Comparative Genomics of Stenotrophomonas maltophilia and Stenotrophomonas rhizophila Revealed Characteristic Features of Both Species





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
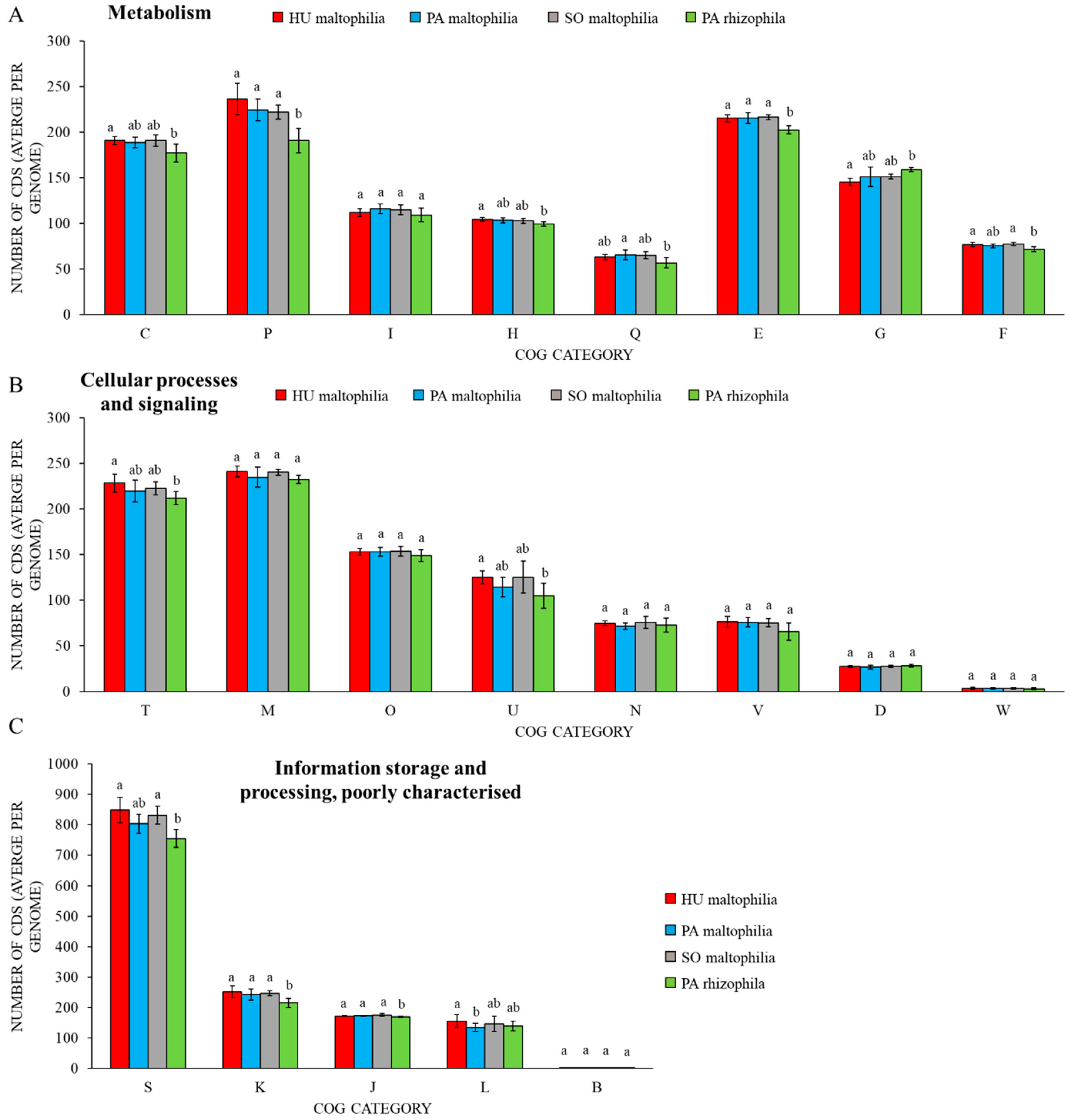
2. Results
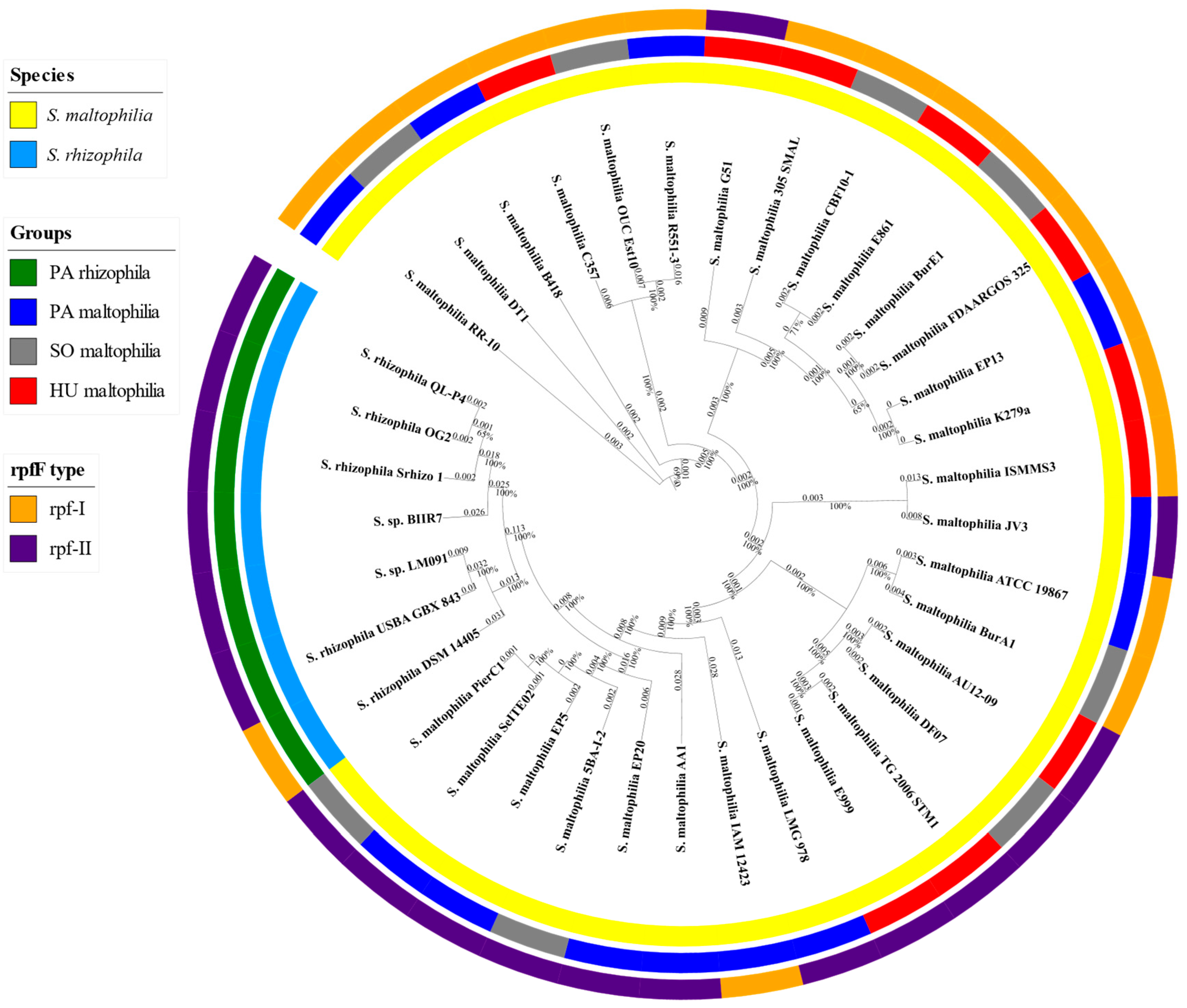
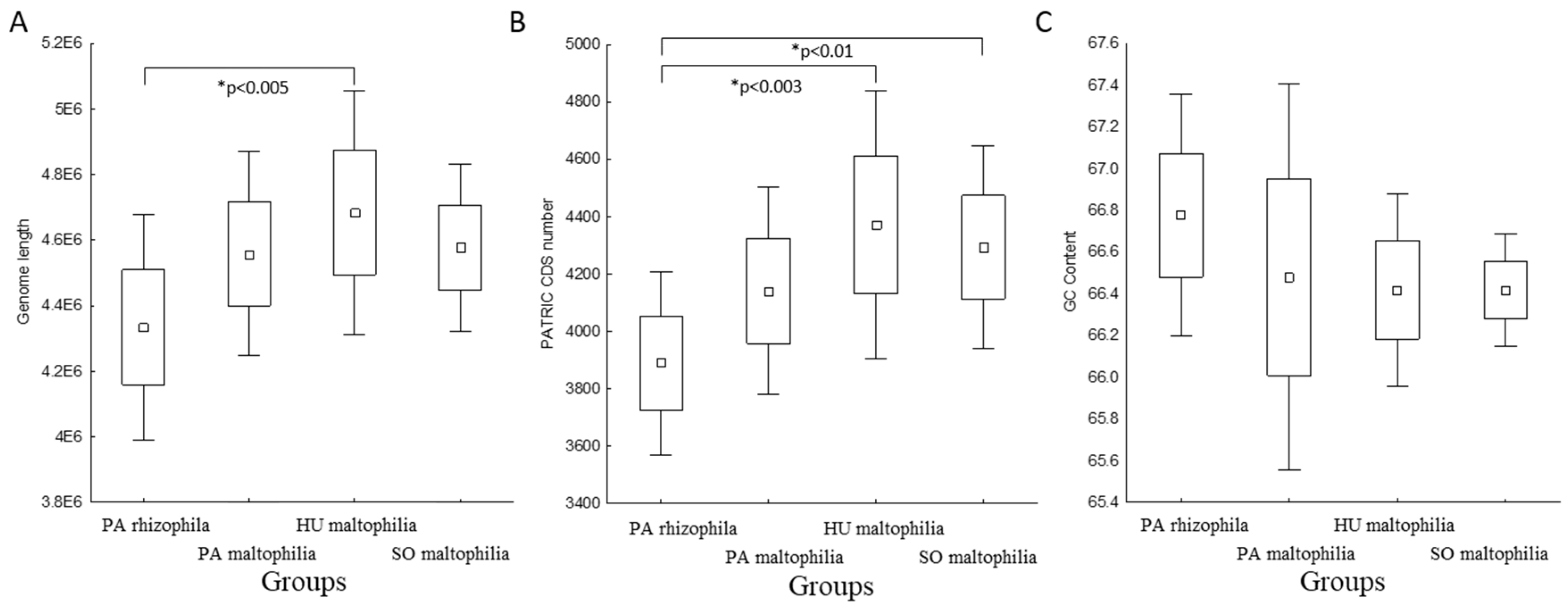
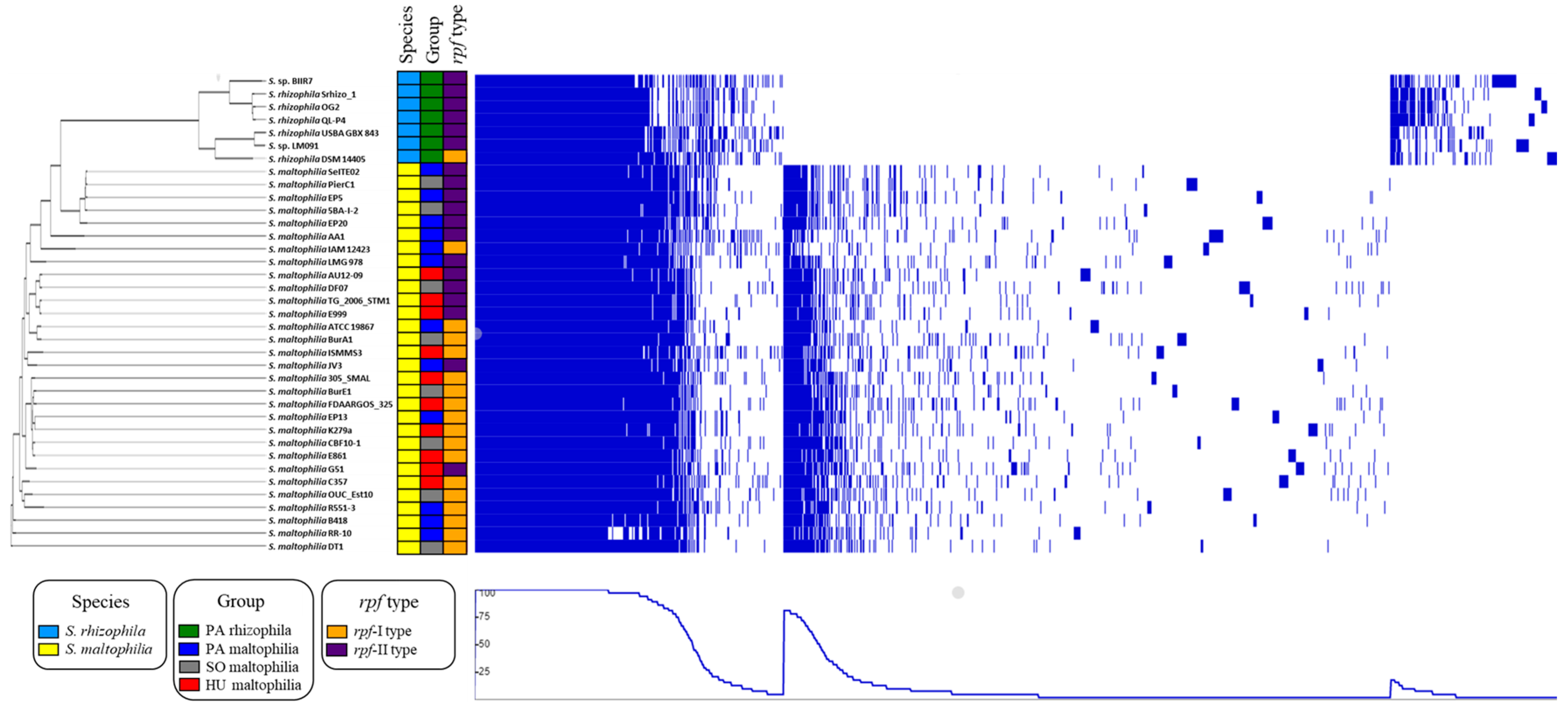
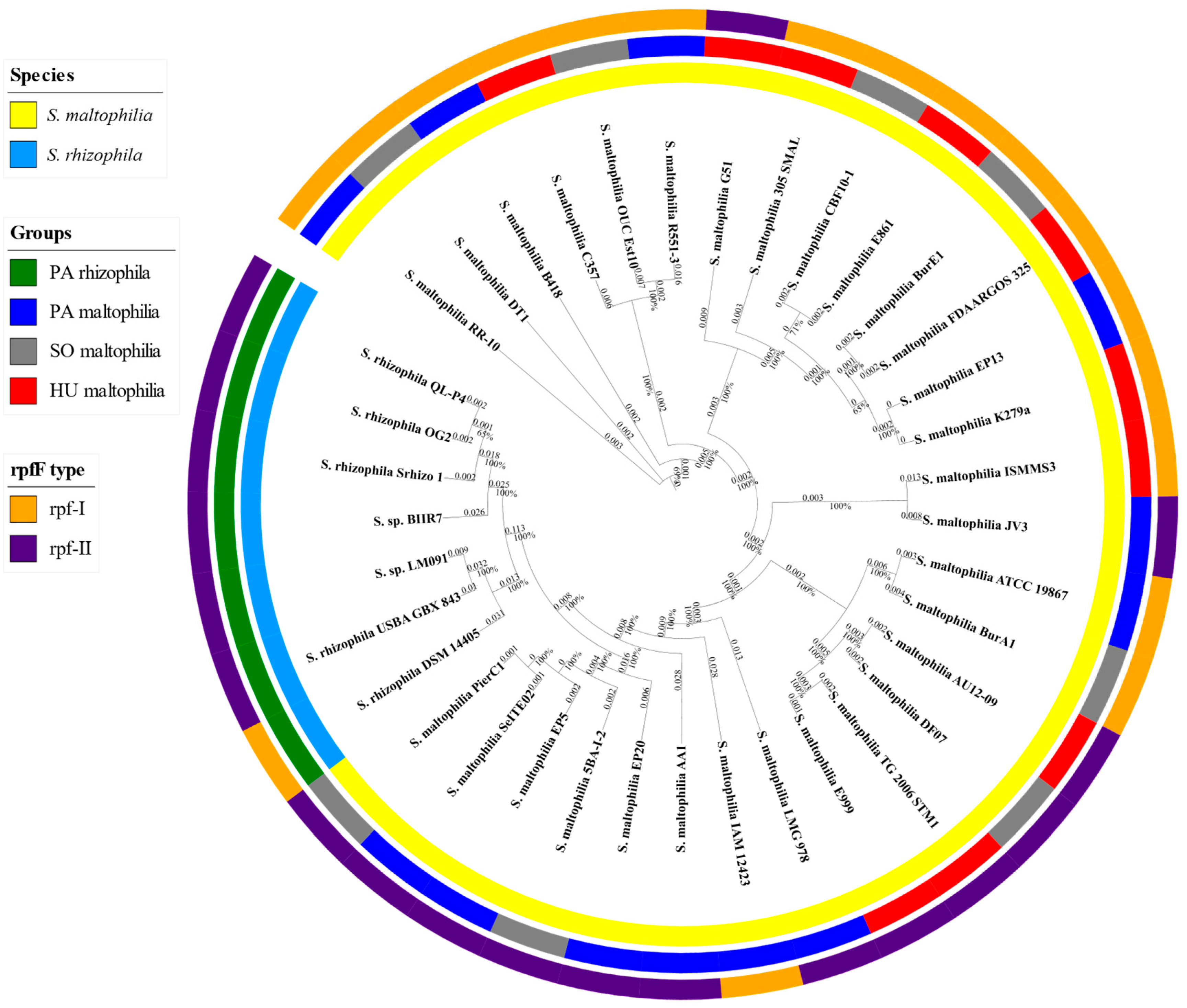
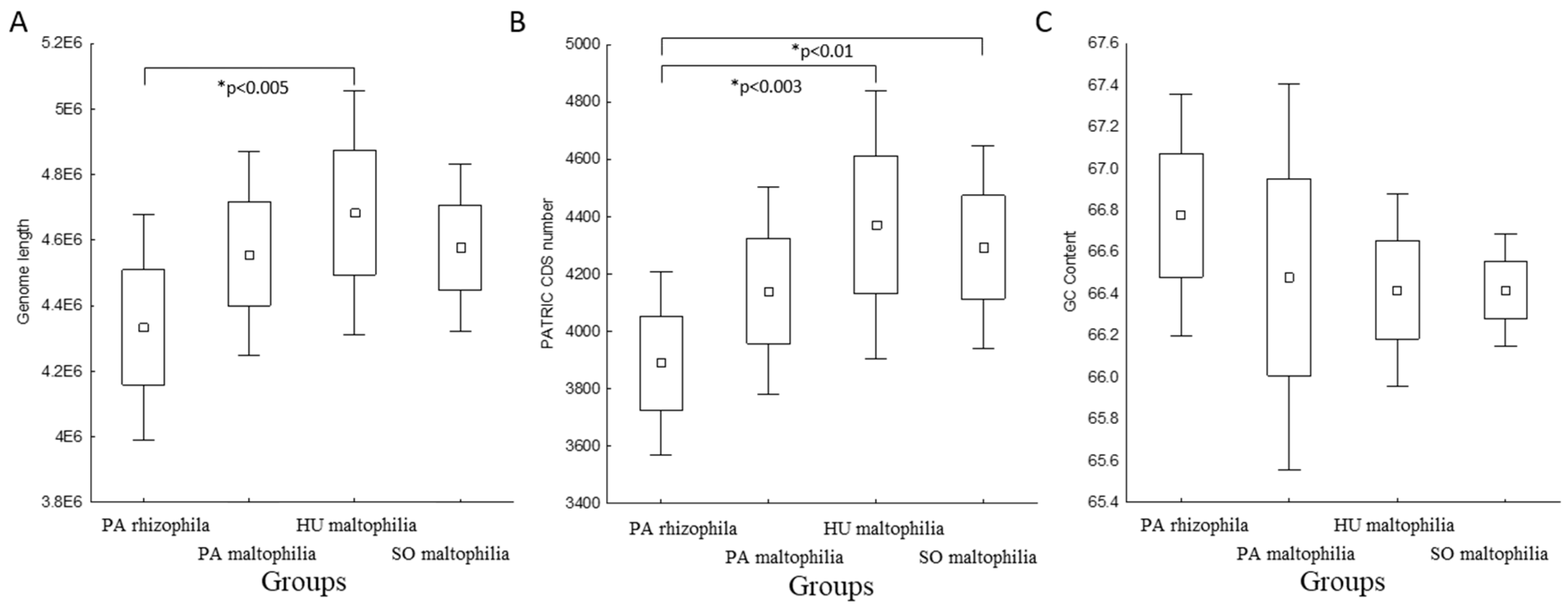
2.1. General Characteristics of S. maltophilia and S. rhizophila Genomes
2.2. Characteristic Features of S. maltophilia
2.3. Characteristic Features of S. rhizophila
2.4. Genes Encoding the Proteins that Are Involved in Plant Growth Promotion
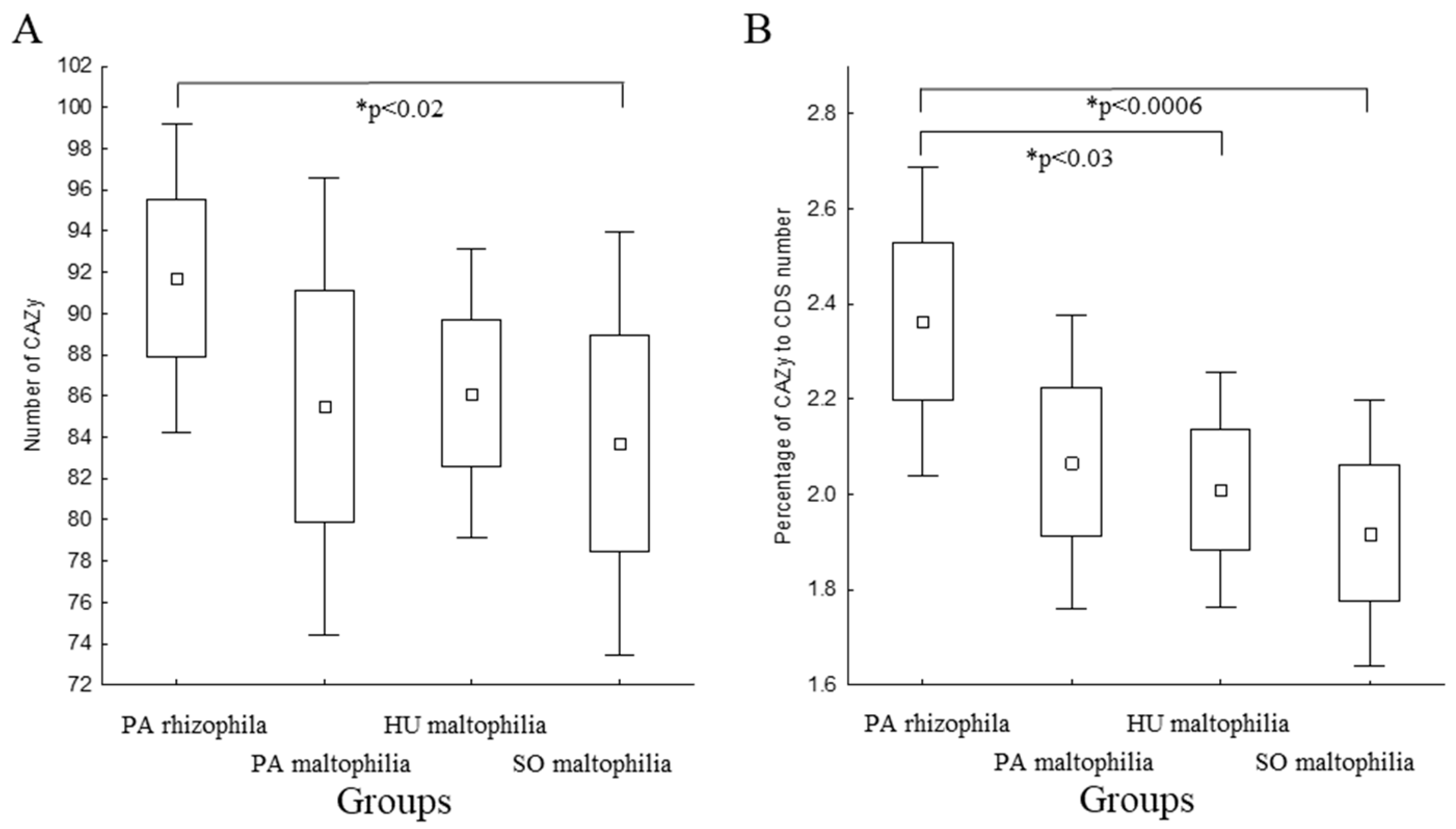
2.5. Degradation of the Xenobiotics
3. Discussion
4. Materials and Method
4.1. Genome Collection
4.2. Phylogenetic Analysis
4.3. Comparative Genomics and Bioinformatic Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CAZy | carbohydrate-active enzyme |
| c-di-GMP | cyclic diguanylate |
| CDS | coding sequence |
| COG | cluster of orthologous genes |
| CSP | cold shock protein |
| DSF | diffusible signal factor |
| HU maltophilia | maltophilia isolated from human |
| PA maltophilia | plant-associated maltophilia |
| PA rhizophila | plant-associated rhizophila |
| rpf | regulation of pathogenicity factors |
| SO maltophilia | maltophilia isolated from soil |
| T2SS | type II secretion system |
Appendix A

References
- Wolf, A. Stenotrophomonas rhizophila sp. nov., a novel plant-associated bacterium with antifungal properties. Int. J. Syst. Evol. Microbiol. 2002, 52, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Hauben, L.; Vauterin, L.; Moore, E.; Hoste, B.; Swings, J. Genomic diversity of the genus Stenotrophomonas. Int. J. Syst. Evol. Microbiol. 1999, 49, 1749–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.Q.; Berg, G. Stenotrophomonas maltophilia. Trends Microbiol. 2018, 26, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Alavi, P.; Starcher, M.R.; Thallinger, G.G.; Zachow, C.; Müller, H.; Berg, G. Stenotrophomonas comparative genomics reveals genes and functions that differentiate beneficial and pathogenic bacteria. BMC Genom. 2014, 15, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Martinez, J.L. Friends or foes: Can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front. Microbiol. 2015, 6, 241. [Google Scholar] [CrossRef]
- Ahmadi, E.; Kowsari, M.; Azadfar, D.; Salehi Jouzani, G. Bacillus pumilus and Stenotrophomonas maltophilia as two potentially causative agents involved in Persian oak decline in Zagros forests (Iran). Forest Pathol. 2019, 49. [Google Scholar] [CrossRef]
- Morris, C.E.; Bardin, M.; Kinkel, L.L.; Moury, B.; Nicot, P.C.; Sands, D.C. Expanding the paradigms of plant pathogen life history and evolution of parasitic fitness beyond agricultural boundaries. PLoS Pathog 2009, 5, e1000693. [Google Scholar] [CrossRef] [Green Version]
- Muresu, R.; Polone, E.; Sorbolini, S.; Squartini, A. Characterization of endophytic and symbiotic bacteria within plants of the endemic association Centaureetum horridae Mol. Plant Biosyst. 2011, 145, 478–484. [Google Scholar] [CrossRef]
- Fox, A.; Kwapinski, W.; Griffiths, B.S.; Schmalenberger, A. The role of sulfur- and phosphorus-mobilizing bacteria in biochar-induced growth promotion of Lolium perenne. FEMS Microbiol. Ecol. 2014, 90, 78–91. [Google Scholar] [CrossRef] [Green Version]
- Mattarozzi, M.; Manfredi, M.; Montanini, B.; Gosetti, F.; Sanangelantoni, A.M.; Marengo, E.; Careri, M.; Visioli, G. A metaproteomic approach dissecting major bacterial functions in the rhizosphere of plants living in serpentine soil. Anal. Bioanal. Chem. 2017, 409, 2327–2339. [Google Scholar] [CrossRef]
- Tian, B.; Zhang, C.; Ye, Y.; Wen, J.; Wu, Y.; Wang, H.; Li, H.; Cai, S.; Cai, W.; Cheng, Z. Beneficial traits of bacterial endophytes belonging to the core communities of the tomato root microbiome. Agric. Ecosyst. Environ. 2017, 247, 149–156. [Google Scholar] [CrossRef]
- McEwan, N.R.; Wilkinson, T.; Girdwood, S.E.; Snelling, T.J.; Collins, T.; Dougal, K.; Jones, D.L.; Godbold, D.L. Evaluation of the microbiome of decaying alder nodules by next generation sequencing. Endocytobiosis Cell Res. 2017, 28, 14–19. [Google Scholar]
- Patterson, S.B.; Mende, K.; Li, P.; Lu, D.; Carson, M.L.; Murray, C.K.; Tribble, D.R.; Blyth, D.M.; Group, I.T. Stenotrophomonas maltophilia infections: Clinical characteristics in a military trauma population. Diagn. Microbiol. Infect. Dis. 2020, 96, 114953. [Google Scholar] [CrossRef] [PubMed]
- Ribbeck-Busch, K.; Roder, A.; Hasse, D.; de Boer, W.; Martinez, J.L.; Hagemann, M.; Berg, G. A molecular biological protocol to distinguish potentially human pathogenic Stenotrophomonas maltophilia from plant-associated Stenotrophomonas rhizophila. Environ. Microbiol. 2005, 7, 1853–1858. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Fu, J.; Wu, J.; Jia, X.; Zhou, Y.; Li, C.; Dong, M.; Wang, S.; Zhang, J.; Chen, F. Bioinformatics analysis and characterization of highly efficient polyvinyl alcohol (PVA)-degrading enzymes from the novel PVA degrader Stenotrophomonas rhizophila QL-P4. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef]
- Wojcieszyńska, D.; Hupert-Kocurek, K.; Jankowska, A.; Guzik, U. Properties of catechol 2,3-dioxygenase from crude extract of Stenotrophomonas maltophilia strain KB2 immobilized in calcium alginate hydrogels. Biochem. Eng. J. 2012, 66, 1–7. [Google Scholar] [CrossRef]
- Afzal, I.; Shinwari, Z.K.; Iqrar, I. Selective isolation and characterization of agriculturally beneficial endophytic bacteria from wild hemp using canola. Pak J. Bot. 2015, 47, 1999–2008. [Google Scholar]
- Egamberdieva, D.; Jabborova, D.; Berg, G. Synergistic interactions between Bradyrhizobium japonicum and the endophyte Stenotrophomonas rhizophila and their effects on growth, and nodulation of soybean under salt stress. Plant Soil 2015, 405, 35–45. [Google Scholar] [CrossRef]
- Pinski, A.; Betekhtin, A.; Hupert-Kocurek, K.; Mur, L.A.J.; Hasterok, R. Defining the genetic basis of plant-endophytic bacteria interactions. Int. J. Mol. Sci. 2019, 20, 1947. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huedo, P.; Yero, D.; Martínez-Servat, S.; Estibariz, I.; Planell, R.; Martínez, P.; Ruyra, À.; Roher, N.; Roca, I.; Vila, J. Two different rpf clusters distributed among a population of Stenotrophomonas maltophilia clinical strains display differential DSF production and virulence regulation. J. Bacteriol. 2014, 196, 2431–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haswell, E.S.; Phillips, R.; Rees, D.C. Mechanosensitive channels: What can they do and how do they do it? Structure 2011, 19, 1356–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, V.K.; East, A.K.; Karunakaran, R.; Downie, J.A.; Poole, P.S. Adaptation of Rhizobium leguminosarum to pea, alfalfa and sugar beet rhizospheres investigated by comparative transcriptomics. Genome Biol. 2011, 12, R106. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Kathariou, S. Differentiation of epidemic-associated strains of Listeria monocytogenes by restriction fragment length polymorphism in a gene region essential for growth at low temperatures (4 degrees C). Appl. Environ. Microbiol. 1995, 61, 4310–4314. [Google Scholar] [CrossRef] [Green Version]
- Robbe-Saule, V.; Lopes, M.D.; Kolb, A.; Norel, F. Physiological effects of Crl in Salmonella are modulated by sigma(s) level and promoter specificity. J. Bacteriol. 2007, 189, 2976–2987. [Google Scholar] [CrossRef] [Green Version]
- Tupin, A.; Gualtieri, M.; Roquet-Baneres, F.; Morichaud, Z.; Brodolin, K.; Leonetti, J.P. Resistance to rifampicin: At the crossroads between ecological, genomic and medical concerns. Int. J. Antimicrob. Agents 2010, 35, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Baysarowich, J.; Koteva, K.; Hughes, D.W.; Ejim, L.; Griffiths, E.; Zhang, K.; Junop, M.; Wright, G.D. Rifamycin antibiotic resistance by ADP-ribosylation: Structure and diversity of Arr. Proc. Natl. Acad. Sci. USA 2008, 105, 4886–4891. [Google Scholar] [CrossRef] [Green Version]
- Porcheron, G.; Dozois, C.M. Interplay between iron homeostasis and virulence: Fur and RyhB as major regulators of bacterial pathogenicity. Vet. Microbiol. 2015, 179, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, D.Y.; Reedy, R.M.; Bick, J.; Oudemans, P.V. Characterization of a chitinase gene from Stenotrophomonas maltophilia strain 34S1 and its involvement in biological control. Appl. Environ. Microbiol. 2002, 68, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, Z.; Nelson, C.E.; Dalhus, B.; Mekasha, S.; Loose, J.S.; Crouch, L.I.; Rohr, A.K.; Gardner, J.G.; Eijsink, V.G.; Vaaje-Kolstad, G. Structural and functional analysis of a lytic polysaccharide monooxygenase important for efficient utilization of chitin in Cellvibrio japonicus. J. Biol. Chem. 2016, 291, 7300–7312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, E.R.; Mecsas, J. Bacterial secretion systems: An overview. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.W.; Walker, G.C. The exoD gene of Rhizobium meliloti encodes a novel function needed for alfalfa nodule invasion. J. Bacteriol. 1991, 173, 664–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.L. Contribution of integrons, and SmeABC and SmeDEF efflux pumps to multidrug resistance in clinical isolates of Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2004, 53, 518–521. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.B.; Ampomah, O.Y.; Darrah, R.; Peters, N.K.; Bhuvaneswari, T. Role of trehalose transport and utilization in Sinorhizobium meliloti-alfalfa interactions. Mol. Plant Microbe Interact. 2005, 18, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Ampomah, O.Y.; Avetisyan, A.; Hansen, E.; Svenson, J.; Huser, T.; Jensen, J.B.; Bhuvaneswari, T.V. The thuEFGKAB operon of rhizobia and Agrobacterium tumefaciens codes for transport of trehalose, maltitol, and isomers of sucrose and their assimilation through the formation of their 3-keto derivatives. J. Bacteriol. 2013, 195, 3797–3807. [Google Scholar] [CrossRef] [Green Version]
- He, Y.-W.; Zhang, L.-H. Quorum sensing and virulence regulation in Xanthomonas campestris. FEMS Microbiol. Rev. 2008, 32, 842–857. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Jiang, W.; Zhao, M.; Ling, J.; Zeng, X.; Deng, J.; Jin, D.; Dow, J.M.; Sun, W. A systematic analysis of the role of GGDEF-EAL domain proteins in virulence and motility in Xanthomonas oryzae pv. oryzicola. Sci. Rep. 2016, 6, 23769. [Google Scholar] [CrossRef] [Green Version]
- Shidore, T.; Dinse, T.; Ohrlein, J.; Becker, A.; Reinhold-Hurek, B. Transcriptomic analysis of responses to exudates reveal genes required for rhizosphere competence of the endophyte Azoarcus sp. strain BH72. Environ. Microbiol. 2012, 14, 2775–2787. [Google Scholar] [CrossRef]
- Castiglioni, P.; Warner, D.; Bensen, R.J.; Anstrom, D.C.; Harrison, J.; Stoecker, M.; Abad, M.; Kumar, G.; Salvador, S.; D’Ordine, R.; et al. Bacterial RNA chaperones confer abiotic stress tolerance in plants and improved grain yield in maize under water-limited conditions. Plant Physiol. 2008, 147, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Taghavi, S.; van der Lelie, D.; Hoffman, A.; Zhang, Y.B.; Walla, M.D.; Vangronsveld, J.; Newman, L.; Monchy, S. Genome sequence of the plant growth promoting endophytic bacterium Enterobacter sp. 638. PLoS Genet. 2010, 6, e1000943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zeng, Q.i.n.; Wood, A.J. The stress-responsive Tortula ruralis gene ALDH21A1 describes a novel eukaryotic aldehyde dehydrogenase protein family. J. Plant Physiol. 2002, 159, 677–684. [Google Scholar] [CrossRef]
- Messiha, N.A.S.; van Diepeningen, A.D.; Farag, N.S.; Abdallah, S.A.; Janse, J.D.; van Bruggen, A.H.C. Stenotrophomonas maltophilia: A new potential biocontrol agent of Ralstonia solanacearum, causal agent of potato brown rot. Eur. J. Plant Pathol. 2007, 118, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Zhang, J.; Liu, B.; Jiang, L.; Du, G.; Chen, J. Cloning, heterologous expression and characterization of two keratinases from Stenotrophomonas maltophilia BBE11-1. Process Biochem. 2014, 49, 647–654. [Google Scholar] [CrossRef]
- Manjeet, K.; Purushotham, P.; Neeraja, C.; Podile, A.R. Bacterial chitin binding proteins show differential substrate binding and synergy with chitinases. Microbiol. Res. 2013, 168, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Glick, B.R. Plant growth-promoting bacteria: Mechanisms and applications. Scientifica (Cairo) 2012, 2012, 963401. [Google Scholar] [CrossRef] [Green Version]
- Rimmele, M.; Boos, W. Trehalose-6-Phosphate hydrolase of Escherichia coli. J. Bacteriol. 1994, 176, 5654–5664. [Google Scholar] [CrossRef] [Green Version]
- Higo, A.; Katoh, H.; Ohmori, K.; Ikeuchi, M.; Ohmori, M. The role of a gene cluster for trehalose metabolism in dehydration tolerance of the filamentous cyanobacterium Anabaena sp. PCC 7120. Microbiology 2006, 152, 979–987. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Liu, Y.; Wu, G.; Zhang, N.; Shen, Q.; Zhang, R. Beneficial rhizobacterium Bacillus amyloliquefaciens SQR9 induces plant salt tolerance through spermidine production. Mol. Plant Microbe Interact. 2017, 30, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.S.; Wu, H.J.; Zang, H.Y.; Wu, L.M.; Zhu, Q.Q.; Gao, X.W. Plant growth promotion by spermidine-producing Bacillus subtilis OKB105. Mol. Plant Microbe Interact. 2014, 27, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Olanrewaju, O.S.; Glick, B.R.; Babalola, O.O. Mechanisms of action of plant growth promoting bacteria. World J. Microbiol. Biotechnol. 2017, 33, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nas, M.Y.; Cianciotto, N.P. Stenotrophomonas maltophilia produces an EntC-dependent catecholate siderophore that is distinct from enterobactin. Microbiology 2017, 163, 1590–1603. [Google Scholar] [CrossRef]
- Sharma, S.B.; Sayyed, R.Z.; Trivedi, M.H.; Gobi, T.A. Phosphate solubilizing microbes: Sustainable approach for managing phosphorus deficiency in agricultural soils. SpringerPlus 2013, 2, 587. [Google Scholar] [CrossRef] [Green Version]
- Youenou, B.; Favre-Bonte, S.; Bodilis, J.; Brothier, E.; Dubost, A.; Muller, D.; Nazaret, S. Comparative genomics of environmental and clinical Stenotrophomonas maltophilia strains with different antibiotic resistance profiles. Genome Biol. Evol. 2015, 7, 2484–2505. [Google Scholar] [CrossRef] [Green Version]
- Esposito, A.; Pompilio, A.; Bettua, C.; Crocetta, V.; Giacobazzi, E.; Fiscarelli, E.; Jousson, O.; Di Bonaventura, G. Evolution of Stenotrophomonas maltophilia in cystic fibrosis lung over chronic infection: A genomic and phenotypic population study. Front. Microbiol. 2017, 8, 1590. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, J.; Mamat, U.; Abda, E.M.; Kirchhoff, L.; Streit, W.R.; Schaible, U.E.; Niemann, S.; Kohl, T.A. Analysis of phylogenetic variation of Stenotrophomonas maltophilia reveals human-specific branches. Front. Microbiol. 2018, 9, 806. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Leon, G.; Hernandez, A.; Hernando-Amado, S.; Alavi, P.; Berg, G.; Martinez, J.L. A function of SmeDEF, the major quinolone resistance determinant of Stenotrophomonas maltophilia, is the colonization of plant roots. Appl. Environ. Microbiol. 2014, 80, 4559–4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.J.; Feltcher, M.E.; Waters, R.J.; Wetmore, K.M.; Mucyn, T.S.; Ryan, E.M.; Wang, G.; Ul-Hasan, S.; McDonald, M.; Yoshikuni, Y.; et al. Genome-wide identification of bacterial plant colonization genes. PLoS Biol. 2017, 15, e2002860. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, J.; Wang, E.; Wang, N. Mechanisms underlying the rhizosphere-to-rhizoplane enrichment of cellvibrio unveiled by genome-centric metagenomics and metatranscriptomics. Microorganisms 2020, 8, 583. [Google Scholar] [CrossRef] [Green Version]
- Huedo, P.; Yero, D.; Martinez-Servat, S.; Ruyra, A.; Roher, N.; Daura, X.; Gibert, I. Decoding the genetic and functional diversity of the DSF quorum-sensing system in Stenotrophomonas maltophilia. Front. Microbiol. 2015, 6, 761. [Google Scholar] [CrossRef] [Green Version]
- Alavi, P.; Muller, H.; Cardinale, M.; Zachow, C.; Sanchez, M.B.; Martinez, J.L.; Berg, G. The DSF quorum sensing system controls the positive influence of Stenotrophomonas maltophilia on plants. PLoS ONE 2013, 8, e67103. [Google Scholar] [CrossRef] [PubMed]
- Alavi, P.; Starcher, M.R.; Zachow, C.; Muller, H.; Berg, G. Root-microbe systems: The effect and mode of interaction of Stress Protecting Agent (SPA) Stenotrophomonas rhizophila DSM14405(T.). Front. Plant Sci. 2013, 4, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhou, L.; Venturi, V.; He, Y.W.; Kojima, M.; Sakakibari, H.; Hofte, M.; De Vleesschauwer, D. Phytohormone-mediated interkingdom signaling shapes the outcome of rice-Xanthomonas oryzae pv. oryzae interactions. BMC Plant Biol. 2015, 15, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lira, F.; Berg, G.; Martinez, J.L. Double-face meets the bacterial world: The opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 2017, 8, 2190. [Google Scholar] [CrossRef] [PubMed]
- Jankiewicz, U.; Larkowska, E.; Swiontek Brzezinska, M. Production, characterization, gene cloning, and nematocidal activity of the extracellular protease from Stenotrophomonas maltophilia N4. J. Biosci. Bioeng. 2016, 121, 614–618. [Google Scholar] [CrossRef]
- Yue, X.Y.; Zhang, B.; Jiang, D.D.; Liu, Y.J.; Niu, T.G. Separation and purification of a keratinase as pesticide against root-knot nematodes. World J. Microbiol. Biotechnol. 2011, 27, 2147–2153. [Google Scholar] [CrossRef]
- Dunne, C.; Crowley, J.J.; Moënne-Loccoz, Y.; Dowling, D.N.; O’Gara, F. Biological control of Pythium ultimum by Stenotrophomonas maltophilia W81 is mediated by an extracellular proteolytic activity. Microbiology 1997, 143, 3921–3931. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Liu, J.; Ding, J.; He, Q.; Xiong, R.; Zhang, K. The investigation of nematocidal activity in Stenotrophomonas maltophilia G2 and characterization of a novel virulence serine protease. Can. J. Microbiol. 2009, 55, 934–942. [Google Scholar] [CrossRef]
- Kobayashi, D.Y.; Guglielmoni, M.; Clarke, B.B. Isolation of the chitinolytic bacteria Xanthomonas maltophilia and Serratia marcescens as biological control agents for summer patch disease of turfgrass. Soil Biol. Biochem. 1995, 27, 1479–1487. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuen, G.Y.; Sarath, G.; Penheiter, A.R. Chitinases from the plant disease biocontrol agent, Stenotrophomonas maltophilia C3. Phytopathology 2001, 91, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankiewicz, U.; Brzezinska, M.S.; Saks, E. Identification and characterization of a chitinase of Stenotrophomonas maltophilia, a bacterium that is antagonistic towards fungal phytopathogens. J. Biosci. Bioeng. 2012, 113, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Jankiewicz, U.; Baranowski, B.; Swiontek Brzezinska, M.; Frak, M. Purification, characterization and cloning of a chitinase from Stenotrophomonas rhizophila G22. 3 Biotech 2020, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Kalidasan, V.; Joseph, N.; Kumar, S.; Awang Hamat, R.; Neela, V.K. Iron and virulence in Stenotrophomonas maltophilia: All we know so far. Front. Cell. Infect. Microbiol. 2018, 8, 401. [Google Scholar] [CrossRef]
- Da Silva, A.R.; Ferro, J.A.; Reinach, F.; Farah, C.; Furlan, L.; Quaggio, R.; Monteiro-Vitorello, C.; Van Sluys, M.; Almeida, N.A.; Alves, L. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 2002, 417, 459. [Google Scholar] [CrossRef]
- Piński, A.; Hupert-Kocurek, K. Genomic analysis of plant-associated bacteria and their potential in enhancing phytoremediation efficiency. J. Ecol. Eng. 2017, 18, 152–159. [Google Scholar] [CrossRef]
- Praveen Kumar, S.V.; Manjunatha, B.K. Studies on hydrocarbon degradation by the bacterial isolate Stenotrophomonas rhizophila (PM-1) from oil spilled regions of Western Ghats of Karnataka. Sci. Technol. Arts Res. J. 2016, 4, 139. [Google Scholar] [CrossRef] [Green Version]
- Saarela, M.; Berlin, M.; Nygren, H.; Lahtinen, P.; Honkapää, K.; Lantto, R.; Maukonen, J. Characterization of feather-degrading bacterial populations from birds’ nests—Potential strains for biomass production for animal feed. Int. Biodeterior. Biodegrad. 2017, 123, 262–268. [Google Scholar] [CrossRef]
- Guzik, U.; Hupert-Kocurek, K.; Sitnik, M.; Wojcieszynska, D. High activity catechol 1,2-dioxygenase from Stenotrophomonas maltophilia strain KB2 as a useful tool in cis,cis-muconic acid production. Antonie Van Leeuwenhoek 2013, 103, 1297–1307. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rossello-Mora, R.; Oliver Glockner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Avram, O.; Rapoport, D.; Portugez, S.; Pupko, T. M1CR0B1AL1Z3R-a user-friendly web server for the analysis of large-scale microbial genomics data. Nucleic Acids Res. 2019, 47, W88–W92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinski, A.; Zur, J.; Hasterok, R.; Hupert-Kocurek, K. Comparative Genomics of Stenotrophomonas maltophilia and Stenotrophomonas rhizophila Revealed Characteristic Features of Both Species. Int. J. Mol. Sci. 2020, 21, 4922. https://doi.org/10.3390/ijms21144922
Pinski A, Zur J, Hasterok R, Hupert-Kocurek K. Comparative Genomics of Stenotrophomonas maltophilia and Stenotrophomonas rhizophila Revealed Characteristic Features of Both Species. International Journal of Molecular Sciences. 2020; 21(14):4922. https://doi.org/10.3390/ijms21144922
Chicago/Turabian StylePinski, Artur, Joanna Zur, Robert Hasterok, and Katarzyna Hupert-Kocurek. 2020. "Comparative Genomics of Stenotrophomonas maltophilia and Stenotrophomonas rhizophila Revealed Characteristic Features of Both Species" International Journal of Molecular Sciences 21, no. 14: 4922. https://doi.org/10.3390/ijms21144922
APA StylePinski, A., Zur, J., Hasterok, R., & Hupert-Kocurek, K. (2020). Comparative Genomics of Stenotrophomonas maltophilia and Stenotrophomonas rhizophila Revealed Characteristic Features of Both Species. International Journal of Molecular Sciences, 21(14), 4922. https://doi.org/10.3390/ijms21144922