.; Sestini, P.; Bargagli, E. Antithrombin III as predictive indicator of survival in IPF patients treated with Nintedanib: A preliminary study. Intern. Med. J. 2020. Online ahead of print. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Carleo, A.; Cameli, P.; Bergantini, L.; Perrone, A.; Vietri, L.; Lanzarone, N.; Vagaggini, C.; Sestini, P.; Bargagli, E. BAL biomarkers’ panel for differential diagnosis of interstitial lung diseases. Clin. Exp. Med. 2020, 20, 207–216. [Google Scholar] [CrossRef]
- Vietri, L.; Bennett, D.; Cameli, P.; Bergantini, L.; Cillis, G.; Sestini, P.; Bargagli, E.; Rottoli, P. Serum amyloid A in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2019, 57, 430–434. [Google Scholar] [CrossRef]
- Vietri, L.; Fui, A.; Bergantini, L.; d’Alessandro, M.; Cameli, P.; Sestini, P.; Rottoli, P.; Bargagli, E. Serum amyloid A: A potential biomarker of lung disorders. Respir. Investig. 2020, 58, 21–27. [Google Scholar] [CrossRef]
- Bergantini, L.; Cameli, P.; d’Alessandro, M.; Vagaggini, C.; Refini, R.M.; Landi, C.; Pieroni, M.G.; Spalletti, M.; Sestini, P.; Bargagli, E. NK and NKT-like cells in granulomatous and fibrotic lung diseases. Clin. Exp. Med. 2019, 19, 487–494. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Landi, C.; Bargagli, E.; Bianchi, L.; Gagliardi, A.; Carleo, A.; Bennett, D.; Perari, M.G.; Armini, A.; Prasse, A.; Rottoli, P.; et al. Towards a functional proteomics approach to the comprehension of idiopathic pulmonary fibrosis, sarcoidosis, systemic sclerosis and pulmonary Langerhans cell histiocytosis. J. Proteom. 2013, 83, 60–75. [Google Scholar] [CrossRef]
- Landi, C.; Bargagli, E.; Carleo, A.; Bianchi, L.; Gagliardi, A.; Prasse, A.; Perari, M.G.; Refini, R.M.; Bini, L.; Rottoli, P. A system biology study of BALF from patients affected by idiopathic pulmonary fibrosis (IPF) and healthy controls. Proteom. Clin. Appl. 2014, 8, 932–950. [Google Scholar] [CrossRef]
- Carleo, A.; Bargagli, E.; Landi, C.; Bennett, D.; Bianchi, L.; Gagliardi, A.; Carnemolla, C.; Perari, M.G.; Cillis, G.; Armini, A.; et al. Comparative proteomic analysis of bronchoalveolar lavage of familial and sporadic cases of idiopathic pulmonary fibrosis. J. Breath Res. 2016, 10, 026007. [Google Scholar] [CrossRef]
- Carleo, A.; Landi, C.; Prasse, A.; Bergantini, L.; D’Alessandro, M.; Cameli, P.; Janciauskiene, S.; Rottoli, P.; Bini, L.; Bargagli, E. Proteomic characterization of idiopathic pulmonary fibrosis patients: Stable versus acute exacerbation. Monaldi Arch. Chest Dis. 2020, 30, 90. [Google Scholar] [CrossRef]
- Rottoli, P.; Bargagli, E.; Landi, C.; Magi, B. Proteomic analysis in interstitial lung diseases: A review. Curr. Opin. Pulm. Med. 2009, 15, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Wen, Z.; Wang, R.; Luo, W.; Du, Y.; Wang, W.; Chen, X. Identification of the lipid biomarkers from plasma in idiopathic pulmonary fibrosis by Lipidomics. BMC Pulm. Med. 2017, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-Y.; Cheng, X.; Lin, R.-C. Lipidomics applications for discovering biomarkers of diseases in clinical chemistry. Int. Rev. Cell Mol. Biol. 2014, 313, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Zehethofer, N.; Bermbach, S.; Hagner, S.; Garn, H.; Müller, J.; Goldmann, T.; Lindner, B.; Schwudke, D.; König, P. Lipid Analysis of Airway Epithelial Cells for Studying Respiratory Diseases. Chromatographia 2015, 78, 403–413. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L. Mass spectrometry strategies for clinical metabolomics and lipidomics in psychiatry, neurology, and neuro-oncology. Neuropsychopharmacology 2014, 39, 24–33. [Google Scholar] [CrossRef]
- Graessler, J.; Schwudke, D.; Schwarz, P.E.H.; Herzog, R.; Shevchenko, A.; Bornstein, S.R. Top-down lipidomics reveals ether lipid deficiency in blood plasma of hypertensive patients. PLoS ONE 2009, 4, e6261. [Google Scholar] [CrossRef]
- Chu, S.G.; Villalba, J.A.; Liang, X.; Xiong, K.; Tsoyi, K.; Ith, B.; Ayaub, E.A.; Tatituri, R.V.; Byers, D.E.; Hsu, F.-F.; et al. Palmitic Acid-Rich High-Fat Diet Exacerbates Experimental Pulmonary Fibrosis by Modulating Endoplasmic Reticulum Stress. Am. J. Respir. Cell Mol. Biol. 2019, 61, 737–746. [Google Scholar] [CrossRef]
- Romero, F.; Summer, R. Protein Folding and the Challenges of Maintaining Endoplasmic Reticulum Proteostasis in Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2017, 14, S410–S413. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Mao, C.; Yang, R.; Yan, B.; Shi, Y.; Liu, X.; Lai, W.; Liu, Y.; Wang, X.; Xiao, D.; et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 2017, 7, 3293–3305. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, X.; He, Q.; Lang, L.; Xu, P.; Wang, P.; Lee, S.C. Resveratrol attenuates diabetes-associated cell centrosome amplification via inhibiting the PKCα-p38 to c-myc/c-jun pathway. Acta Biochim. Biophys. Sin. 2020, 52, 72–83. [Google Scholar] [CrossRef]
- Puglia, M.; Landi, C.; Gagliardi, A.; Breslin, L.; Armini, A.; Brunetti, J.; Pini, A.; Bianchi, L.; Bini, L. The proteome speciation of an immortalized cystic fibrosis cell line: New perspectives on the pathophysiology of the disease. J. Proteom. 2018, 170, 28–42. [Google Scholar] [CrossRef]
- Landi, C.; Luddi, A.; Bianchi, L.; Pannuzzo, G.; Pavone, V.; Piomboni, P.; Bini, L. Proteostasis network alteration in lysosomal storage disorders: Insights from the mouse model of Krabbe disease. J. Neurosci. Res. 2020, 98, 718–733. [Google Scholar] [CrossRef]
- Andrisse, S.; Childress, S.; Ma, Y.; Billings, K.; Chen, Y.; Xue, P.; Stewart, A.; Sonko, M.L.; Wolfe, A.; Wu, S. Low-Dose Dihydrotestosterone Drives Metabolic Dysfunction via Cytosolic and Nuclear Hepatic Androgen Receptor Mechanisms. Endocrinology 2017, 158, 531–544. [Google Scholar] [CrossRef]
- Hsu, H.-S.; Liu, C.-C.; Lin, J.-H.; Hsu, T.-W.; Hsu, J.-W.; Su, K.; Hung, S.-C. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci. Rep. 2017, 7, 14272. [Google Scholar] [CrossRef]
- Huang, F.; Wang, Q.; Guo, F.; Zhao, Y.; Ji, L.; An, T.; Song, Y.; Liu, Y.; He, Y.; Qin, G. FoxO1-mediated inhibition of STAT1 alleviates tubulointerstitial fibrosis and tubule apoptosis in diabetic kidney disease. EBioMedicine 2019, 48, 491–504. [Google Scholar] [CrossRef]
- Garat, C.V.; Majka, S.M.; Sullivan, T.M.; Crossno, J.T.; Reusch, J.E.B.; Klemm, D.J. CREB depletion in smooth muscle cells promotes medial thickening, adventitial fibrosis and elicits pulmonary hypertension. Pulm. Circ. 2020, 10, 2045894019898374. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, H.; Geng, Y.; Xu, D.; Zhang, L.; Yang, Y.; Wei, Z.; Zhang, B.; Li, S.; Gao, X.; et al. Dibutyryl-cAMP attenuates pulmonary fibrosis by blocking myofibroblast differentiation via PKA/CREB/CBP signaling in rats with silicosis. Respir. Res. 2017, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Eijgelaar, W.J.; Daemen, M.J.A.P.; Newby, A.C. Foam Cell Formation In Vivo Converts Macrophages to a Pro-Fibrotic Phenotype. PLoS ONE 2015, 10, e0128163. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.C.; Eijgelaar, W.J.; Daemen, M.J.A.P.; Newby, A.C. The pro-fibrotic and anti-inflammatory foam cell macrophage paradox. Genom. Data 2015, 6, 136–138. [Google Scholar] [CrossRef]
- Landi, C.; Bargagli, E.; Carleo, A.; Refini, R.M.; Bennett, D.; Bianchi, L.; Cillis, G.; Prasse, A.; Bini, L.; Rottoli, P. Bronchoalveolar lavage proteomic analysis in pulmonary fibrosis associated with systemic sclerosis: S100A6 and 14-3-3ε as potential biomarkers. Rheumatology 2019, 58, 165–178. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharmacother. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Deschepper, C.F. Angiotensinogen: Hormonal regulation and relative importance in the generation of angiotensin II. Kidney Int. 1994, 46, 1561–1563. [Google Scholar] [CrossRef]
- Figueiredo, V.P.; Barbosa, M.A.; de Castro, U.G.M.; Zacarias, A.C.; Bezerra, F.S.; de Sá, R.G.; de Lima, W.G.; Dos Santos, R.A.S.; Alzamora, A.C. Antioxidant Effects of Oral Ang-(1-7) Restore Insulin Pathway and RAS Components Ameliorating Cardiometabolic Disturbances in Rats. Oxid. Med. Cell Longev. 2019, 2019, 5868935. [Google Scholar] [CrossRef]
- Abdul-Hafez, A.; Mohamed, T.; Omar, H.; Shemis, M.; Uhal, B.D. The renin angiotensin system in liver and lung: Impact and therapeutic potential in organ fibrosis. J. Lung Pulm. Respir. Res. 2018, 5, 00160. [Google Scholar]
- Li, Y.; Yan, Z.; Chaudhry, K.; Kazlauskas, A. The Renin-Angiotensin-Aldosterone System (RAAS) Is One of the Effectors by Which Vascular Endothelial Growth Factor (VEGF)/Anti-VEGF Controls the Endothelial Cell Barrier. Am. J. Pathol. 2020, 23. [Google Scholar] [CrossRef]
- Landi, C.; Bergantini, L.; Cameli, P.; d’Alessandro, M.; Carleo, A.; Shaba, E.; Rottoli, P.; Bini, L.; Bargagli, E. Idiopathic Pulmonary Fibrosis Serum proteomic analysis before and after nintedanib therapy. Sci. Rep. 2020, 10, 9378. [Google Scholar] [CrossRef] [PubMed]
- Krskova, K.; Filipcik, P.; Zilka, N.; Olszanecki, R.; Korbut, R.; Gajdosechova, L.; Zorad, S. Angiotensinogen and angiotensin-converting enzyme mRNA decrease and AT1 receptor mRNA and protein increase in epididymal fat tissue accompany age-induced elevation of adiposity and reductions in expression of GLUT4 and peroxisome proliferator-activated receptor (PPARγ). J. Physiol. Pharmacol. 2011, 62, 403–410. [Google Scholar] [PubMed]
- Dalan, R.; Bornstein, S.R.; El-Armouche, A.; Rodionov, R.N.; Markov, A.; Wielockx, B.; Beuschlein, F.; Boehm, B.O. The ACE-2 in COVID-19: Foe or Friend? Horm. Metab. Res. 2020, 52, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Yatabe, J.; Yoneda, M.; Yatabe, M.S.; Watanabe, T.; Felder, R.A.; Jose, P.A.; Sanada, H. Angiotensin III stimulates aldosterone secretion from adrenal gland partially via angiotensin II type 2 receptor but not angiotensin II type 1 receptor. Endocrinology 2011, 152, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Petkova, D.K.; Clelland, C.A.; Ronan, J.E.; Lewis, S.; Knox, A.J. Reduced expression of cyclooxygenase (COX) in idiopathic pulmonary fibrosis and sarcoidosis. Histopathology 2003, 43, 381–386. [Google Scholar] [CrossRef]
- Wright, W.R.; Kirkby, N.S.; Galloway-Phillipps, N.A.; Reed, D.M.; Paul-Clark, M.J.; Mitchell, J.A. Cyclooxygenase and cytokine regulation in lung fibroblasts activated with viral versus bacterial pathogen associated molecular patterns. Prostaglandins Other Lipid Mediat. 2013, 107, 4–12. [Google Scholar] [CrossRef]
- Balakumar, P.; Sambathkumar, R.; Mahadevan, N.; Muhsinah, A.B.; Alsayari, A.; Venkateswaramurthy, N.; Jagadeesh, G. A potential role of the renin-angiotensin-aldosterone system in epithelial-to-mesenchymal transition-induced renal abnormalities: Mechanisms and therapeutic implications. Pharmacol. Res. 2019, 146, 104314. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, H.; Zhang, J.; Xie, C.; Fan, C.; Zhang, H.; Wu, P.; Wei, Q.; Tan, W.; Xu, L.; et al. Inflammation and Fibrosis in Perirenal Adipose Tissue of Patients With Aldosterone-Producing Adenoma. Endocrinology 2018, 159, 227–237. [Google Scholar] [CrossRef]
- Ramalingam, L.; Menikdiwela, K.; LeMieux, M.; Dufour, J.M.; Kaur, G.; Kalupahana, N.; Moustaid-Moussa, N. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1106–1114. [Google Scholar] [CrossRef]
- Huang, H.; Fan, Y.; Gao, Z.; Wang, W.; Shao, N.; Zhang, L.; Yang, Y.; Zhu, W.; Chen, Z.; Hu, J.; et al. HIF-1α contributes to Ang II-induced inflammatory cytokine production in podocytes. BMC Pharmacol. Toxicol. 2019, 20, 59. [Google Scholar] [CrossRef]
- Contreras-Lopez, R.; Elizondo-Vega, R.; Paredes, M.J.; Luque-Campos, N.; Torres, M.J.; Tejedor, G.; Vega-Letter, A.M.; Figueroa-Valdés, A.; Pradenas, C.; Oyarce, K.; et al. HIF1α-dependent metabolic reprogramming governs mesenchymal stem/stromal cell immunoregulatory functions. FASEB J. 2020, 34, 8250–8264. [Google Scholar] [CrossRef] [PubMed]
- Dabral, S.; Muecke, C.; Valasarajan, C.; Schmoranzer, M.; Wietelmann, A.; Semenza, G.L.; Meister, M.; Muley, T.; Seeger-Nukpezah, T.; Samakovlis, C.; et al. A RASSF1A-HIF1α loop drives Warburg effect in cancer and pulmonary hypertension. Nat. Commun. 2019, 10, 2130. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell. Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.-F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef]
- Stock, C.J.W.; Michaeloudes, C.; Leoni, P.; Durham, A.L.; Mumby, S.; Wells, A.U.; Chung, K.F.; Adcock, I.M.; Renzoni, E.A.; Lindahl, G.E. Bromodomain and Extraterminal (BET) Protein Inhibition Restores Redox Balance and Inhibits Myofibroblast Activation. BioMed Res. Int. 2019, 2019, 1484736. [Google Scholar] [CrossRef]
- Zhang, S.; Yin, Z.; Qin, W.; Ma, X.; Zhang, Y.; Liu, E.; Chu, Y. Pirfenidone Inhibits Hypoxic Pulmonary Hypertension through the NADPH/ROS/p38 Pathway in Adventitial Fibroblasts in the Pulmonary Artery. Mediat. Inflamm. 2020, 2020, 2604967. [Google Scholar] [CrossRef]
- Cruz, F.F.; Rocco, P.R.M. The potential of mesenchymal stem cell therapy for chronic lung disease. Expert Rev. Respir. Med. 2020, 14, 31–39. [Google Scholar] [CrossRef]
- Tsitoura, E.; Vasarmidi, E.; Bibaki, E.; Trachalaki, A.; Koutoulaki, C.; Papastratigakis, G.; Papadogiorgaki, S.; Chalepakis, G.; Tzanakis, N.; Antoniou, K.M. Accumulation of damaged mitochondria in alveolar macrophages with reduced OXPHOS related gene expression in IPF. Respir. Res. 2019, 20, 264. [Google Scholar] [CrossRef]
- Sangiuolo, F.; Puxeddu, E.; Pezzuto, G.; Cavalli, F.; Longo, G.; Comandini, A.; Di Pierro, D.; Pallante, M.; Sergiacomi, G.; Simonetti, G.; et al. HFE gene variants and iron-induced oxygen radical generation in idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 483–490. [Google Scholar] [CrossRef]
- Lee, J.; Arisi, I.; Puxeddu, E.; Mramba, L.K.; Amicosante, M.; Swaisgood, C.M.; Pallante, M.; Brantly, M.L.; Sköld, C.M.; Saltini, C. Bronchoalveolar lavage (BAL) cells in idiopathic pulmonary fibrosis express a complex pro-inflammatory, pro-repair, angiogenic activation pattern, likely associated with macrophage iron accumulation. PLoS ONE 2018, 13, e0194803. [Google Scholar] [CrossRef]
- Liu, G.; Ding, M.; Chiuve, S.E.; Rimm, E.B.; Franks, P.W.; Meigs, J.B.; Hu, F.B.; Sun, Q. Plasma Levels of Fatty Acid-Binding Protein 4, Retinol-Binding Protein 4, High-Molecular-Weight Adiponectin, and Cardiovascular Mortality Among Men With Type 2 Diabetes: A 22-Year Prospective Study. Arter. Thromb. Vasc. Biol. 2016, 36, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Goh, G.Y.S.; Winter, J.J.; Bhanshali, F.; Doering, K.R.S.; Lai, R.; Lee, K.; Veal, E.A.; Taubert, S. NHR-49/HNF4 integrates regulation of fatty acid metabolism with a protective transcriptional response to oxidative stress and fasting. Aging Cell 2018, 17, e12743. [Google Scholar] [CrossRef] [PubMed]
- Rosell, M.; Hondares, E.; Iwamoto, S.; Gonzalez, F.J.; Wabitsch, M.; Staels, B.; Olmos, Y.; Monsalve, M.; Giralt, M.; Iglesias, R.; et al. Peroxisome proliferator-activated receptors-α and -γ, and cAMP-mediated pathways, control retinol-binding protein-4 gene expression in brown adipose tissue. Endocrinology 2012, 153, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Noy, N. Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim. Biophys. Acta 2012, 1821, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT—Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef]
- Lescoat, A.; Lelong, M.; Jeljeli, M.; Piquet-Pellorce, C.; Morzadec, C.; Ballerie, A.; Jouneau, S.; Jego, P.; Vernhet, L.; Batteux, F.; et al. Combined anti-fibrotic and anti-inflammatory properties of JAK-inhibitors on macrophages in vitro and in vivo: Perspectives for scleroderma-associated interstitial lung disease. Biochem. Pharmacol. 2020, 178, 114103. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Perillo, F.; Metella Refini, R.; Bergantini, L.; Bellisai, F.; Selvi, E.; Cameli, P.; Manganelli, S.; Conticini, E.; Cantarini, L.; et al. Efficacy of baricitinib in treating rheumatoid arthritis: Modulatory effects on fibrotic and inflammatory biomarkers in a real-life setting. Int. Immunopharmacol. 2020, 86, 106748. [Google Scholar] [CrossRef]
- Korman, B.; Marangoni, R.G.; Lord, G.; Olefsky, J.; Tourtellotte, W.; Varga, J. Adipocyte-specific Repression of PPAR-gamma by NCoR Contributes to Scleroderma Skin Fibrosis. Arthritis Res. Ther. 2018, 20, 145. [Google Scholar] [CrossRef]
- Kheirollahi, V.; Wasnick, R.M.; Biasin, V.; Vazquez-Armendariz, A.I.; Chu, X.; Moiseenko, A.; Weiss, A.; Wilhelm, J.; Zhang, J.-S.; Kwapiszewska, G.; et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat. Commun. 2019, 10, 2987. [Google Scholar] [CrossRef]
- Spagnolo, P.; Kreuter, M.; Maher, T.M.; Wuyts, W.; Bonella, F.; Corte, T.J.; Kopf, S.; Weycker, D.; Kirchgaessler, K.-U.; Ryerson, C.J. Metformin Does Not Affect Clinically Relevant Outcomes in Patients with Idiopathic Pulmonary Fibrosis. Respiration 2018, 96, 314–322. [Google Scholar] [CrossRef]
- MacKenzie, B.; Korfei, M.; Henneke, I.; Sibinska, Z.; Tian, X.; Hezel, S.; Dilai, S.; Wasnick, R.; Schneider, B.; Wilhelm, J.; et al. Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Suh, J.M.; Atkins, A.R.; Ahmadian, M.; Li, P.; Whyte, J.; He, M.; Juguilon, H.; Yin, Y.-Q.; Phillips, C.T.; et al. A PPARγ-FGF1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature 2012, 485, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Rehan, V.K.; Torday, J.S. The lung alveolar lipofibroblast: An evolutionary strategy against neonatal hyperoxic lung injury. Antioxid. Redox Signal. 2014, 21, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Walsh, K. Obesity, adiponectin and vascular inflammatory disease. Curr. Opin. Lipidol. 2003, 14, 561–566. [Google Scholar] [CrossRef]
- Guo, M.; Li, C.; Lei, Y.; Xu, S.; Zhao, D.; Lu, X.-Y. Role of the adipose PPARγ-adiponectin axis in susceptibility to stress and depression/anxiety-related behaviors. Mol. Psychiatry 2017, 22, 1056–1068. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Bergantini, L.; Cameli, P.; Lanzarone, N.; Perillo, F.; Perrone, A.; Bargagli, E. BAL and serum multiplex lipid profiling in idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Life Sci. 2020, 20. [Google Scholar] [CrossRef]
- Vansaun, M.N. Molecular pathways: Adiponectin and leptin signaling in cancer. Clin. Cancer Res. 2013, 19, 1926–1932. [Google Scholar] [CrossRef]
- Gui, X.; Chen, H.; Cai, H.; Sun, L.; Gu, L. Leptin promotes pulmonary fibrosis development by inhibiting autophagy via PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 498, 660–666. [Google Scholar] [CrossRef]
- Venosa, A.; Smith, L.C.; Murray, A.; Banota, T.; Gow, A.J.; Laskin, J.D.; Laskin, D.L. Regulation of Macrophage Foam Cell Formation During Nitrogen Mustard (NM)-Induced Pulmonary Fibrosis by Lung Lipids. Toxicol. Sci. 2019, 172, 344–358. [Google Scholar] [CrossRef]
- Chung, K.-P.; Hsu, C.-L.; Fan, L.-C.; Huang, Z.; Bhatia, D.; Chen, Y.-J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.C.; Thomas, M.C. Angiotensin II and Its Role in Tubular Epithelial to Mesenchymal Transition Associated with Chronic Kidney Disease. Cells Tissues Organs 2011, 193, 74–84. [Google Scholar] [CrossRef]
- de Gómez Dumm, N.T.; Giammona, A.M.; Touceda, L.A.; Raimondi, C. Lipid abnormalities in chronic renal failure patients undergoing hemodialysis. Medicina 2001, 61, 142–146. [Google Scholar]
- Rottoli, P.; Magi, B.; Cianti, R.; Bargagli, E.; Vagaggini, C.; Nikiforakis, N.; Pallini, V.; Bini, L. Carbonylated proteins in bronchoalveolar lavage of patients with sarcoidosis, pulmonary fibrosis associated with systemic sclerosis and idiopathic pulmonary fibrosis. Proteomics 2005, 5, 2612–2618. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Javad-Moosavi, S.A.; Reiter, R.J.; Yarahmadi, R.; Ghaznavi, H.; Mehrzadi, S. Oxidative/nitrosative stress, autophagy and apoptosis as therapeutic targets of melatonin in idiopathic pulmonary fibrosis. Expert Opin. Ther. Targets 2018, 22, 1049–1061. [Google Scholar] [CrossRef]
- Fessler, M.B.; Summer, R.S. Surfactant Lipids at the Host-Environment Interface. Metabolic Sensors, Suppressors, and Effectors of Inflammatory Lung Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 624–635. [Google Scholar] [CrossRef]
- Castelino, F.V.; Dellaripa, P.F. Recent progress in systemic sclerosis-interstitial lung disease. Curr. Opin. Rheumatol. 2018, 30, 570–575. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathways Dysregulation in IPF | Dysregulated Proteins Found by Proteomics | Influencing or Influenced Mechanisms | Diseases and Effects | References |
|---|---|---|---|---|
| Renin-angiotensin-aldosterone system (RAAS) | ANGT | ACE-2/Ang 1-7/Mas axis vs. ACE/Ang II/AT1-R Adipocyte modulation Small insulin-sensitive adipocytes modulation AngII has intracrine/autocrine/paracrine roles Secretion of aldosteron Release of cyclooxygenase (COX) 1-derived prostaglandin E(2) Hormone imbalance | Metabolic syndrome Diabetes mellitus type 2 Lung fibrosis Vascular dysfunction Myocardial fibrosis Nephropathy Hypertension Inflammation by IL-6 and TNF-α Fibrosis by TGF-β. | [46,47,48,49] [50,51,52,53] [54,55,56,57,58] |
| Hypoxia, oxidative stress and iron metabolism | HPT, TRFE, PRDX1 PRDX5, GSTP1, CERU, ALBU | RAAS increase oxidative stress HIF-1α and HIF-1β determine transcription of HREs HREs modify glycolysis HREs modify angiogenesis HREs modify pro-survival signalling, cell proliferation and cell migration HIF-1α promotes TGF-β-induced myofibroblast differentiation Extra Cellular Matrix (ECM) deposition Deregulation of mitochondrial oxidative phosphorylation (OXPHOS) Excessive extracellular iron and macrophage haemosiderin | Aging Warburg effect Recurring microscopic injury and fibrosing damage Worse fibrosis evolution | [59,60] [61,62,63,64,65] [66,67,68,69,70] |
| Lipid metabolism dysregulation | FABP4, RBP4, HP, APOAI, ZA2G, APOC3, S100A8 TRFE, C3, PEDF, A1AT A1BG, ALBU, SFPA2, SFTPD | PPAR- ϒ modulation FXR modulation LXR modulation Modulation of JAK/STAT pathway Modulation of adipogenesis and fibrosis by TGF-β | Obesity Metabolic diseases Skin, lung and heart fibrosis Changes in lipid metabolism Regulation of glucose metabolism and insulin resistance | [71,72,73] [74,75,76,77,78] [79,80,81,82,83] [84,85,86,87,88] [20,51,89,90,91,92] |
| Mitochondrial alterations | FABP4, RBP4, HP, APOAI, ZA2G, APOC3, S100A8 TRFE, C3, PEDF, A1AT A1BG, ALBU, SFPA2, SFTPD | Oxidative phosphorylation Mitochondrial DNA biogenesis Mitophagy regulation Metabolic adaptation Synthesis of fatty acid | Insulin-resistance Defective autophagy Telomere attrition Altered proteostasis Cell senescence Increased production of reactive oxygen species | [93,94,95,96] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bargagli, E.; Refini, R.M.; d’Alessandro, M.; Bergantini, L.; Cameli, P.; Vantaggiato, L.; Bini, L.; Landi, C. Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 5663. https://doi.org/10.3390/ijms21165663
Bargagli E, Refini RM, d’Alessandro M, Bergantini L, Cameli P, Vantaggiato L, Bini L, Landi C. Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences. 2020; 21(16):5663. https://doi.org/10.3390/ijms21165663
Chicago/Turabian StyleBargagli, Elena, Rosa Metella Refini, Miriana d’Alessandro, Laura Bergantini, Paolo Cameli, Lorenza Vantaggiato, Luca Bini, and Claudia Landi. 2020. "Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis" International Journal of Molecular Sciences 21, no. 16: 5663. https://doi.org/10.3390/ijms21165663
APA StyleBargagli, E., Refini, R. M., d’Alessandro, M., Bergantini, L., Cameli, P., Vantaggiato, L., Bini, L., & Landi, C. (2020). Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis. International Journal of Molecular Sciences, 21(16), 5663. https://doi.org/10.3390/ijms21165663