Differential Effects of MS Therapeutics on B Cells—Implications for Their Use and Failure in AQP4-Positive NMOSD Patients


Abstract
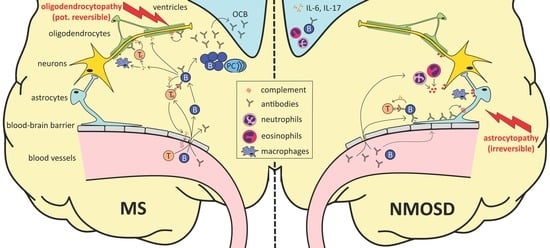
1. Introduction
2. Effects of MS Medications on Peripheral B Cells in Humans
2.1. Inhibition of Proliferation
2.2. Inhibition of Migration
2.3. Depletion
2.4. Immunomodulation
3. MS Medications in NMOSD
3.1. Inhibition of Proliferation
3.2. Inhibition of Migration
3.3. Depletion
3.4. Immunomodulation
4. Ineffectiveness and Failure of MS Therapeutics in NMOSD
4.1. Elevation of the Total B Cell Count
4.2. Increase of Memory B Cells
4.3. Elevated Immunoglobulin Levels
4.4. Elevated Plasmablast Count
4.5. Elevated Serum Interleukin-6 Levels
4.6. Elevated BAFF Levels in Non-Depleting Agents
4.7. Others
5. Novel and Future NMOSD Treatments
5.1. Complement Factor C5 Antibodies
5.2. B Cell-Depleting Antibodies
5.3. Interleukin-6 Receptor Antibodies
5.4. Others
6. Conclusions
- Increase of the total B cell count;
- Elevated proportion of memory B cells;
- Elevated proportion of plasmablasts;
- Increased immunoglobulin production;
- Elevated serum interleukin-6 levels;
- Elevated serum BAFF levels.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AQP4 | aquaporin 4 |
| BAFF | B cell activating factor |
| CD | cluster of differentiation |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| EDSS | expanded disability status scale |
| IgG | immunoglobulin G |
| MOG | myelin oligodendrocyte glycoprotein |
| MS | multiple sclerosis |
| NMOSD | neuromyelitis optica spectrum disorders |
References
- Franciotta, D.; Salvetti, M.; Lolli, F.; Serafini, B.; Aloisi, F. B cells and multiple sclerosis. Lancet Neurol. 2008, 7, 852–858. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct effector cytokine profiles of memory and naive human b cell subsets and implication in multiple sclerosis. J. Immunol. (Baltimore, Md. 1950) 2007, 178, 6092–6099. [Google Scholar] [CrossRef] [PubMed]
- Constant, S.; Schweitzer, N.; West, J.; Ranney, P.; Bottomly, K. B lymphocytes can be competent antigen-presenting cells for priming cd4+ t cells to protein antigens in vivo. J. Immunol. 1995, 155, 3734–3741. [Google Scholar] [PubMed]
- Lowenthal, A.; van Sande, M.; Karcher, D. The differential diagnosis of neurological diseases by fractionating electro-phoretically the csf ? J. Neurochem. 1960, 6, 51–56. [Google Scholar] [CrossRef]
- Obermeier, B.; Lovato, L.; Mentele, R.; Brück, W.; Forne, I.; Imhof, A.; Lottspeich, F.; Turk, K.W.; Willis, S.N.; Wekerle, H.; et al. Related b cell clones that populate the csf and cns of patients with multiple sclerosis produce csf immunoglobulin. J. Neuroimmunol. 2011, 233, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Von Budingen, H.C.; Gulati, M.; Kuenzle, S.; Fischer, K.; Rupprecht, T.A.; Goebels, N. Clonally expanded plasma cells in the cerebrospinal fluid of patients with central nervous system autoimmune demyelination produce “oligoclonal bands”. J. Neuroimmunol. 2010, 218, 134–139. [Google Scholar] [CrossRef]
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999, 5, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Bonnan, M. Intrathecal igg synthesis: A resistant and valuable target for future multiple sclerosis treatments. Mult. Scler. Int. 2015, 2015, 296184. [Google Scholar] [CrossRef] [PubMed]
- Mathey, E.K.; Derfuss, T.; Storch, M.K.; Williams, K.R.; Hales, K.; Woolley, D.R.; Al-Hayani, A.; Davies, S.N.; Rasband, M.N.; Olsson, T.; et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J. Exp. Med. 2007, 204, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Derfuss, T.; Parikh, K.; Velhin, S.; Braun, M.; Mathey, E.; Krumbholz, M.; Kumpfel, T.; Moldenhauer, A.; Rader, C.; Sonderegger, P.; et al. Contactin-2/tag-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc. Natl. Acad. Sci. USA 2009, 106, 8302–8307. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.G.; Catz, I. Relative frequency of autoantibodies to myelin basic protein and proteolipid protein in optic neuritis and multiple sclerosis cerebrospinal fluid. J. Neurol. Sci. 1994, 121, 66–73. [Google Scholar] [CrossRef]
- Kinzel, S.; Weber, M.S. The role of peripheral cns-directed antibodies in promoting inflammatory cns demyelination. Brain. Sci. 2017, 7, 70. [Google Scholar]
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis optica: Clinical features, immunopathogenesis and treatment. Clin. Exp. Immunol. 2014, 176, 149–164. [Google Scholar] [CrossRef]
- Misu, T.; Fujihara, K.; Kakita, A.; Konno, H.; Nakamura, M.; Watanabe, S.; Takahashi, T.; Nakashima, I.; Takahashi, H.; Itoyama, Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: Distinction from multiple sclerosis. Brain 2007, 130, 1224–1234. [Google Scholar]
- Levy, M.; Wildemann, B.; Jarius, S.; Orellano, B.; Sasidharan, S.; Weber, M.S.; Stuve, O. Immunopathogenesis of neuromyelitis optica. Adv. Immunol. 2014, 121, 213–242. [Google Scholar]
- Chihara, N.; Aranami, T.; Sato, W.; Miyazaki, Y.; Miyake, S.; Okamoto, T.; Ogawa, M.; Toda, T.; Yamamura, T. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc. Natl. Acad. Sci. USA 2011, 108, 3701–3706. [Google Scholar] [CrossRef]
- Bennett, J.L.; Lam, C.; Kalluri, S.R.; Saikali, P.; Bautista, K.; Dupree, C.; Glogowska, M.; Case, D.; Antel, J.P.; Owens, G.P.; et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann. Neurol. 2009, 66, 617–629. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Franciotta, D.; Waters, P.; Zipp, F.; Hohlfeld, R.; Vincent, A.; Wildemann, B. Mechanisms of disease: Aquaporin-4 antibodies in neuromyelitis optica. Nat. Clin. Pract. Neurol. 2008, 4, 202–214. [Google Scholar]
- Ghezzi, A.; Bergamaschi, R.; Martinelli, V.; Trojano, M.; Tola, M.R.; Merelli, E.; Mancardi, L.; Gallo, P.; Filippi, M.; Zaffaroni, M.; et al. Clinical characteristics, course and prognosis of relapsing devic’s neuromyelitis optica. J. Neurol. 2004, 251, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Kleiter, I.; Gahlen, A.; Borisow, N.; Fischer, K.; Wernecke, K.D.; Wegner, B.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann. Neurol. 2016, 79, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Saikali, P.; Cayrol, R.; Roth, A.D.; Bar-Or, A.; Prat, A.; Antel, J.P. Functional consequences of neuromyelitis optica-igg astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J. Immunol. 2008, 181, 5730–5737. [Google Scholar] [CrossRef]
- Mitsdoerffer, M.; Kuchroo, V.; Korn, T. Immunology of neuromyelitis optica: A t cell-b cell collaboration. Ann N Y Acad Sci 2013, 1283, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Pitarokoili, K.; Gold, R. Dimethyl fumarate for patients with neuromyelitis optica spectrum disorder. Mult. Scler. 2018, 24, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Selmaj, K.; Selmaj, I. Novel emerging treatments for nmosd. Neurol. Neurochir. Pol. 2019, 53, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Lana-Peixoto, M.A.; Talim, N. Neuromyelitis optica spectrum disorder and anti-mog syndromes. Biomedicines 2019, 7, 42. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Aktas, O.; Asgari, N.; Dale, R.C.; de Seze, J.; Franciotta, D.; Fujihara, K.; Jacob, A.; Kim, H.J.; et al. Mog encephalomyelitis: International recommendations on diagnosis and antibody testing. J. Neuroinflammation 2018, 15, 134. [Google Scholar] [CrossRef]
- Hinson, S.R.; Pittock, S.J.; Lucchinetti, C.F.; Roemer, S.F.; Fryer, J.P.; Kryzer, T.J.; Lennon, V.A. Pathogenic potential of igg binding to water channel extracellular domain in neuromyelitis optica. Neurology 2007, 69, 2221–2231. [Google Scholar] [CrossRef]
- Daclizumab withdrawn from the market worldwide. Drug. Ther. Bull. 2018, 56, 38.
- Blair, P.A.; Noreña, L.Y.; Flores-Borja, F.; Rawlings, D.J.; Isenberg, D.A.; Ehrenstein, M.R.; Mauri, C. Cd19(+)cd24(hi)cd38(hi) b cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity 2010, 32, 129–140. [Google Scholar] [CrossRef]
- Baker, D.; Marta, M.; Pryce, G.; Giovannoni, G.; Schmierer, K. Memory b cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine 2017, 16, 41–50. [Google Scholar] [CrossRef]
- Kreuzaler, M.; Rauch, M.; Salzer, U.; Birmelin, J.; Rizzi, M.; Grimbacher, B.; Plebani, A.; Lougaris, V.; Quinti, I.; Thon, V.; et al. Soluble baff levels inversely correlate with peripheral b cell numbers and the expression of baff receptors. J. Immunol. 2012, 188, 497–503. [Google Scholar] [CrossRef]
- Carter, L.M.; Isenberg, D.A.; Ehrenstein, M.R. Elevated serum baff levels are associated with rising anti-double-stranded DNA antibody levels and disease flare following b cell depletion therapy in systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 2672–2679. [Google Scholar]
- Giovannoni, G. Cladribine to treat relapsing forms of multiple sclerosis. Neurotherapeutics 2017, 14, 874–887. [Google Scholar] [CrossRef]
- Jacobs, B.M.; Ammoscato, F.; Giovannoni, G.; Baker, D.; Schmierer, K. Cladribine: Mechanisms and mysteries in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1266–1271. [Google Scholar] [CrossRef]
- Ceronie, B.; Jacobs, B.M.; Baker, D.; Dubuisson, N.; Mao, Z.; Ammoscato, F.; Lock, H.; Longhurst, H.J.; Giovannoni, G.; Schmierer, K. Cladribine treatment of multiple sclerosis is associated with depletion of memory b cells. J. Neurol. 2018, 265, 1199–1209. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Sellebjerg, F. Pulsed immune reconstitution therapy in multiple sclerosis. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419836913. [Google Scholar] [CrossRef]
- Korsen, M.; Bragado Alonso, S.; Peix, L.; Broker, B.M.; Dressel, A. Cladribine exposure results in a sustained modulation of the cytokine response in human peripheral blood mononuclear cells. PLoS ONE 2015, 10, e0129182. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Merlini, G.; Leblond, V.; Anagnostopoulos, A.; Alexanian, R. How we treat waldenstrom’s macroglobulinemia. Haematologica 2005, 90, 117–125. [Google Scholar]
- Bar-Or, A.; Pachner, A.; Menguy-Vacheron, F.; Kaplan, J.; Wiendl, H. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs 2014, 74, 659–674. [Google Scholar] [CrossRef]
- Gandoglia, I.; Ivaldi, F.; Laroni, A.; Benvenuto, F.; Solaro, C.; Mancardi, G.; Kerlero de Rosbo, N.; Uccelli, A. Teriflunomide treatment reduces b cells in patients with ms. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e403. [Google Scholar] [CrossRef]
- Medina, S.; Sainz de la Maza, S.; Villarrubia, N.; Alvarez-Lafuente, R.; Costa-Frossard, L.; Arroyo, R.; Monreal, E.; Tejeda-Velarde, A.; Rodriguez-Martin, E.; Roldan, E.; et al. Teriflunomide induces a tolerogenic bias in blood immune cells of ms patients. Ann. Clin. Transl. Neurol. 2019, 6, 355–363. [Google Scholar] [CrossRef]
- Zivadinov, R.; Ramanathan, M.; Hagemeier, J.; Bergsland, N.; Ramasamy, D.P.; Durfee, J.; Kolb, C.; Weinstock-Guttman, B. Teriflunomide’s effect on humoral response to epstein-barr virus and development of cortical gray matter pathology in multiple sclerosis. Mult. Scler Relat. Disord. 2019, 36, 101388. [Google Scholar] [CrossRef]
- Mazerski, J.; Martelli, S.; Borowski, E. The geometry of intercalation complex of antitumor mitoxantrone and ametantrone with DNA: Molecular dynamics simulations. Acta Biochim. Pol. 1998, 45, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, O.; Wiendl, H.; Kieseier, B.C.; Archelos, J.J.; Hemmer, B.; Stuve, O.; Hartung, H.P. Multiple sclerosis: Mitoxantrone promotes differential effects on immunocompetent cells in vitro. J. Neuroimmunol. 2005, 168, 128–137. [Google Scholar] [CrossRef]
- Chanvillard, C.; Millward, J.M.; Lozano, M.; Hamann, I.; Paul, F.; Zipp, F.; Dorr, J.; Infante-Duarte, C. Mitoxantrone induces natural killer cell maturation in patients with secondary progressive multiple sclerosis. PLoS ONE 2012, 7, e39625. [Google Scholar] [CrossRef]
- Fidler, J.M.; DeJoy, S.Q.; Gibbons, J.J., Jr. Selective immunomodulation by the antineoplastic agent mitoxantrone. I. Suppression of b lymphocyte function. J. Immunol. 1986, 137, 727–732. [Google Scholar] [PubMed]
- Fox, E.J. Mechanism of action of mitoxantrone. Neurology 2004, 63, S15–S18. [Google Scholar] [CrossRef]
- Gbadamosi, J.; Buhmann, C.; Tessmer, W.; Moench, A.; Haag, F.; Heesen, C. Effects of mitoxantrone on multiple sclerosis patients’ lymphocyte subpopulations and production of immunoglobulin, tnf-alpha and il-10. Eur. Neurol. 2003, 49, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Kannel, K.; Alnek, K.; Vahter, L.; Gross-Paju, K.; Uibo, R.; Kisand, K.V. Changes in blood b cell-activating factor (baff) levels in multiple sclerosis: A sign of treatment outcome. PLoS ONE 2015, 10, e0143393. [Google Scholar] [CrossRef]
- Evans, W.E. Pharmacogenetics of thiopurine s-methyltransferase and thiopurine therapy. Ther. Drug Monit. 2004, 26, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Maltzman, J.S.; Koretzky, G.A. Azathioprine: Old drug, new actions. J. Clin. Invest. 2003, 111, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Leandro, M.J.; Cambridge, G.; Edwards, J.C.; Ehrenstein, M.R.; Isenberg, D.A. B-cell depletion in the treatment of patients with systemic lupus erythematosus: A longitudinal analysis of 24 patients. Rheumatology (Oxford) 2005, 44, 1542–1545. [Google Scholar] [CrossRef]
- Hayry, P.; von Willebrand, E.; Ahonen, J.; Eklund, B.; Salmela, K.; Hockerstedt, K.; Pettersson, E.; Koskimies, S. Effects of cyclosporine, azathioprine, and steroids on the renal transplant, on the cytologic patterns of intragraft inflammation, and on concomitant rejection-associated changes in recipient blood. Transplant. Proc. 1988, 20, 153–162. [Google Scholar]
- Bottomley, M.J.; Chen, M.; Fuggle, S.; Harden, P.N.; Wood, K.J. Application of operational tolerance signatures are limited by variability and type of immunosuppression in renal transplant recipients: A cross-sectional study. Transplant. Direct. 2017, 3, e125. [Google Scholar] [CrossRef]
- Thiel, J.; Salzer, U.; Hassler, F.; Effelsberg, N.M.; Hentze, C.; Sic, H.; Bartsch, M.; Miehle, N.; Peter, H.H.; Warnatz, K.; et al. B cell homeostasis is disturbed by immunosuppressive therapies in patients with anca-associated vasculitides. Autoimmunity 2013, 46, 429–438. [Google Scholar] [CrossRef]
- Roekevisch, E.; Szegedi, K.; Hack, D.P.; Schram, M.E.; Res, P.; Bos, J.D.; Leeflang, M.M.G.; Luiten, R.M.; Kezic, S.; Spuls, P.I.; et al. Effect of immunosuppressive treatment on biomarkers in adult atopic dermatitis patients. J. Eur. Acad. Dermatol. Venereol. 2019. online ahead of print. [Google Scholar] [CrossRef]
- Salinas-Carmona, M.C.; Perez, L.I.; Galan, K.; Vazquez, A.V. Immunosuppressive drugs have different effect on b lymphocyte subsets and igm antibody production in immunized balb/c mice. Autoimmunity 2009, 42, 537–544. [Google Scholar] [CrossRef]
- Sokollik, C.; McLin, V.A.; Vergani, D.; Terziroli Beretta-Piccoli, B.; Mieli-Vergani, G. Juvenile autoimmune hepatitis: A comprehensive review. J. Autoimmun. 2018, 95, 69–76. [Google Scholar] [CrossRef]
- Hernandez-Breijo, B.; Gomez, A.; Soukka, S.; Johansson, P.; Parodis, I. Antimalarial agents diminish while methotrexate, azathioprine and mycophenolic acid increase baff levels in systemic lupus erythematosus. Autoimmun. Rev. 2019, 18, 102372. [Google Scholar] [CrossRef]
- Hall, A.G.; Tilby, M.J. Mechanisms of action of, and modes of resistance to, alkylating agents used in the treatment of haematological malignancies. Blood. Rev. 1992, 6, 163–173. [Google Scholar] [CrossRef]
- Brodsky, R.A. High-dose cyclophosphamide for autoimmunity and alloimmunity. Immunol. Res. 2010, 47, 179–184. [Google Scholar] [CrossRef]
- Zhu, L.P.; Cupps, T.R.; Whalen, G.; Fauci, A.S. Selective effects of cyclophosphamide therapy on activation, proliferation, and differentiation of human b cells. J. Clin. Invest. 1987, 79, 1082–1090. [Google Scholar] [CrossRef]
- Fassbinder, T.; Saunders, U.; Mickholz, E.; Jung, E.; Becker, H.; Schluter, B.; Jacobi, A.M. Differential effects of cyclophosphamide and mycophenolate mofetil on cellular and serological parameters in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 92. [Google Scholar] [CrossRef]
- Dorner, T.; Jacobi, A.M.; Lipsky, P.E. B cells in autoimmunity. Arthritis Res. Ther. 2009, 11, 247. [Google Scholar] [CrossRef]
- Tsokos, G.C.; Smith, P.L.; Balow, J.E. Development of hypogammaglobulinemia in a patient with systemic lupus erythematosus. Am. J. Med. 1986, 81, 1081–1084. [Google Scholar] [CrossRef]
- Moschella, F.; Torelli, G.F.; Valentini, M.; Urbani, F.; Buccione, C.; Petrucci, M.T.; Natalino, F.; Belardelli, F.; Foa, R.; Proietti, E. Cyclophosphamide induces a type i interferon-associated sterile inflammatory response signature in cancer patients’ blood cells: Implications for cancer chemoimmunotherapy. Clin. Cancer Res. 2013, 19, 4249–4261. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.; Proschmann, U.; Ziemssen, T. Fingolimod hydrochloride for the treatment of relapsing remitting multiple sclerosis. Expert. Opin. Pharmacother. 2017, 18, 1649–1660. [Google Scholar] [CrossRef]
- Kaufmann, M.; Haase, R.; Proschmann, U.; Ziemssen, T.; Akgun, K. Real world lab data: Patterns of lymphocyte counts in fingolimod treated patients. Front. Immunol. 2018, 9, 2669. [Google Scholar] [CrossRef] [PubMed]
- Muls, N.; Dang, H.A.; Sindic, C.J.; van Pesch, V. Fingolimod increases cd39-expressing regulatory t cells in multiple sclerosis patients. PLoS ONE 2014, 9, e113025. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Niino, M.; Fukazawa, T.; Takahashi, E.; Nonaka, T.; Amino, I.; Tashiro, J.; Minami, N.; Fujiki, N.; Doi, S.; et al. Suppressed pro-inflammatory properties of circulating b cells in patients with multiple sclerosis treated with fingolimod, based on altered proportions of b-cell subpopulations. Clin. Immunol. 2014, 151, 127–135. [Google Scholar] [CrossRef]
- Blumenfeld-Kan, S.; Staun-Ram, E.; Miller, A. Fingolimod reduces cxcr4-mediated b cell migration and induces regulatory b cells-mediated anti-inflammatory immune repertoire. Mult. Scler Relat. Disord. 2019, 34, 29–37. [Google Scholar] [CrossRef]
- Blumenfeld, S.; Staun-Ram, E.; Miller, A. Fingolimod therapy modulates circulating b cell composition, increases b regulatory subsets and production of il-10 and tgfbeta in patients with multiple sclerosis. J. Autoimmun. 2016, 70, 40–51. [Google Scholar] [CrossRef]
- Grutzke, B.; Hucke, S.; Gross, C.C.; Herold, M.V.; Posevitz-Fejfar, A.; Wildemann, B.T.; Kieseier, B.C.; Dehmel, T.; Wiendl, H.; Klotz, L. Fingolimod treatment promotes regulatory phenotype and function of b cells. Ann. Clin. Transl. Neurol. 2015, 2, 119–130. [Google Scholar] [CrossRef]
- Nakamura, M.; Matsuoka, T.; Chihara, N.; Miyake, S.; Sato, W.; Araki, M.; Okamoto, T.; Lin, Y.; Ogawa, M.; Murata, M.; et al. Differential effects of fingolimod on b-cell populations in multiple sclerosis. Mult. Scler. 2014, 20, 1371–1380. [Google Scholar] [CrossRef]
- Zandi-Esfahan, S.; Fazeli, M.; Shaygannejad, V.; Hasheminia, J.; Badihian, S.; Aghayerashti, M.; Maghzi, H. Evaluating the effect of adding fish oil to fingolimod on tnf-alpha, il1beta, il6, and ifn-gamma in patients with relapsing-remitting multiple sclerosis: A double-blind randomized placebo-controlled trial. Clin. Neurol. Neurosurg. 2017, 163, 173–178. [Google Scholar] [CrossRef]
- Zoehner, G.; Miclea, A.; Salmen, A.; Kamber, N.; Diem, L.; Friedli, C.; Bagnoud, M.; Ahmadi, F.; Briner, M.; Sedille-Mostafaie, N.; et al. Reduced serum immunoglobulin g concentrations in multiple sclerosis: Prevalence and association with disease-modifying therapy and disease course. Ther. Adv. Neurol. Disord. 2019, 12, 1756286419878340. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Niino, M.; Takahashi, E.; Suzuki, M.; Mizuno, M.; Hisahara, S.; Fukazawa, T.; Amino, I.; Nakano, F.; Nakamura, M.; et al. Fingolimod induces baff and expands circulating transitional b cells without activating memory b cells and plasma cells in multiple sclerosis. Clin. Immunol. 2018, 187, 95–101. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (expand): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Wu, Q.; Mills, E.A.; Wang, Q.; Dowling, C.A.; Fisher, C.; Kirch, B.; Lundy, S.K.; Fox, D.A.; Mao-Draayer, Y.; Group, A.M.S.S. Siponimod enriches regulatory t and b lymphocytes in secondary progressive multiple sclerosis. JCI Insight 2020, 5, e134251. [Google Scholar] [CrossRef]
- Pelz, A.; Schaffert, H.; Diallo, R.; Hiepe, F.; Meisel, A.; Kohler, S. S1p receptor antagonists fingolimod and siponimod do not improve the outcome of experimental autoimmune myasthenia gravis mice after disease onset. Eur. J. Immunol. 2018, 48, 498–508. [Google Scholar] [CrossRef]
- O’Sullivan, C.; Schubart, A.; Mir, A.K.; Dev, K.K. The dual s1pr1/s1pr5 drug baf312 (siponimod) attenuates demyelination in organotypic slice cultures. J. Neuroinflammation 2016, 13, 31. [Google Scholar] [CrossRef]
- Ufer, M.; Shakeri-Nejad, K.; Gardin, A.; Su, Z.; Paule, I.; Marbury, T.C.; Legangneux, E. Impact of siponimod on vaccination response in a randomized, placebo-controlled study. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e398. [Google Scholar] [CrossRef]
- Comi, G.; Kappos, L.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.P.; Montalban, X.; Kubala Havrdova, E.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (sunbeam): A multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019, 18, 1009–1020. [Google Scholar] [CrossRef]
- Cohen, J.A.; Comi, G.; Selmaj, K.W.; Bar-Or, A.; Arnold, D.L.; Steinman, L.; Hartung, H.P.; Montalban, X.; Kubala Havrdova, E.; Cree, B.A.C.; et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (radiance): A multicentre, randomised, 24-month, phase 3 trial. Lancet Neurol. 2019, 18, 1021–1033. [Google Scholar] [CrossRef]
- Delbue, S.; Comar, M.; Ferrante, P. Natalizumab treatment of multiple sclerosis: New insights. Immunotherapy 2017, 9, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.C.; Castro, A.C. Very late antigen 4 (vla4) antagonists as anti-inflammatory agents. Curr. Opin. Chem. Biol. 1998, 2, 453–457. [Google Scholar] [CrossRef]
- Traub, J.W.; Pellkofer, H.L.; Grondey, K.; Seeger, I.; Rowold, C.; Bruck, W.; Husseini, L.; Hausser-Kinzel, S.; Weber, M.S. Natalizumab promotes activation and pro-inflammatory differentiation of peripheral b cells in multiple sclerosis patients. J. Neuroinflammation 2019, 16, 228. [Google Scholar] [CrossRef] [PubMed]
- Saraste, M.; Penttila, T.L.; Airas, L. Natalizumab treatment leads to an increase in circulating cxcr3-expressing b cells. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e292. [Google Scholar] [CrossRef]
- Krumbholz, M.; Meinl, I.; Kumpfel, T.; Hohlfeld, R.; Meinl, E. Natalizumab disproportionately increases circulating pre-b and b cells in multiple sclerosis. Neurology 2008, 71, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Planas, R.; Jelcic, I.; Schippling, S.; Martin, R.; Sospedra, M. Natalizumab treatment perturbs memory- and marginal zone-like b-cell homing in secondary lymphoid organs in multiple sclerosis. Eur. J. Immunol. 2012, 42, 790–798. [Google Scholar] [CrossRef]
- Gahlen, A.; Trampe, A.K.; Haupeltshofer, S.; Ringelstein, M.; Aktas, O.; Berthele, A.; Wildemann, B.; Gold, R.; Jarius, S.; Kleiter, I. Aquaporin-4 antibodies in patients treated with natalizumab for suspected ms. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e363. [Google Scholar] [CrossRef] [PubMed]
- Warnke, C.; Stettner, M.; Lehmensiek, V.; Dehmel, T.; Mausberg, A.K.; von Geldern, G.; Gold, R.; Kumpfel, T.; Hohlfeld, R.; Maurer, M.; et al. Natalizumab exerts a suppressive effect on surrogates of b cell function in blood and csf. Mult. Scler. 2015, 21, 1036–1044. [Google Scholar] [CrossRef]
- Harrer, A.; Tumani, H.; Niendorf, S.; Lauda, F.; Geis, C.; Weishaupt, A.; Kleinschnitz, C.; Rauer, S.; Kuhle, J.; Stangel, M.; et al. Cerebrospinal fluid parameters of b cell-related activity in patients with active disease during natalizumab therapy. Mult. Scler. 2013, 19, 1209–1212. [Google Scholar] [CrossRef]
- Selter, R.C.; Biberacher, V.; Grummel, V.; Buck, D.; Eienbroker, C.; Oertel, W.H.; Berthele, A.; Tackenberg, B.; Hemmer, B. Natalizumab treatment decreases serum igm and igg levels in multiple sclerosis patients. Mult. Scler. 2013, 19, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Dooley, J.; Pauwels, I.; Franckaert, D.; Smets, I.; Garcia-Perez, J.E.; Hilven, K.; Danso-Abeam, D.; Terbeek, J.; Nguyen, A.T.; De Muynck, L.; et al. Immunologic profiles of multiple sclerosis treatments reveal shared early b cell alterations. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e240. [Google Scholar] [CrossRef] [PubMed]
- Havrdova, E.; Horakova, D.; Kovarova, I. Alemtuzumab in the treatment of multiple sclerosis: Key clinical trial results and considerations for use. Ther. Adv. Neurol. Disord. 2015, 8, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, G.; Shin, H.J.; Hyun, J.W.; Kim, S.H.; Lee, E.; Kim, H.J. Restoration of regulatory b cell deficiency following alemtuzumab therapy in patients with relapsing multiple sclerosis. J. Neuroinflamm. 2018, 15, 300. [Google Scholar] [CrossRef]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C.; Giovannoni, G.; Schmierer, K. Interpreting lymphocyte reconstitution data from the pivotal phase 3 trials of alemtuzumab. JAMA Neurol. 2017, 74, 961–969. [Google Scholar] [CrossRef]
- Mohn, N.; Pfeuffer, S.; Ruck, T.; Gross, C.C.; Skripuletz, T.; Klotz, L.; Wiendl, H.; Stangel, M.; Meuth, S.G. Alemtuzumab therapy changes immunoglobulin levels in peripheral blood and csf. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e654. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.; Chang, Z.; Pauly, K.; Kwun, J.; Fechner, J.; Hayes, C.; Samaniego, M.; Knechtle, S. Baff is increased in renal transplant patients following treatment with alemtuzumab. Am. J. Transplant. 2009, 9, 1835–1845. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thompson, S.A.; Jones, J.L.; Cox, A.L.; Compston, D.A.; Coles, A.J. B-cell reconstitution and baff after alemtuzumab (campath-1h) treatment of multiple sclerosis. J. Clin. Immunol. 2010, 30, 99–105. [Google Scholar] [CrossRef] [PubMed]
- McGinley, M.P.; Moss, B.P.; Cohen, J.A. Safety of monoclonal antibodies for the treatment of multiple sclerosis. Expert Opin. Drug Saf. 2017, 16, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Ellrichmann, G.; Bolz, J.; Peschke, M.; Duscha, A.; Hellwig, K.; Lee, D.H.; Linker, R.A.; Gold, R.; Haghikia, A. Peripheral cd19(+) b-cell counts and infusion intervals as a surrogate for long-term b-cell depleting therapy in multiple sclerosis and neuromyelitis optica/neuromyelitis optica spectrum disorders. J. Neurol. 2019, 266, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Milo, R. Therapies for multiple sclerosis targeting b cells. Croat. Med. J. 2019, 60, 87–98. [Google Scholar] [CrossRef]
- Jakimovski, D.; Weinstock-Guttman, B.; Ramanathan, M.; Kolb, C.; Hojnacki, D.; Minagar, A.; Zivadinov, R. Ocrelizumab: A b-cell depleting therapy for multiple sclerosis. Expert Opin. Biol. Ther. 2017, 17, 1163–1172. [Google Scholar] [CrossRef]
- Dorner, T.; Burmester, G.R. New approaches of b-cell-directed therapy: Beyond rituximab. Curr. Opin. Rheumatol. 2008, 20, 263–268. [Google Scholar] [CrossRef]
- Vallerskog, T.; Heimburger, M.; Gunnarsson, I.; Zhou, W.; Wahren-Herlenius, M.; Trollmo, C.; Malmstrom, V. Differential effects on baff and april levels in rituximab-treated patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R167. [Google Scholar] [CrossRef]
- Ehrenstein, M.R.; Wing, C. The baffling effects of rituximab in lupus: Danger ahead? Nat. Rev. Rheumatol. 2016, 12, 367–372. [Google Scholar] [CrossRef]
- Jones, J.D.; Hamilton, B.J.; Skopelja, S.; Rigby, W.F. Induction of interleukin-6 production by rituximab in human b cells. Arthritis Rheumatol. 2014, 66, 2938–2946. [Google Scholar] [CrossRef]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of il-6-producing b cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef]
- Wang, C.; Ning, Q.; Jin, K.; Xie, J.; Ye, J. Does rituximab improve clinical outcomes of patients with thyroid-associated ophthalmopathy? A systematic review and meta-analysis. BMC Ophthalmol. 2018, 18, 46. [Google Scholar] [CrossRef]
- Kasper, L.H.; Reder, A.T. Immunomodulatory activity of interferon-beta. Ann. Clin. Transl. Neurol. 2014, 1, 622–631. [Google Scholar] [CrossRef]
- Walther, E.U.; Hohlfeld, R. Multiple sclerosis: Side effects of interferon beta therapy and their management. Neurology 1999, 53, 1622–1627. [Google Scholar] [CrossRef]
- Marziniak, M.; Meuth, S. Current perspectives on interferon beta-1b for the treatment of multiple sclerosis. Adv. Ther. 2014, 31, 915–931. [Google Scholar] [CrossRef]
- Calabresi, P.A.; Pelfrey, C.M.; Tranquill, L.R.; Maloni, H.; McFarland, H.F. Vla-4 expression on peripheral blood lymphocytes is downregulated after treatment of multiple sclerosis with interferon beta. Neurology 1997, 49, 1111–1116. [Google Scholar] [CrossRef]
- Schubert, R.D.; Hu, Y.; Kumar, G.; Szeto, S.; Abraham, P.; Winderl, J.; Guthridge, J.M.; Pardo, G.; Dunn, J.; Steinman, L.; et al. Ifn-beta treatment requires b cells for efficacy in neuroautoimmunity. J. Immunol. 2015, 194, 2110–2116. [Google Scholar] [CrossRef]
- Kalled, S.L. Impact of the baff/br3 axis on b cell survival, germinal center maintenance and antibody production. Semin. Immunol. 2006, 18, 290–296. [Google Scholar] [CrossRef]
- Krumbholz, M.; Faber, H.; Steinmeyer, F.; Hoffmann, L.A.; Kumpfel, T.; Pellkofer, H.; Derfuss, T.; Ionescu, C.; Starck, M.; Hafner, C.; et al. Interferon-beta increases baff levels in multiple sclerosis: Implications for b cell autoimmunity. Brain 2008, 131, 1455–1463. [Google Scholar] [CrossRef]
- Ramgolam, V.S.; Sha, Y.; Marcus, K.L.; Choudhary, N.; Troiani, L.; Chopra, M.; Markovic-Plese, S. B cells as a therapeutic target for ifn-beta in relapsing-remitting multiple sclerosis. J. Immunol. 2011, 186, 4518–4526. [Google Scholar] [CrossRef]
- Rizzo, F.; Giacomini, E.; Mechelli, R.; Buscarinu, M.C.; Salvetti, M.; Severa, M.; Coccia, E.M. Interferon-β therapy specifically reduces pathogenic memory b cells in multiple sclerosis patients by inducing a fas-mediated apoptosis. Immunol. Cell Biol. 2016, 94, 886–894. [Google Scholar] [CrossRef]
- Nakatsuji, Y.; Nakano, M.; Moriya, M.; Kishigami, H.; Tatsumi, C.; Tada, S.; Sadahiro, S.; Naka, T.; Mitani, K.; Funauchi, M.; et al. Beneficial effect of interferon-beta treatment in patients with multiple sclerosis is associated with transient increase in serum il-6 level in response to interferon-beta injection. Cytokine 2006, 36, 69–74. [Google Scholar] [CrossRef]
- Dubucquoi, S.; de Seze, J.; Lefranc, D.; Almeras, L.; Dutoit, V.; Prin, L.; Vermersch, P. Interferon beta in multiple sclerosis: Relationship between sustained serum igg levels and clinical outcome. J. Neuroimmunol. 2002, 129, 232. [Google Scholar] [CrossRef]
- Hedegaard, C.J.; Sellebjerg, F.; Krakauer, M.; Hesse, D.; Bendtzen, K.; Nielsen, C.H. Interferon-beta increases systemic baff levels in multiple sclerosis without increasing autoantibody production. Mult. Scler. 2011, 17, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets gapdh and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef]
- Traub, J.; Traffehn, S.; Ochs, J.; Hausser-Kinzel, S.; Stephan, S.; Scannevin, R.; Bruck, W.; Metz, I.; Weber, M.S. Dimethyl fumarate impairs differentiated b cells and fosters central nervous system integrity in treatment of multiple sclerosis. Brain. Pathol. 2019, 29, 640–657. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Ghadiri, M.; Luessi, F.; Zipp, F.; Li, H.; Giacomini, P.S.; Antel, J.; Bar-Or, A. Dimethyl fumarate treatment mediates an anti-inflammatory shift in b cell subsets of patients with multiple sclerosis. J. Immunol. (Baltimore, Md. : 1950) 2017, 198, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Martin, K.A.; Calabresi, P.A.; Bhargava, P. Dimethyl fumarate alters b-cell memory and cytokine production in ms patients. Ann. Clin. Transl. Neurol. 2017, 4, 351–355. [Google Scholar] [CrossRef]
- Montes Diaz, G.; Fraussen, J.; van Wijmeersch, B.; Hupperts, R.; Somers, V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci. Rep. 2018, 8, 8194. [Google Scholar] [CrossRef]
- Mehta, D.; Miller, C.; Arnold, D.L.; Bame, E.; Bar-Or, A.; Gold, R.; Hanna, J.; Kappos, L.; Liu, S.; Matta, A.; et al. Effect of dimethyl fumarate on lymphocytes in rrms: Implications for clinical practice. Neurology 2019, 92, e1724–e1738. [Google Scholar] [PubMed]
- Staun-Ram, E.; Najjar, E.; Volkowich, A.; Miller, A. Dimethyl fumarate as a first- vs second-line therapy in ms: Focus on b cells. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e508. [Google Scholar] [CrossRef]
- Holm Hansen, R.; Hojsgaard Chow, H.; Sellebjerg, F.; Rode von Essen, M. Dimethyl fumarate therapy suppresses b cell responses and follicular helper t cells in relapsing-remitting multiple sclerosis. Mult. Scler. 2019, 25, 1289–1297. [Google Scholar] [CrossRef]
- Cunill, V.; Massot, M.; Clemente, A.; Calles, C.; Andreu, V.; Nunez, V.; Lopez-Gomez, A.; Diaz, R.M.; Jimenez, M.L.R.; Pons, J.; et al. Relapsing-remitting multiple sclerosis is characterized by a t follicular cell pro-inflammatory shift, reverted by dimethyl fumarate treatment. Front. Immunol. 2018, 9, 1097. [Google Scholar] [CrossRef]
- McKeage, K. Glatiramer acetate 40 mg/ml in relapsing-remitting multiple sclerosis: A review. CNS Drugs 2015, 29, 425–432. [Google Scholar]
- Hausler, D.; Hajiyeva, Z.; Traub, J.W.; Zamvil, S.S.; Lalive, P.H.; Bruck, W.; Weber, M.S. Glatiramer acetate immune modulates b-cell antigen presentation in treatment of ms. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e698. [Google Scholar] [CrossRef]
- Ireland, S.J.; Guzman, A.A.; O’Brien, D.E.; Hughes, S.; Greenberg, B.; Flores, A.; Graves, D.; Remington, G.; Frohman, E.M.; Davis, L.S.; et al. The effect of glatiramer acetate therapy on functional properties of b cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol. 2014, 71, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, P.B.; Carbone, F.; Perna, F.; Bruzzese, D.; La Rocca, C.; Galgani, M.; Montella, S.; Petracca, M.; Florio, C.; Maniscalco, G.T.; et al. Longitudinal assessment of immuno-metabolic parameters in multiple sclerosis patients during treatment with glatiramer acetate. Metabolism 2015, 64, 1112–1121. [Google Scholar] [CrossRef] [PubMed]
- Kuerten, S.; Jackson, L.J.; Kaye, J.; Vollmer, T.L. Impact of glatiramer acetate on b cell-mediated pathogenesis of multiple sclerosis. CNS Drugs 2018, 32, 1039–1051. [Google Scholar] [CrossRef]
- Amrouche, K.; Pers, J.O.; Jamin, C. Glatiramer acetate stimulates regulatory b cell functions. J. Immunol. 2019, 202, 1970–1980. [Google Scholar] [CrossRef]
- Vaknin-Dembinsky, A.; Brill, L.; Orpaz, N.; Abramsky, O.; Karussis, D. Preferential increase of b-cell activating factor in the cerebrospinal fluid of neuromyelitis optica in a white population. Mult. Scler. 2010, 16, 1453–1457. [Google Scholar] [CrossRef] [PubMed]
- Sahraian, M.A.; Moghadasi, A.N.; Azimi, A.R.; Asgari, N.; F, H.A.; Abolfazli, R.; Alaie, S.; Ashtari, F.; Ayromlou, H.; Baghbanian, S.M.; et al. Diagnosis and management of neuromyelitis optica spectrum disorder (nmosd) in iran: A consensus guideline and recommendations. Mult. Scler. Relat. Disord. 2017, 18, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Trebst, C.; Jarius, S.; Berthele, A.; Paul, F.; Schippling, S.; Wildemann, B.; Borisow, N.; Kleiter, I.; Aktas, O.; Kumpfel, T.; et al. Update on the diagnosis and treatment of neuromyelitis optica: Recommendations of the neuromyelitis optica study group (nemos). J. Neurol. 2014, 261, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Weinstock-Guttman, B.; Ramanathan, M.; Lincoff, N.; Napoli, S.Q.; Sharma, J.; Feichter, J.; Bakshi, R. Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (devic disease). Arch. Neurol. 2006, 63, 957–963. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, W.; Park, M.S.; Sohn, E.H.; Li, X.F.; Kim, H.J. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch. Neurol. 2011, 68, 473–479. [Google Scholar] [CrossRef]
- Cabre, P.; Olindo, S.; Marignier, R.; Jeannin, S.; Merle, H.; Smadja, D.; Aegis of French National Observatory of Multiple, S. Efficacy of mitoxantrone in neuromyelitis optica spectrum: Clinical and neuroradiological study. J. Neurol. Neurosurg. Psychiatry 2013, 84, 511–516. [Google Scholar] [CrossRef]
- Sellner, J.; Boggild, M.; Clanet, M.; Hintzen, R.Q.; Illes, Z.; Montalban, X.; Du Pasquier, R.A.; Polman, C.H.; Sorensen, P.S.; Hemmer, B. Efns guidelines on diagnosis and management of neuromyelitis optica. Eur. J. Neurol. 2010, 17, 1019–1032. [Google Scholar] [CrossRef]
- Kimbrough, D.J.; Fujihara, K.; Jacob, A.; Lana-Peixoto, M.A.; Leite, M.I.; Levy, M.; Marignier, R.; Nakashima, I.; Palace, J.; de Seze, J.; et al. Treatment of neuromyelitis optica: Review and recommendations. Mult. Scler. Relat. Disord. 2012, 1, 180–187. [Google Scholar] [CrossRef]
- Torres, J.; Pruitt, A.; Balcer, L.; Galetta, S.; Markowitz, C.; Dahodwala, N. Analysis of the treatment of neuromyelitis optica. J. Neurol. Sci. 2015, 351, 31–35. [Google Scholar] [CrossRef]
- Elsone, L.; Kitley, J.; Luppe, S.; Lythgoe, D.; Mutch, K.; Jacob, S.; Brown, R.; Moss, K.; McNeillis, B.; Goh, Y.Y.; et al. Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: A multicentre retrospective observational study from the UK. Mult. Scler. 2014, 20, 1533–1540. [Google Scholar] [CrossRef]
- Awad, A.; Stuve, O. Cyclophosphamide in multiple sclerosis: Scientific rationale, history and novel treatment paradigms. Ther. Adv. Neurol. Disord. 2009, 2, 50–61. [Google Scholar]
- Yaguchi, H.; Sakushima, K.; Takahashi, I.; Nishimura, H.; Yashima-Yamada, M.; Nakamura, M.; Tsuzaka, K.; Maruo, Y.; Takahashi, T.; Yabe, I.; et al. Efficacy of intravenous cyclophosphamide therapy for neuromyelitis optica spectrum disorder. Int. Med. 2013, 52, 969–972. [Google Scholar]
- Mok, C.C.; To, C.H.; Mak, A.; Poon, W.L. Immunoablative cyclophosphamide for refractory lupus-related neuromyelitis optica. J. Rheumatol. 2008, 35, 172–174. [Google Scholar]
- Saida, T. Treatment of nmo. Rinsho Shinkeigaku 2009, 49, 902–905. [Google Scholar] [CrossRef][Green Version]
- Yoshii, F.; Moriya, Y.; Ohnuki, T.; Ryo, M.; Takahashi, W. Fingolimod-induced leukoencephalopathy in a patient with neuromyelitis optica spectrum disorder. Mult. Scler. Relat. Disord. 2016, 7, 53–57. [Google Scholar] [CrossRef]
- Izaki, S.; Narukawa, S.; Kubota, A.; Mitsui, T.; Fukaura, H.; Nomura, K. A case of neuromyelitis optica spectrum disorder developing a fulminant course with multiple white-matter lesions following fingolimod treatment. Rinsho Shinkeigaku 2013, 53, 513–517. [Google Scholar] [CrossRef][Green Version]
- Wagner, F.; Grunder, L.; Hakim, A.; Kamber, N.; Horn, M.P.; Muellner, J.; Hoepner, R.; Wiest, R.; Metz, I.; Chan, A.; et al. Rebound after fingolimod and a single daclizumab injection in a patient retrospectively diagnosed with nmo spectrum disorder-mri apparent diffusion coefficient maps in differential diagnosis of demyelinating cns disorders. Front. Neurol. 2018, 9, 782. [Google Scholar]
- Kleiter, I.; Hellwig, K.; Berthele, A.; Kumpfel, T.; Linker, R.A.; Hartung, H.P.; Paul, F.; Aktas, O.; Neuromyelitis Optica Study, G. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch. Neurol. 2012, 69, 239–245. [Google Scholar] [CrossRef]
- Kitley, J.; Evangelou, N.; Kuker, W.; Jacob, A.; Leite, M.I.; Palace, J. Catastrophic brain relapse in seronegative nmo after a single dose of natalizumab. J. Neurol. Sci. 2014, 339, 223–225. [Google Scholar]
- Barnett, M.H.; Prineas, J.W.; Buckland, M.E.; Parratt, J.D.; Pollard, J.D. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult. Scler. 2012, 18, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Hoshi, M.; Hemmer, B.; Berthele, A. Failure of alemtuzumab as a rescue in a nmosd patient treated with rituximab. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e208. [Google Scholar] [CrossRef]
- Tsourdi, E.; Gruber, M.; Rauner, M.; Blankenburg, J.; Ziemssen, T.; Hofbauer, L.C. Graves’ disease after treatment with alemtuzumab for multiple sclerosis. Hormones (Athens) 2015, 14, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Cotter, J.; Klingman, J.; Huang, E.J.; Cree, B.A. Massive cns monocytic infiltration at autopsy in an alemtuzumab-treated patient with nmo. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e34. [Google Scholar] [CrossRef] [PubMed]
- Cabre, P.; Mejdoubi, M.; Jeannin, S.; Merle, H.; Plumelle, Y.; Cavillon, G.; Smadja, D.; Marignier, R.; Francophone Society of Multiple, S.; Investigators, O. Treatment of neuromyelitis optica with rituximab: A 2-year prospective multicenter study. J. Neurol. 2018, 265, 917–925. [Google Scholar] [CrossRef]
- Alldredge, B.; Jordan, A.; Imitola, J.; Racke, M.K. Safety and efficacy of rituximab: Experience of a single multiple sclerosis center. Clin. Neuropharmacol. 2018, 41, 56–59. [Google Scholar] [CrossRef]
- Gao, F.; Chai, B.; Gu, C.; Wu, R.; Dong, T.; Yao, Y.; Zhang, Y. Effectiveness of rituximab in neuromyelitis optica: A meta-analysis. BMC Neurol. 2019, 19, 36. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, W.; Li, X.F.; Jung, I.J.; Kim, H.J. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult. Scler. 2012, 18, 1480–1483. [Google Scholar] [CrossRef]
- Jarernsook, B.; Siritho, S.; Prayoonwiwat, N. Efficacy and safety of beta-interferon in thai patients with demyelinating diseases. Mult. Scler. 2013, 19, 585–592. [Google Scholar] [CrossRef]
- Asgari, N.; Kyvik, K.O.; Steenstrup, T.; Stenager, E.; Lillevang, S.T. Antibodies against interferon-beta in neuromyelitis optica patients. J. Neurol. Sci. 2014, 339, 52–56. [Google Scholar] [CrossRef]
- Palace, J.; Leite, M.I.; Nairne, A.; Vincent, A. Interferon beta treatment in neuromyelitis optica: Increase in relapses and aquaporin 4 antibody titers. Arch. Neurol. 2010, 67, 1016–1017. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Tanaka, K.; Komori, M. Interferon-beta(1b) treatment in neuromyelitis optica. Eur. Neurol. 2009, 62, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Lin, K.H.; Lee, T.C.; Lee, C.L.; Chen, S.Y.; Chen, S.J.; Chin, L.T.; Tsai, C.P. Poor responses to interferon-beta treatment in patients with neuromyelitis optica and multiple sclerosis with long spinal cord lesions. PLoS ONE 2014, 9, e98192. [Google Scholar] [CrossRef] [PubMed]
- Uzawa, A.; Mori, M.; Hayakawa, S.; Masuda, S.; Kuwabara, S. Different responses to interferon beta-1b treatment in patients with neuromyelitis optica and multiple sclerosis. Eur. J. Neurol. 2010, 17, 672–676. [Google Scholar] [CrossRef]
- Warabi, Y.; Matsumoto, Y.; Hayashi, H. Interferon beta-1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J. Neurol. Sci. 2007, 252, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Popiel, M.; Psujek, M.; Bartosik-Psujek, H. Severe disease exacerbation in a patient with neuromyelitis optica spectrum disorder during treatment with dimethyl fumarate. Mult. Scler. Relat. Disord. 2018, 26, 204–206. [Google Scholar] [CrossRef]
- Yamout, B.I.; Beaini, S.; Zeineddine, M.M.; Akkawi, N. Catastrophic relapses following initiation of dimethyl fumarate in two patients with neuromyelitis optica spectrum disorder. Mult. Scler. 2017, 23, 1297–1300. [Google Scholar] [CrossRef]
- Ayzenberg, I.; Schollhammer, J.; Hoepner, R.; Hellwig, K.; Ringelstein, M.; Aktas, O.; Kumpfel, T.; Krumbholz, M.; Trebst, C.; Paul, F.; et al. Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: A multicenter retrospective study. J. Neurol. 2016, 263, 575–582. [Google Scholar]
- Stellmann, J.P.; Krumbholz, M.; Friede, T.; Gahlen, A.; Borisow, N.; Fischer, K.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Immunotherapies in neuromyelitis optica spectrum disorder: Efficacy and predictors of response. J. Neurol. Neurosurg. Psychiatry 2017, 88, 639–647. [Google Scholar] [CrossRef]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Hausler, D.; Nessler, S.; Kruse, N.; Bruck, W.; Metz, I. Natalizumab analogon therapy is effective in a b cell-dependent multiple sclerosis model. Neuropathol. Appl. Neurobiol. 2015, 41, 814–831. [Google Scholar] [CrossRef] [PubMed]
- Paty, D.W.; Li, D.K. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. Ii. Mri analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. Ubc ms/mri study group and the ifnb multiple sclerosis study group. Neurology 1993, 43, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; Yu, H.; Qiao, J.; Xiao, B.; Zhao, G.; Wu, Z.; Li, Z.; Lu, C. Impaired regulatory function and enhanced intrathecal activation of b cells in neuromyelitis optica: Distinct from multiple sclerosis. Mult. Scler. 2013, 19, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Quan, C.; ZhangBao, J.; Lu, J.; Zhao, C.; Cai, T.; Wang, B.; Yu, H.; Qiao, J.; Lu, C. The immune balance between memory and regulatory b cells in nmo and the changes of the balance after methylprednisolone or rituximab therapy. J. Neuroimmunol. 2015, 282, 45–53. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, W.; Li, X.F.; Jung, I.J.; Kim, H.J. Repeated treatment with rituximab based on the assessment of peripheral circulating memory b cells in patients with relapsing neuromyelitis optica over 2 years. Arch. Neurol. 2011, 68, 1412–1420. [Google Scholar] [CrossRef]
- Cohen, M.; Romero, G.; Bas, J.; Ticchioni, M.; Rosenthal, M.; Lacroix, R.; Brunet, C.; Rico, A.; Pelletier, J.; Audoin, B.; et al. Monitoring cd27+ memory b-cells in neuromyelitis optica spectrum disorders patients treated with rituximab: Results from a bicentric study. J. Neurol. Sci. 2017, 373, 335–338. [Google Scholar] [CrossRef]
- Lebrun, C.; Cohen, M.; Rosenthal-Allieri, M.A.; Bresch, S.; Benzaken, S.; Marignier, R.; Seitz-Polski, B.; Ticchioni, M. Only follow-up of memory b cells helps monitor rituximab administration to patients with neuromyelitis optica spectrum disorders. Neurol. Ther. 2018, 7, 373–383. [Google Scholar] [CrossRef]
- Kim, S.H.; Huh, S.Y.; Lee, S.J.; Joung, A.; Kim, H.J. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013, 70, 1110–1117. [Google Scholar] [CrossRef]
- Lanzillotta, M.; Della-Torre, E.; Milani, R.; Bozzolo, E.; Bozzalla-Cassione, E.; Rovati, L.; Arcidiacono, P.G.; Partelli, S.; Falconi, M.; Ciceri, F.; et al. Effects of glucocorticoids on b-cell subpopulations in patients with igg4-related disease. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 118), 159–166. [Google Scholar]
- Lin, W.; Zhang, P.; Chen, H.; Chen, Y.; Yang, H.; Zheng, W.; Zhang, X.; Zhang, F.; Zhang, W.; Lipsky, P.E. Circulating plasmablasts/plasma cells: A potential biomarker for igg4-related disease. Arthritis Res. Ther. 2017, 19, 25. [Google Scholar] [CrossRef]
- Kowarik, M.C.; Astling, D.; Gasperi, C.; Wemlinger, S.; Schumann, H.; Dzieciatkowska, M.; Ritchie, A.M.; Hemmer, B.; Owens, G.P.; Bennett, J.L. Cns aquaporin-4-specific b cells connect with multiple b-cell compartments in neuromyelitis optica spectrum disorder. Ann. Clin. Transl. Neurol. 2017, 4, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. Il-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]
- Lavie, F.; Miceli-Richard, C.; Ittah, M.; Sellam, J.; Gottenberg, J.E.; Mariette, X. Increase of b cell-activating factor of the tnf family (baff) after rituximab treatment: Insights into a new regulating system of baff production. Ann. Rheum. Dis. 2007, 66, 700–703. [Google Scholar] [CrossRef]
- Jarius, S.; Ruprecht, K.; Kleiter, I.; Borisow, N.; Asgari, N.; Pitarokoili, K.; Pache, F.; Stich, O.; Beume, L.A.; Hummert, M.W.; et al. Mog-igg in nmo and related disorders: A multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J. Neuroinflamm. 2016, 13, 280. [Google Scholar] [CrossRef]
- Nakashima, I.; Takahashi, T.; Cree, B.A.; Kim, H.J.; Suzuki, C.; Genain, C.P.; Vincent, T.; Fujihara, K.; Itoyama, Y.; Bar-Or, A. Transient increases in anti-aquaporin-4 antibody titers following rituximab treatment in neuromyelitis optica, in association with elevated serum baff levels. J. Clin. Neurosci. 2011, 18, 997–998. [Google Scholar] [CrossRef] [PubMed]
- Rowland, S.L.; Leahy, K.F.; Halverson, R.; Torres, R.M.; Pelanda, R. Baff receptor signaling aids the differentiation of immature b cells into transitional b cells following tonic bcr signaling. J. Immunol. 2010, 185, 4570–4581. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, K.; Zhong, X.; Qiu, W.; Dai, Y.; Wu, A.; Hu, X. Cerebrospinal fluid baff and april levels in neuromyelitis optica and multiple sclerosis patients during relapse. J. Clin. Immunol. 2012, 32, 1007–1011. [Google Scholar] [CrossRef]
- Lesley, R.; Xu, Y.; Kalled, S.L.; Hess, D.M.; Schwab, S.R.; Shu, H.B.; Cyster, J.G. Reduced competitiveness of autoantigen-engaged b cells due to increased dependence on baff. Immunity 2004, 20, 441–453. [Google Scholar] [CrossRef]
- Vincent, F.B.; Morand, E.F.; Schneider, P.; Mackay, F. The baff/april system in sle pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Makris, N. Neuromyelitis optica: A distinct demyelinating disease of the central nervous system. Acta Neurol. Scand. 2008, 118, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.W.; Davidson, A. Baff inhibition in sle-is tolerance restored? Immunol. Rev. 2019, 292, 102–119. [Google Scholar] [CrossRef]
- Huntington, N.D.; Tomioka, R.; Clavarino, C.; Chow, A.M.; Linares, D.; Mana, P.; Rossjohn, J.; Cachero, T.G.; Qian, F.; Kalled, S.L.; et al. A baff antagonist suppresses experimental autoimmune encephalomyelitis by targeting cell-mediated and humoral immune responses. Int. Immunol. 2006, 18, 1473–1485. [Google Scholar] [CrossRef]
- Zhou, X.; Xia, Z.; Lan, Q.; Wang, J.; Su, W.; Han, Y.P.; Fan, H.; Liu, Z.; Stohl, W.; Zheng, S.G. Baff promotes th17 cells and aggravates experimental autoimmune encephalomyelitis. PLoS ONE 2011, 6, e23629. [Google Scholar] [CrossRef]
- Kappos, L.; Hartung, H.P.; Freedman, M.S.; Boyko, A.; Radu, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (atams): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Sergott, R.C.; Bennett, J.L.; Rieckmann, P.; Montalban, X.; Mikol, D.; Freudensprung, U.; Plitz, T.; van Beek, J.; Group, A.T. Aton: Results from a phase ii randomized trial of the b-cell-targeting agent atacicept in patients with optic neuritis. J. Neurol. Sci. 2015, 351, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.L.; Phuah, C.L.; Cox, A.L.; Thompson, S.A.; Ban, M.; Shawcross, J.; Walton, A.; Sawcer, S.J.; Compston, A.; Coles, A.J. Il-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (campath-1h). J. Clin. Invest. 2009, 119, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Heckers, S.; Pedreiturria, X.; Hess, D.; Trampe, A.K.; Chan, A.; Gold, R. Low dose fumaric acid esters are effective in a mouse model of spontaneous chronic encephalomyelitis. J. Neuroimmunol. 2015, 285, 16–21. [Google Scholar] [CrossRef]
- Gold, R.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Giovannoni, G.; Selmaj, K.; Tornatore, C.; Sweetser, M.T.; Yang, M.; Sheikh, S.I.; et al. Placebo-controlled phase 3 study of oral bg-12 for relapsing multiple sclerosis. N. Engl. J. Med. 2012, 367, 1098–1107. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Miller, D.H.; Phillips, J.T.; Hutchinson, M.; Havrdova, E.; Kita, M.; Yang, M.; Raghupathi, K.; Novas, M.; Sweetser, M.T.; et al. Placebo-controlled phase 3 study of oral bg-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 2012, 367, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.A.; Donofrio, P.; Bril, V.; Dalakas, M.C.; Deng, C.; Hanna, K.; Hartung, H.P.; Latov, N.; Merkies, I.S.; van Doorn, P.A.; et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ice study): A randomised placebo-controlled trial. Lancet Neurol. 2008, 7, 136–144. [Google Scholar] [CrossRef]
- Magraner, M.J.; Coret, F.; Casanova, B. The effect of intravenous immunoglobulin on neuromyelitis optica. Neurologia 2013, 28, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Elsone, L.; Panicker, J.; Mutch, K.; Boggild, M.; Appleton, R.; Jacob, A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: Experience in 10 patients. Mult. Scler. 2014, 20, 501–504. [Google Scholar] [CrossRef]
- Abboud, H.; Petrak, A.; Mealy, M.; Sasidharan, S.; Siddique, L.; Levy, M. Treatment of acute relapses in neuromyelitis optica: Steroids alone versus steroids plus plasma exchange. Mult. Scler. 2016, 22, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Nakashima, I.; Misu, T.; Miyazawa, I.; Shiga, Y.; Fujihara, K.; Itoyama, Y. Therapeutic efficacy of plasma exchange in nmo-igg-positive patients with neuromyelitis optica. Mult. Scler. 2007, 13, 128–132. [Google Scholar] [CrossRef]
- Bonnan, M.; Valentino, R.; Debeugny, S.; Merle, H.; Ferge, J.L.; Mehdaoui, H.; Cabre, P. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of nmo spectrum disorders. J. Neurol. Neurosurg. Psychiatry 2018, 89, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Pardo, S.; Giovannoni, G.; Hawkes, C.; Lechner-Scott, J.; Waubant, E.; Levy, M. Editorial on: Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. Mult. Scler. Relat. Disord. 2019, 33, A1–A2. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, T.; Kleiter, I.; Fujihara, K.; Palace, J.; Greenberg, B.; Zakrzewska-Pniewska, B.; Patti, F.; Tsai, C.P.; Saiz, A.; Yamazaki, H.; et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 2114–2124. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (n-momentum): A double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Brachet, G.; Bourquard, T.; Gallay, N.; Reiter, E.; Gouilleux-Gruart, V.; Poupon, A.; Watier, H. Eculizumab epitope on complement c5: Progress towards a better understanding of the mechanism of action. Mol. Immunol. 2016, 77, 126–131. [Google Scholar] [CrossRef]
- Soltys, J.; Liu, Y.; Ritchie, A.; Wemlinger, S.; Schaller, K.; Schumann, H.; Owens, G.P.; Bennett, J.L. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J. Clin. Invest. 2019, 129, 2000–2013. [Google Scholar] [CrossRef]
- Dhillon, S. Eculizumab: A review in generalized myasthenia gravis. Drugs 2018, 78, 367–376. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Weitz, I.C.; Razavi, P.; Rochanda, L.; Zwicker, J.; Furie, B.; Manly, D.; Mackman, N.; Green, R.; Liebman, H.A. Eculizumab therapy results in rapid and sustained decreases in markers of thrombin generation and inflammation in patients with pnh independent of its effects on hemolysis and microparticle formation. Thromb. Res. 2012, 130, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Alfinito, F.; Ruggiero, G.; Sica, M.; Udhayachandran, A.; Rubino, V.; Della Pepa, R.; Palatucci, A.T.; Annunziatella, M.; Notaro, R.; Risitano, A.M.; et al. Eculizumab treatment modifies the immune profile of pnh patients. Immunobiology 2012, 217, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Ravulizumab. In Drugs and Lactation Database (Lactmed); National Library of Medicine: Bethesda, MD, USA, 2006.
- Chen, D.; Gallagher, S.; Monson, N.L.; Herbst, R.; Wang, Y. Inebilizumab, a b cell-depleting anti-cd19 antibody for the treatment of autoimmune neurological diseases: Insights from preclinical studies. J. Clin. Med. 2016, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A.; Grove, R.A.; Austin, D.J.; Tolson, J.M.; VanMeter, S.A.; Lewis, E.W.; Derosier, F.J.; Lopez, M.C.; Kavanagh, S.T.; Miller, A.E.; et al. Subcutaneous ofatumumab in patients with relapsing-remitting multiple sclerosis: The mirror study. Neurology 2018, 90, e1805–e1814. [Google Scholar] [CrossRef] [PubMed]
- Ayzenberg, I.; Kleiter, I.; Schroder, A.; Hellwig, K.; Chan, A.; Yamamura, T.; Gold, R. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-cd20 therapy. JAMA Neurol. 2013, 70, 394–397. [Google Scholar] [CrossRef]
- Uchida, T.; Mori, M.; Uzawa, A.; Masuda, H.; Muto, M.; Ohtani, R.; Kuwabara, S. Increased cerebrospinal fluid metalloproteinase-2 and interleukin-6 are associated with albumin quotient in neuromyelitis optica: Their possible role on blood-brain barrier disruption. Mult. Scler. 2017, 23, 1072–1084. [Google Scholar] [CrossRef]
- Kimura, A.; Naka, T.; Kishimoto, T. Il-6-dependent and -independent pathways in the development of interleukin 17-producing t helper cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12099–12104. [Google Scholar] [CrossRef]
- Araki, M.; Matsuoka, T.; Miyamoto, K.; Kusunoki, S.; Okamoto, T.; Murata, M.; Miyake, S.; Aranami, T.; Yamamura, T. Efficacy of the anti-il-6 receptor antibody tocilizumab in neuromyelitis optica: A pilot study. Neurology 2014, 82, 1302–1306. [Google Scholar] [CrossRef]
- Ringelstein, M.; Ayzenberg, I.; Harmel, J.; Lauenstein, A.S.; Lensch, E.; Stogbauer, F.; Hellwig, K.; Ellrichmann, G.; Stettner, M.; Chan, A.; et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol. 2015, 72, 756–763. [Google Scholar] [CrossRef]
- Ectrims 2019 committees. Mult. Scler. 2019, 25, 1–2. [CrossRef]
- Moura, R.A.; Quaresma, C.; Vieira, A.R.; Goncalves, M.J.; Polido-Pereira, J.; Romao, V.C.; Martins, N.; Canhao, H.; Fonseca, J.E. B-cell phenotype and igd-cd27- memory b cells are affected by tnf-inhibitors and tocilizumab treatment in rheumatoid arthritis. PLoS ONE 2017, 12, e0182927. [Google Scholar] [CrossRef]
- Zhou, L.; Xiao, D.M.; Qin, W.; Xie, B.H.; Wang, T.H.; Huang, H.; Zhao, B.J.; Han, X.; Sun, Q.Q.; Wu, X.D.; et al. The clinical value of hematological markers in rheumatoid arthritis patients treated with tocilizumab. J. Clin. Lab. Anal. 2019, 33, e22862. [Google Scholar] [CrossRef]
- Roll, P.; Muhammad, K.; Schumann, M.; Kleinert, S.; Einsele, H.; Dorner, T.; Tony, H.P. In vivo effects of the anti-interleukin-6 receptor inhibitor tocilizumab on the b cell compartment. Arthritis Rheum. 2011, 63, 1255–1264. [Google Scholar] [CrossRef]
- Choi, I.A.; Lee, S.J.; Park, W.; Park, S.H.; Shim, S.C.; Baek, H.J.; Yoo, D.H.; Kim, H.A.; Lee, S.K.; Lee, Y.J.; et al. Effects of tocilizumab therapy on serum interleukin-33 and interleukin-6 levels in patients with rheumatoid arthritis. Arch Rheumatol. 2018, 33, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Igawa, T.; Ishii, S.; Tachibana, T.; Maeda, A.; Higuchi, Y.; Shimaoka, S.; Moriyama, C.; Watanabe, T.; Takubo, R.; Doi, Y.; et al. Antibody recycling by engineered ph-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 2010, 28, 1203–1207. [Google Scholar] [CrossRef]
- Verkman, A.S.; Phuan, P.W.; Asavapanumas, N.; Tradtrantip, L. Biology of aqp4 and anti-aqp4 antibody: Therapeutic implications for nmo. Brain. Pathol. 2013, 23, 684–695. [Google Scholar] [CrossRef]
- Verkman, A.S.; Smith, A.J.; Phuan, P.W.; Tradtrantip, L.; Anderson, M.O. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin. Ther. Targets 2017, 21, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Duan, T.; Tradtrantip, L.; Phuan, P.W.; Bennett, J.L.; Verkman, A.S. Affinity-matured ‘aquaporumab’ anti-aquaporin-4 antibody for therapy of seropositive neuromyelitis optica spectrum disorders. Neuropharmacology 2020, 162, 107827. [Google Scholar] [CrossRef]
- Saadoun, S.; Waters, P.; MacDonald, C.; Bell, B.A.; Vincent, A.; Verkman, A.S.; Papadopoulos, M.C. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin g-induced damage in mouse brain. Ann. Neurol. 2012, 71, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Herges, K.; de Jong, B.A.; Kolkowitz, I.; Dunn, C.; Mandelbaum, G.; Ko, R.M.; Maini, A.; Han, M.H.; Killestein, J.; Polman, C.; et al. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult. Scler. 2012, 18, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Verkman, A.S. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J. Clin. Invest. 2013, 123, 2306–2316. [Google Scholar] [CrossRef]
- Muroishi, T.; Sakai, K.; Yanase, D.; Ikeda, Y.; Machiya, T.; Kato-Motozaki, Y.; Samuraki, M.; Yamada, M. Serum anti-aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder presenting as acute eosinophilic encephalomyelitis. J. Clin. Neurosci. 2018, 48, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Katz Sand, I.; Fabian, M.T.; Telford, R.; Kraus, T.A.; Chehade, M.; Masilamani, M.; Moran, T.; Farrell, C.; Ebel, S.; Cook, L.J.; et al. Open-label, add-on trial of cetirizine for neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e441. [Google Scholar] [CrossRef]
- Zhang, C.; Tian, D.C.; Yang, C.S.; Han, B.; Wang, J.; Yang, L.; Shi, F.D. Safety and efficacy of bortezomib in patients with highly relapsing neuromyelitis optica spectrum disorder. JAMA Neurol. 2017, 74, 1010–1012. [Google Scholar] [CrossRef]
- Burman, J.; Tolf, A.; Hagglund, H.; Askmark, H. Autologous haematopoietic stem cell transplantation for neurological diseases. J. Neurol. Neurosurg. Psychiatry 2018, 89, 147–155. [Google Scholar] [CrossRef]
- Matiello, M.; Pittock, S.J.; Porrata, L.; Weinshenker, B.G. Failure of autologous hematopoietic stem cell transplantation to prevent relapse of neuromyelitis optica. Arch. Neurol. 2011, 68, 953–955. [Google Scholar] [CrossRef][Green Version]


{kind=link}
{kind=link}
| Agent | Lymphocyte Count | B Cell Count | % Transitional/Regulatory BC | % Memory/Activated BC | Plasmablast Count | Interleukin-6 (Serum) | IgG (Serum) | BAFF (Serum) |
|---|---|---|---|---|---|---|---|---|
| MS: Inhibition of Proliferation | ||||||||
| Cladribine | ↓ | ↓ | ↑ | ↓ | ⇿ | ⇿ | ↓ | – |
| Teriflunomide | ↓ | ↓ | ⇿ | ⇿ | ↓ | – | ↓ | – |
| Mitoxanthrone | ↓ | ↓ | ↑ | ↓ | – | – | ↓ | ↑ |
| (Azathioprine) | ↓ | ↓ | ↓ | ↓ | ⇿ | ↓ | ↓ | ↑ |
| (Cyclophosphamide) | ↓ | ↓ | – | ↓ | ↓ | – | ↓ | ↑ |
| Inhibition of Migration | ||||||||
| Fingolimod | ↓ | ↓ | ↑ | ↓ | ↑1 | ↓ | ↓ | ↑ |
| Siponimod | ↓ | ↓ | ↑ | ↓ | ⇿2 | ↓3 | ⇿ | – |
| Natalizumab | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↓ | ⇿ |
| Depletion | ||||||||
| Alemtuzumab | ↓ | ↑4 | ↑ | ↓ | ↓ | ↑5 | ↓ | ↑ |
| Ocrelizumab | ↓ | ↓ | ↑ | ↓ | ⇿ | ⇿6 | ↓ | ↑6 |
| Immunomodulation | ||||||||
| Interferon-β | ⇿ | ↑ | ↑ | ↓ | ↑ | ↑ | ↓ | ↑ |
| Dimethyl fumarate | ↓ | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | ⇿ |
| Glatiramer acetate | ⇿ | ⇿ | ↑ | ↓ | ↓ | ↑7 | ↑ | ↑ |
| NMOSD: B Cell Depletion | ||||||||
| Rituximab | ↓ | ↓ | ↑ | ↓ | ⇿ | ⇿ | ↓ | ↑ |
| Inebilizumab | ↓ | ↓ | – | – | ↓ | – | ↓ | – |
| Ofatumumab | ↓ | ↓ | – | – | – | – | – | – |
| Interleukin-6 Receptor Antibodies | ||||||||
| Tocilizumab | ⇿8 | ⇿ | ⇿ | ↓8 | ⇿ | ⇿8 | ↓ | ⇿8 |
| Satralizumab | – | – | – | – | – | – | – | – |
| Complement Factor C5 Antibodies | ||||||||
| Eculizumab | ↑9 | ↑9 | – | – | – | ↓9 | – | – |
| Ravulizumab | – | – | – | – | – | – | – | – |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traub, J.; Häusser-Kinzel, S.; Weber, M.S. Differential Effects of MS Therapeutics on B Cells—Implications for Their Use and Failure in AQP4-Positive NMOSD Patients. Int. J. Mol. Sci. 2020, 21, 5021. https://doi.org/10.3390/ijms21145021
Traub J, Häusser-Kinzel S, Weber MS. Differential Effects of MS Therapeutics on B Cells—Implications for Their Use and Failure in AQP4-Positive NMOSD Patients. International Journal of Molecular Sciences. 2020; 21(14):5021. https://doi.org/10.3390/ijms21145021
Chicago/Turabian StyleTraub, Jan, Silke Häusser-Kinzel, and Martin S. Weber. 2020. "Differential Effects of MS Therapeutics on B Cells—Implications for Their Use and Failure in AQP4-Positive NMOSD Patients" International Journal of Molecular Sciences 21, no. 14: 5021. https://doi.org/10.3390/ijms21145021
APA StyleTraub, J., Häusser-Kinzel, S., & Weber, M. S. (2020). Differential Effects of MS Therapeutics on B Cells—Implications for Their Use and Failure in AQP4-Positive NMOSD Patients. International Journal of Molecular Sciences, 21(14), 5021. https://doi.org/10.3390/ijms21145021