Self-Amplifying RNA Viruses as RNA Vaccines

Abstract
1. Introduction
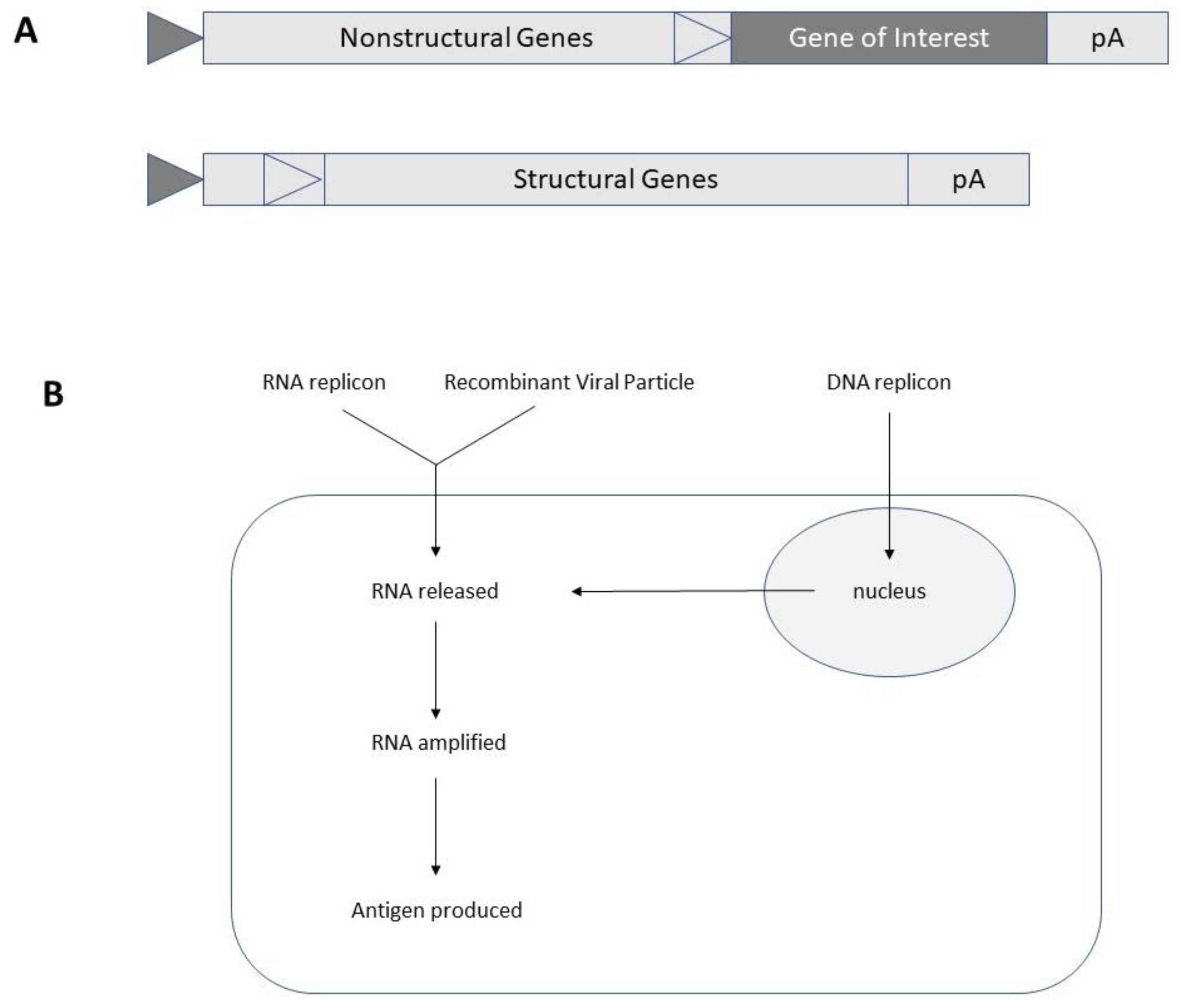
2. Self-Amplifying RNA Virus-Based Expression
3. Vaccines Against Infectious Diseases
4. Vaccines Against Cancer
5. Clinical Trials
6. Conclusions
Funding
Conflicts of Interest
References
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines – a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Schnee, M.; Vogel, A.B.; Voss, D.; Petsch, B.; Baumhof, P.; Kramps, T.; Stitz, L. An mRNA vaccine encoding rabies virus glycoprotein induces protection against lethal infection in mice and correlates of protection in adult and newborn pigs. PLoS Negl. Trop. Dis. 2016, 10, e0004746. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Ö.; Thompson, J.; et al. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Bacckert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Lundstrom, K. Self-amplifying RNA virus vectors: Clinical applications in cancer drug delivery. Exp. Opin Drug Deliv. 2019, 16, 1027–1029. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The Alphaviruses: Gene Expression, Replication and Evolution. Micobiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Banerjee, A.K. Transcription and Replication of Rhabdoviruses. Microbiol. Rev. 1987, 51, 66–87. [Google Scholar] [CrossRef]
- Lundstrom, K. RNA Viruses as Tools in Gene Therapy and Vaccine Development. Genes 2018, 10, E189. [Google Scholar] [CrossRef]
- Liljestrom, P.; Garoff, H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology 1991, 9, 1356–1361. [Google Scholar] [CrossRef]
- Xiong, C.; Levis, R.; Shen, P.; Schlesinger, S.; Rice, C.M.; Huang, H.V. Sindbis virus: An efficient, broad host range vector for gene expression in animal cells. Science 1989, 243, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.L.; Willis, L.V.; Smith, J.F.; Johnston, R.F. In vitro synthesis of infectious Venezuelan equine encephalitis virus RNA from a cDNA clone: Analysis of a viable deletion mutant. Virology 1989, 171, 189–204. [Google Scholar] [CrossRef]
- DiCiommo, D.P.; Bremner, R. Rapid, high level protein production using DNA-based Semliki Forest virus vectors. J. Biol. Chem. 1998, 273, 18060–18066. [Google Scholar] [CrossRef] [PubMed]
- White, C.L.; Thomson, M.; Dimmock, N.J. Deletion analysis of a defective interfering Semliki Forest virus RNA genome defines a region in the nsP2 sequence that is required for efficient packaging of the genome into viral particles. J. Virol. 1998, 72, 4320–4326. [Google Scholar] [CrossRef] [PubMed]
- Berglund, P.; Sjöberg, M.; Garoff, H.; Atkins, G.J.; Sheahan, B.J.; Liljeström, P. Semliki Forest virus expression system: Production of conditionally infectious recombinant particles. Biotechnology 1993, 11, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Smerdou, C.; Liljeström, P. Two-helper system for production of recombinant Semliki Forest virus particles. J. Virol. 1999, 73, 1092–1098. [Google Scholar] [CrossRef]
- Pijlman, G.P.; Suhrbier, A.; Khromykh, A.A. Kunjin virus replicons: An RNA-based, non-cytopathic viral vector system for protein production, vaccine and gene therapy applications. Exp. Opin Biol Ther. 2006, 6, 134–145. [Google Scholar] [CrossRef]
- De Felipe, F. Skipping the co-expression problem: The new 2A ‘CHYSEL’ technology. Genet. Vaccines Ther. 2004, 2. [Google Scholar] [CrossRef]
- Khromykh, A.A.; Varnavski, A.N.; Westaway, E.G. Encapsidation of the flavivirus Kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J. Virol. 1998, 72, 5967–5977. [Google Scholar] [CrossRef]
- Shi, P.Y.; Tilgner, M.; Lo, M.K. Construction and characterization of subgenomic replicons of New York strain of West Nile virus. Virology 2002, 296, 219–233. [Google Scholar] [CrossRef]
- Scholle, I.; Girard, Y.A.; Zhao, Q.; Higgs, S.; Mason, P.W. Trans-packaged West Nile virus-like particles: Infectious properties in vitro and in infected mosquito vectors. J. Virol. 2004, 78, 11605–11614. [Google Scholar] [CrossRef] [PubMed]
- Molenkamp, R.; Kooi, E.A.; Lucassen, M.A.; Greve, S.; Thijssen, J.C.; Spaan, W.J.; Bredenbeek, P.J. Yellow fever virus replicons as an expression system for hepatitis C virus structural proteins. J. Virol. 2003, 77, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.T.; Patkar, C.G.; Kuhn, R.J. Construction and applications of yellow fever virus replicons. Virology 2005, 331, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.; Davidson, A.; Hibbert, L.; Gruenwald, P.; Schlaak, J.; Ball, S.; Foster, G.R.; Jacobs, M. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J. Virol. 2005, 79, 5414–5420. [Google Scholar] [CrossRef]
- Pang, X.; Zhang, M.; Dayton, A.I. Development of dengue virus type 2 replicons capable of prolonged expression in host cells. BMC Microbiol. 2001, 1, 18. [Google Scholar]
- Gherke, R.; Ecker, M.; Aberle, S.W.; Allison, S.L.; Heinz, F.X.; Mandi, C.W. Incorporation of tick-borne encephalitis virus replicons into virus-like particles by a packaging cell line. J. Virol. 2003, 77, 8924–8933. [Google Scholar] [CrossRef]
- Hayasaka, D.; Yoshii, K.; Ueki, T.; Goto, A.; Mizutani, T.; Kariwa, H.; Iwasaki, T.; Gould, E.A.; Takashima, I. Sub-genomic replicons of tick-borne encephalitis virus. Arch. Virol. 2004, 149, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Radecke, F.; Spielhofer, P.; Schneider, H.; Kaelin, K.; Huber, M.; Dötsch, C.; Christiansen, G.; Billeter, M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995, 14, 5773–5784. [Google Scholar] [CrossRef]
- Singh, M.; Cattaneo, R.; Billeter, M.A. A recombinant measles virus expressing hepatitis B surface antigen induces humoral responses in genetically modified mice. J. Virol. 1999, 73, 4823–4828. [Google Scholar] [CrossRef]
- Osakada, F.; Callaway, E.M. Design and generation of recombinant rabies virus vectors. Nat. Protoc. 2013, 8, 1583–1601. [Google Scholar] [CrossRef]
- An, H.; Kim, G.N.; Kang, C.Y. Genetically modified VSV(NJ) vector is capable of accommodating a large foreign gene insert and allows high level gene expression. Virus Res. 2013, 171, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Harty, R.N.; Brown, M.E.; Hayes, F.P.; Wright, N.T.; Schnell, M.J. Vaccinia virus-free recovery of vesicular stomatitis virus. J. Mol. Microbiol Biotechnol. 2001, 3, 513–517. [Google Scholar] [PubMed]
- Ohara, S.; Inoue, K.; Yamada, M.; Yamawaki, T.; Koganezawa, N.; Tsuttsui, K.; Witter, M.P.; Iijima, T. Dual transneural tracing in the rat entorhoinal-hippocampal circuit by intracerebral injection of recombinant rabies virus vectors. Front. Neuroanat. 2009, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Takayama-Ito, M.; Yamada, K.; Hosokawa, J.; Sugiyama, M.; Minamoto, N. Improved recovery of rabies virus from cloned cDNA using a vaccinia virus-free reverse genetics system. Microbiol Immunol. 2003, 47, 613–617. [Google Scholar] [CrossRef]
- Khalil, S.M.; Tonkin, D.R.; Mattocks, M.D.; Snead, A.T.; Johnston, R.E.; White, L.J. A tetravalent alphavirus-vector based dengue vaccine provides effective immunity in an early life mouse model. Vaccine 2014, 32, 4068–4074. [Google Scholar] [CrossRef] [PubMed]
- Harahap-Carrillo, I.S.; Ceballos-Olvera, I.; Valle, J.R. Immunogenic subviral particles displaying domain III of dengue 2 envelope protein vectored by measles virus. Vaccines 2015, 3, 503–518. [Google Scholar] [CrossRef]
- Hu, H.M.; Chen, H.W.; Hsiao, Y.; Wu, S.H.; Chung, H.H.; Hsieh, C.H.; Chong, P.; Leng, C.H.; Pan, C.H. The successful induction of T-cell and antibody responses by a recombinant measles virus-vectored tetravalent dengue vaccine provides partial protection against dengue-2 infection. Hum. Vaccin Immunother. 2016, 12, 1678–1689. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Cage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A nanostructured lipid carrier for delivery of a replicating viral RNA provides single, low-dose protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef]
- Chattopadhyay, A.; Aquilar, P.V.; Bopp, N.E.; Yarovinsky, T.O.; Weaver, S.C.; Rose, J.K. A recombinant virus vaccine that protects both against Chikungunya and Zika virus infections. Vaccine 2018, 36, 3894–3900. [Google Scholar] [CrossRef]
- Pyankov, O.V.; Bodnev, S.A.; Pyankova, O.G.; Solodkyi, V.V.; Pyankov, S.A.; Setoh, Y.X.; Volchokova, V.A.; Suhrbier, A.; Volchikov, V.V.; Agafonov, A.A.; et al. A Kunjin replicon virus-like vaccine provides protection against Ebola virus infection in nonhuman primates. J. Infect. Dis. 2015, 212 (Suppl. S2), S368–S371. [Google Scholar] [CrossRef]
- Marzi, A.; Robertson, S.J.; Haddock, E.; Feldmann, F.; Hanley, P.W.; Scott, D.-P.; Strong, J.E.; Kobinger, G.; Best, S.M.; Feldmann, H. Ebola vaccine. VSV-EBOV rapidly protects macaques against infection with the 2014/2015 Ebola virus outbreak strain. Science 2015, 349, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Feldmann, H. Recombinant vesicular stomatitis virus-based vaccines against Ebola and Marburg infections. J. Infect. Dis. 2011, 204 (Suppl. S3), S1075–S1081. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.S.; Kuehne, A.I.; Barth, J.F.; Ortiz, R.A.; Nichols, D.K.; Zak, S.E.; Stonier, S.W.; Muhammad, M.A.; Bakken, R.R.; Prugar, L.I.; et al. Venezuelan equine encephalitis virus replicon particle vaccine protects nonhuman primates from intramuscular and aerosol challenge with ebolavirus. J. Virol. 2013, 87, 4852–4964. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.A.; Hart, M.K. Protection from Ebola virus mediated by cytotoxic T-lymphocytes specific for the viral nucleoprotein. J. Virol. 2001, 75, 2660–2664. [Google Scholar] [CrossRef] [PubMed]
- Pushko, P.; Bray, M.; Ludwig, G.V.; Parker, M.; Schmaljohn, A.; Sanchez, A.; Jahrling, P.B.; Smith, J.F. Recombinant RNA replicons derived from attenuated Venezuelan equine encephalitis virus protect guinea pigs and mice from Ebola hemorrhagic fever virus. Vaccine 2000, 19, 142–153. [Google Scholar] [CrossRef]
- Öhlund, P.; Garcia-Arriaza, J.; Zusinaite, E.; Szurgot, I.; Mànnik, A.; Kraus, A.; Ustav, M.; Merits, A.; Esteban, M.; Liljestrom, P.; et al. DNA-launched RNA replicon vaccines induce potent anti-Ebola immune responses that can be further improved by a recombinant MVA boost. Sci. Rep. 2018, 8, 12459. [Google Scholar] [CrossRef]
- Safronetz, D.; Mire, C.; Rosenke, K.; Feldmann, F.; Haddock, E.; Geissbert, T.; Feldmann, H. A recombinant vesicular stomatitis virus-based Lassa fever vaccine protects guinea pigs and macaques against challenge with geographically and genetically distinct Lassa viruses. PLoS Negl. Trop. Dis. 2015, 9, e0003736. [Google Scholar] [CrossRef]
- Wang, M.; Jokinen, J.; Tretvakova, I.; Pushko, P.; Lukashevich, I.S. Alphavirus vector-based replicon particles expressing multivalent cross-protective Lassa virus glycoproteins. Vaccine 2018, 36, 683–690. [Google Scholar] [CrossRef]
- Kainulainen, M.H.; Spengler, J.R.; Welch, S.R.; Coleman-McCray, J.D.; Harmon, J.R.; Klena, J.D.; Nichol, S.T.; Albarino, C.G.; Spiropoulou, C.F. Use of a scalable replicon-particle vaccine to protect against lethal Lassa virus infection in the guinea pig model. J. Infect. Dis. 2018, 217, 1957–1966. [Google Scholar] [CrossRef]
- Johnson, D.M.; Jokinen, J.D.; Wang, M.; Pfeiffer, T.; Tretyakova, I.; Carrion, R., Jr.; Griffiths, A.; Pushko, P.; Lukashevich, I.S. Bivalent Junin and Machupo experimental vaccine based on alphavirus RNA replicon vector. Vaccine 2020, 38, 2949–2959. [Google Scholar] [CrossRef]
- Brand, D.; Lemiale, F.; Turbica, I.; Buzelay, L.; Brunet, S.; Barin, F. Comparative analysis of humoral immune responses to HIV type 1 envelope glycoproteins in mice immunized with a DNA vaccine, recombinant Semliki Forest virus RNA, or recombinant Semliki Forest virus particles. AIDS Res. Hum. Retroviruses 1998, 14, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Giraud, A.; Ataman-Onal, Y.; Battail, N. Generation of monoclonal antibodies to native human immunodeficiency virus type 1 envelope glycoprotein by immunization of mice with naked RNA. J. Virol. Methods 1999, 79, 75–84. [Google Scholar] [CrossRef]
- Ajbani, S.P.; Velhal, S.M.; Kadam, R.B.; Patel, V.V.; Lundstrom, K.; Bandivdekar, A.H. Immunogenicity of virus-like Semliki Forest virus replicon particles expressing Indian HIV-1C gag, env and pol RT genes. Immunol Lett. 2017, 190, 221–232. [Google Scholar] [CrossRef]
- Knudsen, M.L.; Ljungberg, K.; Tatoud, R.; Weber, J.; Esteban, M.; Liljestrom, P. Alphavirus replicon DNA expressing HIV antigens is an excellent prime for boosting with recombinant modified vaccinia Ankara (MVA) or with HIV gp140 protein antigen. PLoS ONE 2015, 10, e0117042. [Google Scholar] [CrossRef] [PubMed]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Bannerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic emulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.; Porter, E.; Zhang, Y.; Silva, M.; Li, N.; Dobosh, B.; Liquori, A.; Skog, P.; Landais, E.; Menis, S. Immunogenicity of RNA Replicons Encoding HIV Env Immunogens Designed for Self-Assembly into Nanoparticles. Mol. Ther. 2019, 27, 2080–2090. [Google Scholar] [CrossRef]
- Anraku, I.; Mokhonov, V.V.; Rattanasena, P.; Mokhonova, E.I.; Leung, J.; Pijlman, G.; Cara, A.; Schroder, W.A.; Khromykh, A.A.; Suhrbier, A. Kunjin replicon-based simian immunodeficiency virus gag vaccines. Vaccine 2008, 26, 3268–3276. [Google Scholar] [CrossRef]
- Nilsson, C.; Mäkitalo, B.; Berglund, P.; Bex, F.; Liljeström, P.; Sutter, G. Enhanced simian immunodeficiency virus-specific immune responses in macaques induced by priming with recombinant Semliki Forest virus and boosting with modified vaccinia virus Ankara. Vaccine 2001, 19, 3526–3536. [Google Scholar] [CrossRef]
- Gambhira, R.; Keele, B.F.; Schell, J.B.; Hunter, M.J.; Dufour, J.P.; Montefiori, D.C.; Tang, H.; Rose, J.K.; Rose, N.; Marx, P.A. Transmitter/founder simian immunodeficiency virus envelope sequences in vesicular stomatitis and Semliki Forest virus vector immunized rhesus macaques. PLoS ONE 2014, 9, e109678. [Google Scholar] [CrossRef]
- Malone, J.G.; Berglund, P.J.; Liljestrom, P.; Rhodes, G.H.; Malone, R.W. Mucosal immune responses associated with polynucleotide vaccination. Behring Inst. Mitt. 1997, 98, 63–72. [Google Scholar]
- Schultz-Cherry, S.; Dybing, J.K.; Davis, N.L.; Williamson, C.; Suarez, D.L.; Johnston, R.; Perdue, M.L. Influenza virus (A/HK/156/97) hemagglutinin expressed by an alphavirus replicon system protects against lethal infection with Hong Kong-origin H5N1 viruses. Virology 2000, 278, 55–59. [Google Scholar] [CrossRef]
- Fleeton, M.N.; Chen, M.; Berglund, P.; Rhodes, G.; Parker, S.E.; Murphy, M.; Atkins, G.J.; Liljestrom, P. Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J. Infect. Dis. 2001, 183, 1395–1398. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reufer, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef]
- Khalil, S.M.; Tonkin, D.R.; Snead, A.T.; Parks, G.D.; Johnston, R.E.; White, L.J. An alphavirus-based adjuvant enhances serum and mucosal antibodies, T cells and protective immunity to influenza virus in neonatal mice. J. Virol. 2014, 88, 9182–9196. [Google Scholar] [CrossRef]
- Englezou, P.C.; Sapet, C.; Démoulins, T.; Milona, P.; Ebensen, T.; Schulze, K. Self-amplifying replicon RNA delivery to dendritic cells by cationic lipids. Mol. Ther Nucl Acids 2018, 12, 118–134. [Google Scholar] [CrossRef]
- Deming, D.; Sheahan, T.; Heise, M.; Yount, B.; Davis, N.; Sims, A.; Suthar, M.; Harkema, J.; Whitmore, A.; Pickles, R.; et al. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 2006, 3, e525. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.; Whitmore, A.; Long, K.; Ferris, M.; Rockx, B.; Funkhouser, B.; Donaldson, E.; Gralinski, L.; Collier, M.; Heise, M.; et al. Successful vaccination strategies that protect aged mice from lethal challenge from influenza virus and heterologous severe acute respiratory syndrome coronavirus. J. Virol. 2011, 85, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Malczyk, A.H.; Kupke, A.; Prüfer, S.; Scheuplein, V.A.; Hutzler, S.; Kreuz, D. A highly immunogenic and protective Middle East respiratory syndrome coronavirus vaccine based on a recombinant measles virus vaccine platform. J. Virol. 2015, 89, 11654–11667. [Google Scholar] [CrossRef]
- Del Valle, J.R.; Devaux, P.; Hodge, G.; Wegner, N.J.; McChesney, M.B.; Cattaneo, R. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against virus challenge. J. Virol. 2007, 81, 10597–10605. [Google Scholar] [CrossRef]
- Reynolds, T.D.; Buonocore, L.; Rose, N.F.; Rose, J.K.; Robek, M.D. Virus-like vesicle-based therapeutic vaccine vectors for chronic hepatis B virus infection. J. Virol. 2015, 89, 10407–10415. [Google Scholar] [CrossRef]
- Niedre-Otomere, B.; Bogdanova, A.; Skrastina, D.; Zajakina, A.; Bruvere, R.; Ose, V.; Gerlich, W.H.; Garoff, H.; Pumpens, P.; Glebe, D.; et al. Recombinant Semliki Forest virus vectors encoding hepatitis B small surface and pre-S1 antigens induced broadly reactive neutralizing antibodies. J. Viral Hepat. 2012, 19, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Reed, D.S.; Glass, P.J.; Bakken, R.R.; Barth, J.F.; Lind, C.M.; da Silva, L.; Hart, M.K.; Rayner, J.; Alterson, K.; Custer, M.; et al. Combined alphavirus replicon particle vaccine induces durable and cross-protective immune responses against equine encephalitis virus. J. Virol. 2014, 88, 12077–12086. [Google Scholar] [CrossRef] [PubMed]
- Tretyakova, I.; Tibbens, A.; Jokinen, J.D.; Johnson, D.M.; Lukashevich, J.S.; Pushko, P. Novel DNA-launched Venezuelan equine encephalitis virus vaccine with rearranged genome. Vaccine 2019, 37, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Tretyakova, I.; Plante, K.S.; Rossi, S.L.; Lawrence, W.S.; Peel, J.E.; Gudjohnsen, S.; Wang, E.; Mirchandani, D.; Tibbens, A.; Lamichhane, T.N. Venezuelan equine encephalitis vaccine with rearranged genome resists reversion and protects non-human primates from viremia after aerosol challenge. Vaccine 2020, 38, 3378–3386. [Google Scholar] [CrossRef]
- Kaplan, E.H. Containing 2019-nCoV (Wuhan) coronavirus. Health Care Manag. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Yang, Y.; Peng, F.; Wang, R.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronavirus; The 2003 SARS epidemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020, 3, 102434. [Google Scholar] [CrossRef]
- The World Health Organization. Draft landscape of COVID-19 candidate vaccines. Available online: https://www.who.int/who-documents-detail/draft-landscape-of-covid-19-candidate-vaccines (accessed on 27 May 2020).
- Kamrud, K.I.; Custer, M.; Dudek, J.M.; Owens, G.; Alterson, K.D.; Lee, J.S.; Groebner, J.L.; Smith, J.F. Alphavirus replicon approach to promoterless analysis of IRES elements. Virology 2007, 360, 376–387. [Google Scholar] [CrossRef]
- Paraskevakou, G.; Allen, C.; Nakamura, T.; Zollman, P.; James, C.D.; Peng, K.W. Epidermal growth factor receptor (EGFR)retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol. Ther. 2007, 15, 677–686. [Google Scholar] [CrossRef]
- Yamanaka, R.; Zullo, S.A.; Ramsey, J.; Onodera, M.; Tanaka, R.; Blaese, M. Induction of therapeutic antitumor antiangiogenesis by intratumoral injection of genetically engineered endostatin-producing Semliki Forest virus. Cancer Gene Ther. 2001, 8, 796–802. [Google Scholar] [CrossRef]
- Yamanaka, R.; Tsuchiya, N.; Yajima, N.; Honma, J.; Hasegawa, H.; Tanaka, R.; Ramsey, J.; Blasé, R.M.; Xanthopoulos, K.G. Induction of ab antitumor immunological response by an intratumoral injection of dendritic cells pulsed with genetically engineered Semliki Forest virus to produce interleukin-18 combined with the systemic administration of interleukin-12. J. Neurosurg. 2003, 99, 746–753. [Google Scholar] [CrossRef]
- Martikainen, M.; Niittykoski, M.; von und zu Frauenberg, M.; Immonen, A.; Koponen, S. MicroRNA-attenuated clone of virulent Semliki Forest virus overcomes antiviral type I interferon in resistant mouse CT-2A glioma. J. Virol. 2015, 89, 10637–10647. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mao, G.; van den Pol, A.N. Chikungunya-vesicular stomatitis chimeric virus targets and eliminates brain tumors. Virology 2018, 522, 244–259. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.J.; Erlichman, C.; Ingle, J.N.; Rosales, G.A.; Allen, C.; Greiner, S.M. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast Cancer Res. Treat. 2006, 99, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.P.; Rao, X.M.; Price, J.E.; Zhou, H.S.; Lachman, L.B. Prime-boost vaccination with plasmid and adenovirus gene vaccines control HER2/neu+ metastatic breast cancer in mice. Breast Cancer Res. 2005, 7, R580–R588. [Google Scholar] [CrossRef] [PubMed]
- Lachman, L.B.; Rao, X.M.; Kremer, R.H.; Ozpolat, B.; Kirjakova, G.; Price, J.E. DNA vaccination against neu reduces breast cancer incidence and metastasis in mice. Cancer Gene Ther. 2001, 8, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Moran, T.P.; Burgents, J.E.; Long, B. Alphaviral vector-transduced dendritic cells are successful therapeutic vaccines against neu-overexpressing tumors in wild-type mice. Vaccine 2007, 25, 6604–6612. [Google Scholar] [CrossRef]
- Vasilevska, J.; Skrastina, D.; Spunde, K.; Garoff, H.; Kozlovska, T.; Zajakina, A. Semliki Forest virus biodistribution in tumor-free and 4T1 mammary tumor-bearing mice: A comparison of transgene delivery by recombinant virus particles and naked RNA replicon. Cancer Gene Ther. 2012, 19, 579–587. [Google Scholar] [CrossRef]
- Kramer, M.G.; Masner, M.; Casales, E.; Moreno, M.; Smerdou, C.; Chabalgoity, J.A. Neoadjuvant administration of Semliki Forest virus expressing interleukin-12 combined with Salmonella eradicates breast cancer metastasis and achieves long-term survival in immunocompetent mice. BMC Cancer 2015, 15, 620. [Google Scholar] [CrossRef]
- Chikkanna-Gowda, C.P.; Sheahan, B.J.; Fleeton, M.N.; Atkins, G.J. Regression of mouse tumours and inhibition of metastases following administration of a Semliki Forest virus vector with enhanced expression of IL-12. Gene Ther. 2005, 12, 1253–1263. [Google Scholar] [CrossRef]
- Velders, M.P.; McElhiney, S.; Cassetti, M.C.; Eiben, G.L.; Higgins, T.; Kovacs, G.R. Eradication of established tumors by vaccination with Venezuelan equine encephalitis virus replicon particles delivering human papillomavirus 16 E7 RNA. Cancer Res. 2001, 61, 7861–7867. [Google Scholar]
- Daemen, T.; Riezebos-Brilman, A.; Bungener, L.; Regts, J.; Dontje, B.; Wilschut, J. Eradication of established HPV16-transformed tumours after immunisation with recombinant Semliki Forest virus expressing a fusion protein of E6 and E7. Vaccine 2000, 21, 1082–1088. [Google Scholar] [CrossRef]
- Van de Wall, S.; Ljungberg, K.; Ip, P.P.; Boerma, A.; Knudsen, M.L.; Nijman, H.W.; Liljeström, P.; Daemen, T. Potent therapeutic efficacy of an alphavirus replicon DNA vaccine expressing human papilloma virus E6 and E7 antigens. Oncoimmunology 2018, 7, e1487913. [Google Scholar] [CrossRef]
- Draghiciu, O.; Walczak, M.; Hoogeboom, B.N.; Franken, K.L.; Melief, K.J.; Nijman, H.W. Therapeutic immunization and local low-dose tumor irradiation, a reinforcing combination. Int. J. Cancer 2014, 134, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Draghiciu, O.; Boerma, A.; Hoogeboom, B.N.; Nijman, H.W.; Daemen, T. A rationally designed combined treatment with an alphavirus-based cancer vaccine, sunitinib and low-dose tumor irradiation completely blocks tumor development. Oncoimmunology 2015, 4, e1029699. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zaks, T.Z.; Wang, R.-F.; Irvine, K.R.; Kammula, U.S.; Marincola, F.M. Cancer therapy using a self-replicating RNA vaccine. Nat. Med. 1999, 5, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.A.; Sheahan, B.J.; Galbraith, S.E. Inhibition of angiogenesis by a Semliki Forest virus vector expressing VEGFR-2 reduces tumour growth and metastasis in mice. Gene Ther. 2007, 14, 503–513. [Google Scholar] [CrossRef]
- Granot, T.; Yamanashi, Y.; Meruelo, D. Sindbis viral vectors transiently deliver tumor-associated antigens to lymph nodes and elicit diversified antitumor CD8+ T-cell immunity. Mol. Ther. 2014, 22, 112–122. [Google Scholar] [CrossRef]
- Hoang-Le, D.; Smeenk, L.; Anraku, I.; Pijlman, G.P.; Wang, X.P.; de Vrij, J. A Kunjin replicon vector encoding granulocyte macrophage colony-stimulating factor for intra-tumoral gene therapy. Gene Ther. 2009, 16, 190–199. [Google Scholar] [CrossRef]
- Murphy, A.M.; Morris-Downes, M.M.; Sheahan, B.J.; Atkins, G.J. Inhibition of human lung carcinoma cell growth by apoptosis induction using Semliki Forest virus recombinant particles. Gene Ther. 2000, 7, 1477–1482. [Google Scholar] [CrossRef]
- Määttä, A.M.; Mäkinen, K.; Ketola, A.; Liimatainen, T.; Yongabi, F.N.; Vähä-Koskela, M. Replication competent Semliki Forest virus prolongs survival in experimental lung cancer. Int. J. Cancer 2008, 123, 1704–1711. [Google Scholar] [CrossRef]
- Patel, M.R.; Jacobson, B.A.; Ji, Y.; Drees, J.; Tang, S.; Xiong, K. Vesicular stomatitis virus expressing interferon-β is oncolytic and promotes antitumor immune responses in a syngeneic murine model of non-small cell lung cancer. Oncotarget 2015, 6, 33165–33177. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Jacobson, B.A.; Belgum, H.; Raza, A.; Sadiq, A.; Drees, J.; Wang, H.; Jay-Dixon, J.; Etchison, R.; Federspiel, M.J.; et al. Measles vaccine strains for virotherapy of non-small cell lung carcinoma. J. Thorac. Oncol. 2014, 9, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Fujiyuki, T.; Yoneda, M.; Amagai, Y.; Obayashi, K.; Ikeda, F.; Shoji, K. A measles virus selectively blind to signalling lymphocytic activation molecule shows anti-tumor activity against lung cancer cells. Oncotarget 2015, 6, 24895–24903. [Google Scholar] [CrossRef]
- McAllister, A.; Arbetman, A.E.; Mandl, S.; Pena-Rossi, C.; Andino, R. Recombinant yellow fever viruses are effective therapeutic vaccines for treatment of murine solid tumors and pulmonary metastases. J. Virol. 2000, 74, 9197–9205. [Google Scholar] [CrossRef] [PubMed]
- Avogadri, F.; Merghoub, T.; Maughan, M.F.; Hirschhorn-Cymerman, D.; Morris, J.; Ritter, E. Alphavirus replicon particles expressing TRP-2 provide potent therapeutic effect on melanoma through activation of humoral and cellular immunity. PLoS ONE 2010, 5, e12670. [Google Scholar] [CrossRef]
- Avogadri, F.; Zappasodi, R.; Yang, A.; Budhu, S.; Malandro, N.; Hisrchhorn-Cymerman, D. Combination of alphavirus replicon particle-based vaccination with immunomodulatory antibodies: Therapeutic activity in the B16 melanoma mouse model and immune correlates. Cancer Immunol. Res. 2014, 2, 448–458. [Google Scholar] [CrossRef]
- Yin, X.; Wang, W.; Zhu, X.; Wang, Y.; Wu, S.; Wang, Z. Synergistic antitumor efficacy of combined DNA vaccines targeting tumor cells and angiogenesis. Biochem. Biophys. Res. Comm. 2015, 465, 239–244. [Google Scholar] [CrossRef]
- Allaqui, F.; Achard, C.; Panterne, C.; Combredet, C.; Labarrière, N.; Dréno, B.; Elgaaied, A.B.; Pouliquen, D.; Tangy, F.; Fonteneau, J.F.; et al. Modulation of the Type I Interferon Response Defines the Sensitivity of Human Melanoma Cells to Oncolytic Measles Virus. Curr. Gene Ther. 2017, 16, 419–428. [Google Scholar] [CrossRef]
- Ammour, Y.; Ryabaya, O.; Shchetinina, Y.; Prokofeva, E.; Gavrilova, M.; Khochenkov, D.; Vorobyev, D.; Faizuloev, E.; Shohin, I.; Zverev, V.V.; et al. The Susceptibility of Human Melanoma Cells to Infection with the Leningrad-16 Vaccine Strain of Measles Virus. Viruses 2020, 12, E173. [Google Scholar] [CrossRef]
- Muik, A.; Kneiske, I.; Werbizki, M.; Wilflingseder, D.; Giroglou, T.; Ebert, O. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J. Virol. 2011, 85, 5679–5684. [Google Scholar] [CrossRef]
- Kimpel, J.; Urbiola, C.; Koske, I.; Tober, R.; Banki, Z.; Wollmann, G. The Oncolytic virus VSV-GP is effective against malignant melanoma. Viruses 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.; Kottke, T.; Thompson, J.; Rajani, K.; Zaidi, S.; Evgin, L.; Coffey, M.; Ralph, C.; Diaz, R.; Pandha, H.; et al. Prime-boost using separate oncolytic viruses in combination with checkpoint blockade improves anti-tumour therapy. Gene Ther. 2017, 24, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Dold, C.; Rodriguez Urbiola, C.; Wollmann, G.; Egerer, L.; Muik, A.; Bellmann, L.; Fiegl, H.; Marth, C.; Kimpel, J.; von Laer, D. Application of interferon modulators to overcome partial resistance to ovarian cancers to VSV-GP oncolytic viral therapy. Mol. Ther Oncolytics 2016, 3, 16021. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Nakamura, T.; Harvey, M.; Ikeda, Y.; Oberg, A.; Figini, M.; Canevari, S.; Hartmann, L.C.; Peng, K.W. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006, 12, 6170–6178. [Google Scholar] [CrossRef]
- Granot, T.; Meruelo, D. The role of natural killer cells in combinatorial anti-cancer therapy using Sindbis viral vector and irinotecan. Cancer Gene Ther. 2012, 19, 588–591. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Tsai, Y.C.; Monie, A.; Wu, T.C.; Hung, C.F. Enhancing the therapeutic effect against ovarian cancer through a combination of viral oncolysis and antigen-specific immunotherapy. Mol. Ther. 2010, 18, 692–699. [Google Scholar] [CrossRef]
- Venticinque, L.; Meruelo, D. Sindbis viral vector induced apoptosis requires translational inhibition and signaling through Mcl-1 and Bak. Mol. Cancer 2010, 9, 37. [Google Scholar] [CrossRef]
- McGray, A.J.R.; Huang, R.-Y.; Battaglia, S.; Eppolito, C.; Miliotto, A.; Stephenson, K.B.; Lugade, A.A.; Webster, G.; Lichty, B.D.; Seshadri, M.; et al. Oncolytic Maraba virus armed with tumor antigen boosts vaccine priming and reveals diverse therapeutic response patterns when combined with checkpoint blockade in ovarian cancer. J. Immunother Cancer 2019, 17, 189. [Google Scholar] [CrossRef]
- Murphy, A.M.; Besmer, D.M.; Moerdyk-Schauwecker, M.; Moestl, N.; Ornelles, D.A.; Mukherjee, P. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J. Virol. 2012, 86, 3073–3087. [Google Scholar] [CrossRef]
- Felt, S.A.; Droby, G.N.; Grdzelishvili, V.Z. Ruxolitinib and polycation combination treatment overcomes multiple mechanisms of resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Msaouel, P.; Iankov, I.D.; Allen, C.; Morris, J.C.; von Messling, V.; Cattaneo, R. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate 2009, 69, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Durso, R.J.; Andjelic, S.; Gardner, J.P.; Margitich, D.J.; Donovan, G.P.; Arrigale, R.R. A novel alphavirus vaccine encoding prostate-specific membrane antigen elicits potent cellular and humoral immune responses. Clin. Cancer Res. 2007, 13, 3999–4008. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hernandez, M.L.; Gray, A.; Hubby, B.; Kast, W.M. In vivo effects of vaccination with six-transmembrane epithelial antigen of the prostate: A candidate antigen for treating prostate cancer. Cancer Res. 2007, 67, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hernandez, M.L.; Gray, A.; Hubby, B.; Klinger, O.J.; Kast, W.M. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008, 68, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Riabov, V.; Treykova, I.; Alexander, R.B.; Pushko, P.; Klyushnenkova, E.N. Anti-tumor effect of the alphavirus-based virus-like particle vector expressing prostate-specific antigen in a HLA-DR transgenic mouse model of prostate cancer. Vaccine 2015, 33, 5386–5395. [Google Scholar] [CrossRef]
- Urbiola, C.; Santer, F.R.; Petersson, M.; van der Pluijm, G.; Horninger, W.; Erlmann, P. Oncolytic activity of the rhabdovirus VSV-GP against prostate cancer. Int. J. Cancer 2018, 143, 1786–1796. [Google Scholar] [CrossRef]
- Son, H.A.; Zhang, L.; Cuong, B.K.; Van Tong, H.; Cuong, L.D.; Hang, N.T.; Nhung, H.T.M.; Yamamoto, N.; Toan, N.L. Combination of Vaccine-Strain Measles and Mumps Viruses Enhances Oncolytic Activity against Human Solid Malignancies. Cancer Invest. 2018, 7, 106–117. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Reap, E.A.; Katen, K.; Watson, A.; Smith, K.; Norberg, P. Randomized, double-blind, phase I trial on an alphavirus replicon vaccine for cytomegalovirus in CMV negative volunteers. Vaccine 2010, 28, 484–493. [Google Scholar] [CrossRef]
- Wecker, M.; Gilbert, P.; Russell, N.; Hural, J.; Allen, M.; Pensiero, M. Phase I safety and immunogenicity evaluations of an alphavirus replicon HIV-1 subtype C gag vaccine in healthy HIV-1-uninfected adults. Clin. Vaccine Immunol. 2012, 19, 1651–1660. [Google Scholar] [CrossRef]
- Regules, J.A.; Beigel, J.H.; Paolino, K.M.; Voell, J.; Castellano, A.R.; Hu, Z.; Munoz, P.; Moon, J.E.; Ruck, R.C.; Bennett, J.W.; et al. A recombinant vesicular stomatitis virus ebola vaccine. N. Engl. J. Med. 2017, 376, 330–341. [Google Scholar] [CrossRef]
- ElSherif, M.S.; Brown, C.; MacKinnon-Cameron, D.; Li, L.; Racine, T.; Alimonti, J.; Rudge, T.L.; Sabourin, C.; Silvera, P.; Hooper, J.W.; et al. Assessing the safety and immunogenicity of recombinant vesicular stomatitis virus Ebola vaccine in healthy adults: A randomized clinical trial. CMAJ 2017, 189, E819–E827. [Google Scholar] [CrossRef] [PubMed]
- Dahlke, C.; Kasonta, R.; Lunemann, S.; Krahling, V.; Zinser, M.E.; Biedenkopf, N.; Fehling, S.K.; Ly, M.L.; Rechtien, A.; Stubbe, H.C.; et al. Dose-dependent T-cell dynamics and cytokine cascade following rVSV-ZEBOV immunization. EBioMedicine 2017, 19, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Poetsch, J.H.; Dahlke, C.; Zinser, M.E.; Kasonta, R.; Lunemann, S.; Rechtien, A.; Ly, M.L.; Stubbe, H.C.; Krähling, V.; Biedenkopf, N.; et al. Detectable Vesicular Stomatitis Virus (VSV)-Specific Humoral and Cellular Immune Responses Following VSV-Ebola Virus Vaccination in Humans. J. Infect. Dis. 2019, 219, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Huttner, A.; Dayer, J.A.; Yerly, S.; Combescure, C.; Auderset, F.; Desmeules, J. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: A randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 2015, 15, 1156–1166. [Google Scholar] [CrossRef]
- Kennedy, S.B.; Bolay, F.; Kieh, M.; Grandits, G.; Badio, M.; Ballou, R.; Eckes, R.; Feinberg, M.; Follmann, D.; Grund, B.; et al. Phase 2 placebo-controlled trial of two vaccines to prevent ebola in Liberia. N. Engl. J. Med. 2017, 377, 1438–1447. [Google Scholar] [CrossRef]
- Henao-Restrepo, A.M.; Longini, I.M.; Egger, M.; Dean, N.E.; Edmunds, W.J.; Camacho, A.; Carroll, M.W.; Doumbia, M.; Draguez, B.; Duraffour, S. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: Interim results from the Guinea ring vaccination cluster-randomised trial. Lancet 2015, 386, 857–866. [Google Scholar] [CrossRef]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef]
- Metzger, W.G.; Vivas-Martinez, S. Questionable efficacy of the rVSV-ZEBOV Ebola vaccine. Lancet 2018, 391, 1021. [Google Scholar] [CrossRef]
- Longini, I.M.; Røttingen, J.A.; Kieny, M.P.; Edmunds, W.J.; Henao-Restrepo, A.M. Questionable efficacy of the rVSV-ZEBOV Ebola vaccine – Authors’ reply. Lancet 2018, 391, 1021–1022. [Google Scholar] [CrossRef]
- Widdowson, M.; Schrag, S.; Carter, R.; Carr, W.; Legardy-Williams, J.; Gibson, L.; Lisk, D.R.; Jalloh, M.I.; Bash-Taqi, D.A.; Kargbo, S.A.; et al. Implementing an Ebola vaccine study—Sierra Leone. MMWR Suppl. 2016, 65, 98–106. [Google Scholar] [CrossRef]
- Halperin, S.A.; Arribas, J.R.; Rupp, R.; Andrews, C.P.; Chu, L.; Das, R.; Simon, J.K.; Onorato, M.T.; Liu, K.; Martin, J.; et al. Six-month safety data of recombinant vesicular stomatitis virus-zaire ebola virus envelope glycoprotein vaccine in a phase 3 double-blind, placebo-controlled randomized study in healthy adults. J. Infect. Dis. 2017, 215, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.L.; Coates, E.E.; Plummer, S.H.; Carter, C.A.; Berkowitz, N.; Conan-Cibotti, M.; Cox, J.H.; Beck, A.; O’Callahan, M.; Andrews, C.; et al. Effect of a Chikungunya Virus-Like Particle Vaccine on Safety and Tolerability Outcomes: A Randomized Clinical Trial. JAMA 2020, 323, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Hannaman, D.; Dupuy, L.C.; Ellefsen, B.; Schmaljohn, C.S. A Phase 1 clinical trial of a DNA vaccine for Venezuelan equine encephalitis delivered by intramuscular or intradermal electroporation. Vaccine 2016, 34, 3607–3612. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trials. Gov. Viral Therapy in Treating Patients with Recurrent Glioblastoma Multiforme. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00390299 (accessed on 6 May 2020).
- Lundstrom, K. Biology and application of alphaviruses in gene therapy. Gene Ther. 2005, 12 (Suppl. S1), S92–S97. [Google Scholar] [CrossRef]
- Heinzerling, L.; Kunzi, V.; Oberholzer, P.A.; Kundig, T.; Naim, H.; Dummer, R. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon resistant tumor cells. Blood 2005, 106, 2287–2289. [Google Scholar] [CrossRef]
- Galanis, E.; Hartmann, L.C.; Cliby, W.A.; Long, H.J.; Peethambaram, P.P.; Barrette, B.A.; Kaur, J.S.; Haluska, P.J., Jr.; Aderca, I.; Zollman, P.J.; et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010, 70, 875–882. [Google Scholar] [CrossRef]
- Russell, S.J.; Federspiel, M.J.; Peng, K.W.; Tong, C.; Dingli, D.; Morice, W.G.; Lowe, V.; O’Connor, M.K.; Kyle, R.A.; Leung, N.; et al. Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clinic Proc. 2014, 89, 926–933. [Google Scholar] [CrossRef]
- Morse, M.A.; Hobelka, A.C.; Osada, T.; Berglund, P.; Hubby, B.; Negri, S. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Investig. 2010, 120, 3234–3241. [Google Scholar] [CrossRef]
- Slovin, S.F.; Kehoe, M.; Durso, R.; Fernandez, C.; Olson, W.; Gao, J.P. A phaseI dose escalation trial of vaccine replicon particles (VRP) expressing prostate-specific membrane antigen (PSMA) in subjects with prostate cancer. Vaccine 2013, 31, 943–949. [Google Scholar] [CrossRef]
- Kelvin, A.A. Outbreak of Chikungunya in the Republic of Congo and the global picture. J. Infect. Dev. Ctries. 2011, 5, 441–444. [Google Scholar] [CrossRef]
- Jansen, K.A. The 2005–2007 Chikungunya epidemic in Reunion: Ambiguous etiologies, memories, and meaning-making. Med. Anthropol. 2013, 32, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Salas, R.; Rico-Hesse, R.; Ludwig, G.V.; Oberste, M.S.; Boshell, J.; Tesh, R.B. Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America. VEE Study Group. Lancet 1996, 348, 436–440. [Google Scholar] [CrossRef]
- Stief, A.E.; McCart, J.A. Oncolytic virotherapy for multiple myeloma. Expert. Opin. Biol. Ther. 2008, 8, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W. Measles virus for cancer therapy. Curr. Top. Microbiol. Immunol. 2009, 330, 213–241. [Google Scholar] [PubMed]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van Glabbeke, M.; van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef]
- Markman, M.; Webster, K.; Zanotti, K.; Peterson, G.; Kulp, B.; Belinson, J. Survival following the documentation of platinum and taxane resistance in ovarian cancer: A single institution experience involving multiple phase 2 clinical trials. Gynecol. Oncol. 2004, 93, 699–701. [Google Scholar] [CrossRef]
- Lundstrom, K. Replicon RNA Viral Vectors as Vaccines. Vaccines 2016, 4, 39. [Google Scholar] [CrossRef]
- Kirman, J.R.; Turon, T.; Su, H.; Li, A.; Kraus, C.; Polo, J.M.; Belisle, J.; Morris, S.; Seder, R.A. Enhanced immunogenicity to Mycobacterium tuberculosis by vaccination with an alphavirus plasmid replicon expressing antigen 85A. Infect. Immun. 2003, 71, 575–579. [Google Scholar] [CrossRef]
- Tsuji, M.; Bergmann, C.C.; Takita-Sonoda, Y.; Murata, K.; Rodrigues, E.G.; Nussenzweig, R.S.; Zavala, F. Recombinant Sindbis viruses expressing a cytotoxic T-lymphocyte epitope of a malaria parasite or of influenza virus elicit protection against the corresponding pathogen in mice. J. Virol. 1998, 72, 6907–6910. [Google Scholar] [CrossRef]
- Lundstrom, K. Coronaviruses Pandemic – Therapy and Vaccines. Biomedicines 2020, 8, 109. [Google Scholar] [CrossRef]
- Hariharan, M.J.; Driver, D.A.; Townsend, K.; Brumm, D.; Polo, J.M.; Belli, B.A.; Catton, D.J.; Hsu, D.; Mittelstaedt, D.; McCormack, J.E. DNA immunization against herpes simplex virus: Enhanced efficacy using a Sindbis virus-based vector. J. Virol. 1998, 72, 950–958. [Google Scholar] [CrossRef]
- Nosaki, K.; Hamada, K.; Takashima, Y.; Sagara, M.; Matsumura, Y.; Miayamoto, S.; Hijikata, Y.; Okazaki, T.; Nakanishi, Y.; Tani, K. A novel, polymer-coated oncolytic measles virus overcomes immune suppression and induces robust antitumor activity. Mol. Ther. Oncolytics 2016, 3, 16022. [Google Scholar] [CrossRef] [PubMed]
- Ketola, A.; Schlesinger, S.; Wahlfors, J. Properties of Sindbis virus vectors produced with a chimeric split helper system. Int. J. Mol. Med. 2005, 15, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Pushko, P.; Parker, M.; Ludwig, G.V.; Davis, N.L.; Johnston, R.E.; Smith, J.F. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: Expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 1997, 239, 389–401. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Virus | Target/Antigen | Vector Type | Finding | Ref |
|---|---|---|---|---|
| Flaviviruses | ||||
| DENV | E85 ectodomain | VEE VLPs | Dengue protection in mice | [35] |
| DV2 | MV | Neutralizing antibodies | [36] | |
| DV1-4 | MV | Dengue protection in mice | [37] | |
| Zika virus | prM. E | NLC-VEE RNA | ZIKV protection in mice | [38] |
| M, E | VSV VLPs | ZIKV protection in mice | [39] | |
| Filoviruses | ||||
| EBOV | GP/D637L | KUN VLPs | EBOV protection in 75% of primates | [40] |
| EBOV-GP | VSV VLPs | EBOV protection in macaques | [41] | |
| EBOV-GP | VSV VLPs | EBOV protection in primates | [42] | |
| EBOV-GP | VSV-VLPs | EBOV protection in macaques | [43] | |
| EBOV-NP | VEE VLPs | EBOV protection in mice | [44] | |
| EBOV-GP, NP | VEE VLPs | EBOV protection in mice and guinea pigs | [45] | |
| EBOV-GP, VP40 | SFV DNA | Neutralizing antibodies | [46] | |
| MARV | MARV-GP | VSV VLPs | MARV protection in primates | [42] |
| SUDV | SUDV-GP | VEE VLPs | SUDV protection in macaques | [43] |
| Arenaviruses | ||||
| LASV | LASV-GPC | VSV VLPs | LASV protection in guinea pigs | [47] |
| LASV-GPC/ΔGfib | VEE VLPs | LASV protection in mice | [48] | |
| LASV-GPC | LASV VLPs | LASV protection in guinea pigs | [49] | |
| JUNV | JUNV-GPC | VEE VLPs | JUNV protection in guinea pigs | [50] |
| MACV | MACV-GPC | VEE VLPs | MACV protection in guinea pigs | [50] |
| Lentiviruses | ||||
| HIV-1 | HIV-1 Env gp100 | SFV VLPs | Humoral immune responses | [51] |
| HIV-1 Env | SFV RNA | Antibody responses, mAbs | [52] | |
| Env/Gag/PolRT | SFV VLPs/RNA | Antigen-specific responses: VLPs > RNA | [53] | |
| Env/GagPolNef | SFV DNA | Superior to MVA, HIV gp40 | [54] | |
| TV1 gp140 | VEE* RNA-NPs | Immunogenicity in macaques | [55] | |
| Env gp120 | VEE RNA-NPs | gp120-specific antibodies | [56] | |
| SIV | Gag-pol | KUN VLPs | SIV protection in mice | [57] |
| Env, Gag-pol, Nef, Rev, Tat | SFV + MVA VLPs | Humoral and cellular responses | [58] | |
| Gag-pol | VSV + SFV VLPs | Partial SIV protection in macaques | [59] | |
| Influenza | ||||
| Influenza | NP | SFV VLPs | Mucosal immune response | [60] |
| HA | VEE-VLPs | Protection in chicken | [61] | |
| HA | SFV RNA | Protection in mice | [62] | |
| HA | VEE RNA | Protection in mice | [63] | |
| iFlu | VEE VLPs | Enhanced immune response | [64] | |
| NP | CSFV RNA-NPs | Immune response in mice | [65] | |
| Coronaviruses | ||||
| SARS-CoV | SARS-CoV S | VEE VLPs | Protection in mice | [66] |
| SARS-CoV S | VEE VLPs | Protection in mice | [67] | |
| MERS-CoV | MERS-CoV S | MV | Protection in mice | [68] |
| Hepatotropic | ||||
| HBV | HBsAg | MV | Partial protection | [69] |
| MHB, HBcAg | SFV-G VLPs | Protection in mice by MHB | [70] | |
| HBV S | SFV VLPs | Neutralization of HBV infectivity | [71] | |
| Alphaviruses | ||||
| CHIKV | E1-E3, C | VSV | CHIKV protection in mice | [39] |
| VEE | VEE Replicon | VEE VLPs | Protection in mice, macaques | [72] |
| EEE | EEE Replicon | EEE VLPs | Protection in mice, macaques | [72] |
| WEE | WEE Replicon | WEE VLPs | Weak protection in mice, macaques | [72] |
| VEE | VEE V4020 | VEE DNA | VEE protection in mice | [73] |
| VEE V4020 | VEE DNA | VEE protection in macaques | [74] |
| Cancer | Target/Antigen | Vector | Response | Ref |
|---|---|---|---|---|
| Brain | ||||
| GBM | SLAM, EGFR | MV | Tumor regression, prolonged survival | [79] |
| GBM | Endostatin | SFV VLPs | Tumor regression, prolonged survival | [80] |
| GBM | IL-18, IL-12 | DC-SFV | Enhanced antitumor activity | [81] |
| GBM | miR124 | SFV4 | Viral replication in tumors, tumor growth inhibition, prolonged survival | [82] |
| GBM | Chimeric VLPs | VSVΔG-CHIKV | Tumor cell infection, prolonged survival | [83] |
| Breast | ||||
| MDA-MB | CEA | MV | Prolonged survival in mice | [84] |
| A2L2 | HER2/neu | SIN VLPs | Prolonged survival in mice | [85] |
| A2L2 | HER2/neu | SIN DNA | Reduced tumor incidence and mass, protection with 80% less DNA | [86] |
| A2L2 | neuΔ | VEE VLPs + DCs | Specific Abs, tumor regression in mice | [87] |
| 4T1 | luciferase | SFV-VLPs, -RNA | Tumor targeting, re-administration | [88] |
| 4T1 | IL-12 | SFV-IL12, LVR01 | Long-term survival, lung metastasis prevention | [89] |
| 4T1 | IL-12 | SFV VLPs | Tumor regression, metastases | [90] |
| Cervical | ||||
| HPV | HPV-16 E7 | VEE VLPs | Prevention of tumor development | [91] |
| HPV | HPV E6-E7 | SFV VLPs | Complete elimination of tumors | [92] |
| HPV | HPV E6/7 | SFV DNA | 85% of immunized mice tumor-free | [93] |
| HPV | HPV E6, 7 | SFV VLPs, Rad | Enhanced antitumor activity | [94] |
| HPV | HPV E6, 7 | SFV VLPs, Rad+ sunitinib | 100% tumor-free survival | [95] |
| Colon | ||||
| CT26 | LacZ | SFV RNA | Antibody response, protection in mice | [96] |
| CT26 | VEGFR-2 | SFV VLPs | Inhibition of tumor growth | [97] |
| CT26 | VEGFR-2 + IL-4 | SFV VLPs | Superior survival of mice | [97] |
| CT26 | IL-12 | SFV VLPs | Tumor cell necrosis | [90] |
| CT26 | LacZ | SIN VLPs | Therapeutic effect in mouse model | [98] |
| CT26 | G-CSF | KUN VLPs | Cure in >50% of immunized mice | [99] |
| Lung | ||||
| H358a | EGFP | SFV VLPs | Complete regression in 3 out of 7 mice | [100] |
| A549 | EGFP | SFV (VA7) | Prolonged survival in mice | [101] |
| CT26.CL25 | LacZ | SIN VLPs | Protection against tumor challenges | [98] |
| H2009, A549 | GFP, IFNβ | VSV VLPs | Reduced tumor growth in mice | [102] |
| LM2 | IFNβ | VSV VLPs | Prolonged survival, cure of 30% of mice | [102] |
| A549, LLC | GFP, CEA | MV | Tumor regression in mice | [103] |
| NCI-H441 | EGFP | MV-SLAMblind | Suppression of tumor growth in mice | [104] |
| Melanoma | ||||
| B16-OVA | G-CSF | KUN VLPs | Cure in >50% of immunized mice | [99] |
| B16-OVA | SIINFEKL | YVF VLPs | Protection against malignant melanoma | [105] |
| B16 | TRP-2 | VEE VLPs | Prolonged survival in mice | [106] |
| B16 | TRP-2 | VEE VLPs + anti- CTLA-4/GITR | Tumor regression in mice | [107] |
| B16 | VEGFR-2, IL-12+ Sur, β-hCG | SFV DNA | Prolonged survival in mice | [108] |
| B16 | ASMEL, anti-PD1 | Oncolytic VSV + Reovirus | Extended survival, long-term cure in mice | [113] |
| Mel Z | MV L-16 | MV | Inhibition of tumor growth | [110] |
| A375, B16 | GFP, Luc | VSV-GP-LCMV | Prolonged survival in mice | [112] |
| Ovarian | ||||
| A2780 | Luc | VSV-GP-LCMV | Oncolytic activity in vitro and in vivo | [114] |
| ARH77 | EGFP | MV-alphaFR | Increased survival rate in vivo | [115] |
| ES2 | IL-12 | SIN VLPs, CPT-11 | Long-term survival in SCID mice | [116] |
| MOSEC | OVA | SFV VLPs + VV | Enhanced immune responses in mice | [117] |
| MOSEC | GFP, Luc | SIN VLPs | Induction of cellular stress, apoptosis | [118] |
| ID9-mp1, ID8 | DCT | Oncolytic Maraba | Robust immune response, tumor control | [119] |
| Pancreatic | ||||
| Pan02 | GFP, Luc | SIN VLPs | Induction of cellular stress, apoptosis | [118] |
| Panc-1, | GFP | VSV VLPs | Oncolytic activity in vitro and in vivo | [120] |
| Su.86.86 | GFP | VSV VLPs | Oncolytic activity in vitro and in vivo | [120] |
| KLM1, | SLAM | MV-SLAMblind | Suppression of tumor growth in mice | [122] |
| Capan-2 | SLAM | MV-SLAMblind | Suppression of tumor growth in mice | [122] |
| Prostate | ||||
| PC-3 | [122] | |||
| LnCaP | CEA | MV | Prolonged survival of mice | [123] |
| TRAMP-C | PSMA | VEE VLPs | Robust immune response in mice | [124] |
| TRAMP | STEAP | VEE VLPs | Prolonged survival in mice | [125] |
| TRAMP-PSA | PSCA | VEE VLPs | 90% survival rate in mice | [125] |
| Du145, 22Rv1 | PSA | VEE VLPs | Tumor growth delay in mice | [126] |
| TRAMP, PC-3 | Luc | VSV-GP-LCMV | Long-term remission in mice | [127] |
| Luc | VSV-GP-LCMV | Remission in subcutaneous tumors and bone metastases | [127] | |
| PC-3 | MV, MuV | MV + MuV | Prolonged survival in mice | [128] |
| Indication | Vector/Antigen | Phase | Response | Ref |
|---|---|---|---|---|
| Infections | ||||
| CMV | VEE-gB/p55-IE1 | Phase I | CMV-specific Abs | [129] |
| AIDS | VEE-HIV-gag | Phase I | Modest antibody responses | [130] |
| EBOV | VSV-ZEBOV | Phase I | Anti-ZEBOV Abs | [131] |
| VSV∆G-ZEBOV | Phase I | Sustainable IgG titers for 180 days | [132] | |
| VSV-ZEBOV | Phase I | EBOV-specific neutralizing Abs | [133] | |
| VSV-ZEBOV | Phase I/II | Lower dose, improved tolerability | [135] | |
| VSV∆G-ZEBOV | Phase II | Ab-response in 80% of vaccines | [136] | |
| VSV-ZEBOV | Phase III | 100% protection against EVD | [137] | |
| VSV-ZEBOV | Phase III | Substantial protection against EVD | [138] | |
| VSV-ZEBOV | Phase II/III | No EVD, no vaccine-related AEs | [141] | |
| VSV∆G-ZEBOV | Phase III | Safe, no vaccine-related AEs | [142] | |
| CHIK | CHIK VLPs | Phase II | Safe, well tolerated | [143] |
| VEE | VEE DNA | Phase I | VEE-specific neutralizing Abs | [144] |
| Cancers | ||||
| Glioblastoma | MV-CEA | Phase I | No dose-limiting toxicity | [145] |
| Kidney | LipoVIL12 | Phase I | Safe, tumor targeting | [146] |
| Lymphoma | MVEZ | Phase I | Tumor regression | [147] |
| Melanoma | LipoVIL12 | Phase I | Safe, tumor targeting | [146] |
| Myeloma | MV-NIS | Phase I | Complete remission in one patient | [148] |
| Ovarian | MV-CEA | Phase I/II | Stable disease (high dose treatment) | [149] |
| Pancreas | VEE-CEA | Phase I | Prolonged survival | [150] |
| Prostate | VEE-PSMA | Phase I | Neutralizing Abs against PSMA | [151] |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lundstrom, K. Self-Amplifying RNA Viruses as RNA Vaccines. Int. J. Mol. Sci. 2020, 21, 5130. https://doi.org/10.3390/ijms21145130
Lundstrom K. Self-Amplifying RNA Viruses as RNA Vaccines. International Journal of Molecular Sciences. 2020; 21(14):5130. https://doi.org/10.3390/ijms21145130
Chicago/Turabian StyleLundstrom, Kenneth. 2020. "Self-Amplifying RNA Viruses as RNA Vaccines" International Journal of Molecular Sciences 21, no. 14: 5130. https://doi.org/10.3390/ijms21145130
APA StyleLundstrom, K. (2020). Self-Amplifying RNA Viruses as RNA Vaccines. International Journal of Molecular Sciences, 21(14), 5130. https://doi.org/10.3390/ijms21145130