A Review of T-Cell Related Therapy for Osteosarcoma



Abstract
1. Introduction
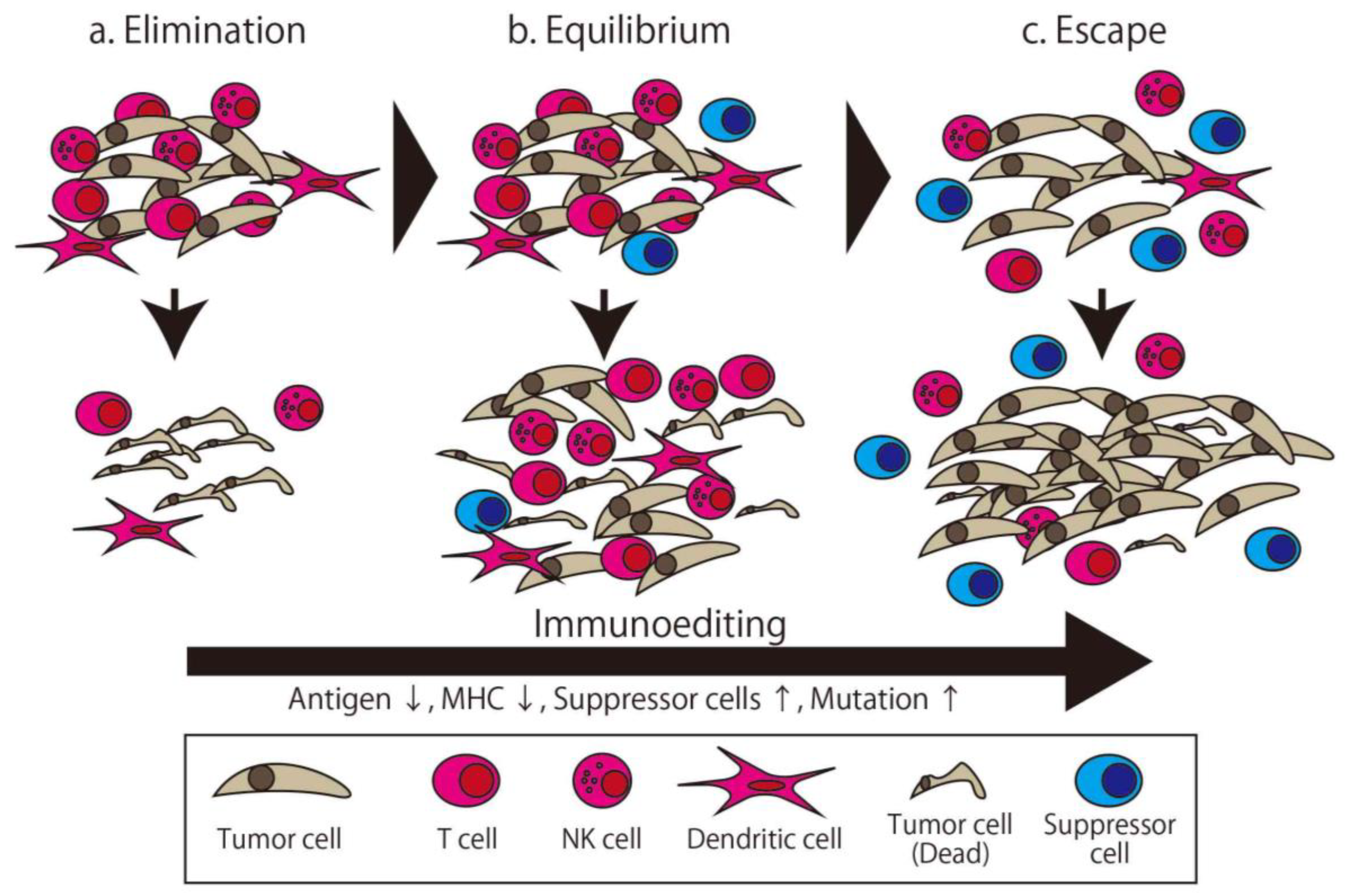
2. Cancer Immune Therapy and Cancer Immunoediting
2.1. Adaptive Immunity
2.1.1. Cancer Vaccine
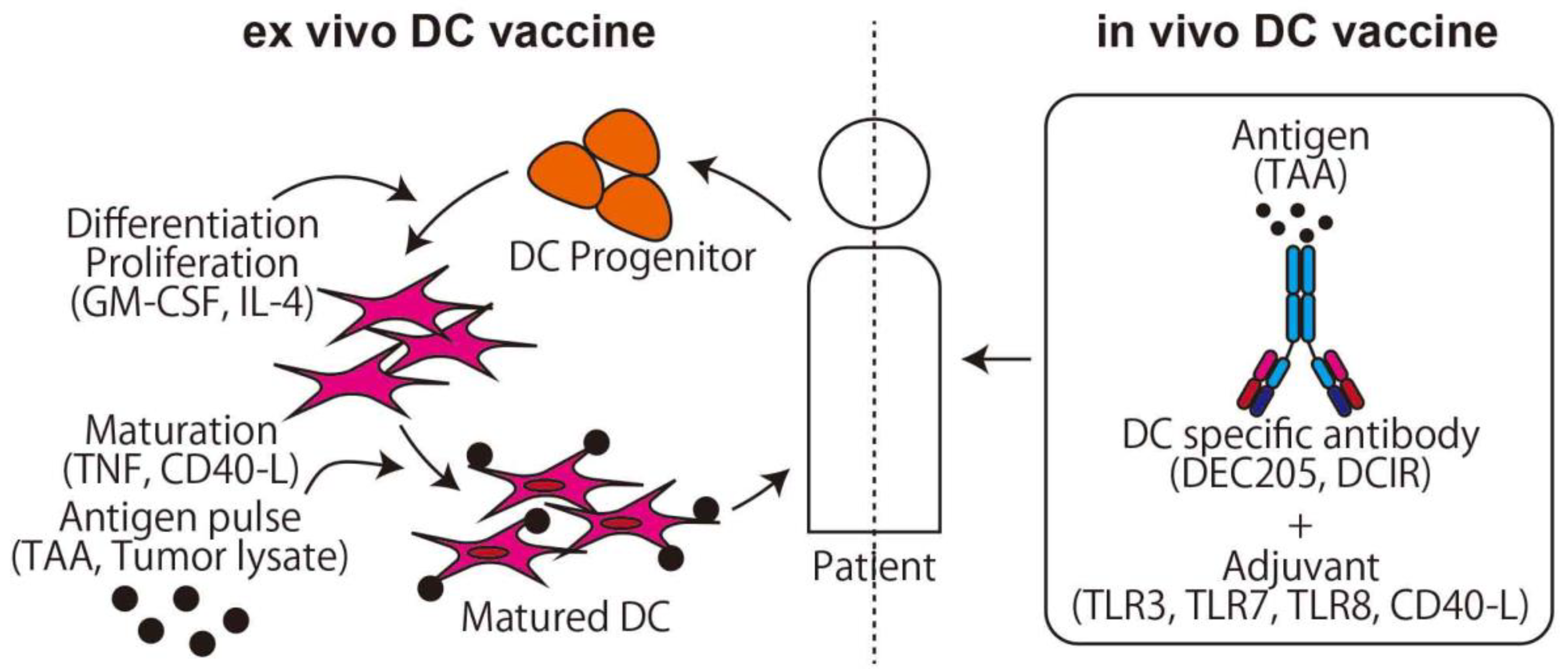
2.1.2. DC Vaccine
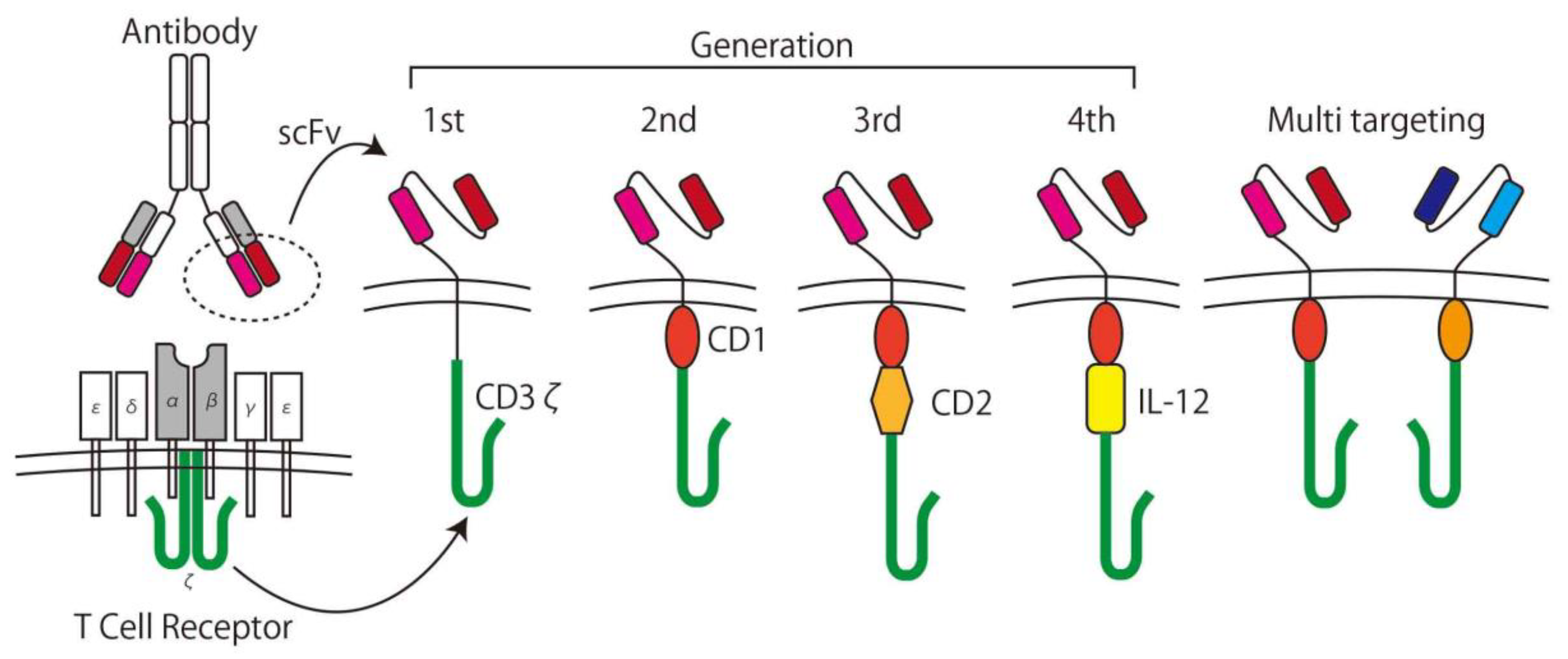
2.1.3. CAR T-cells
2.2. Innate Immunity
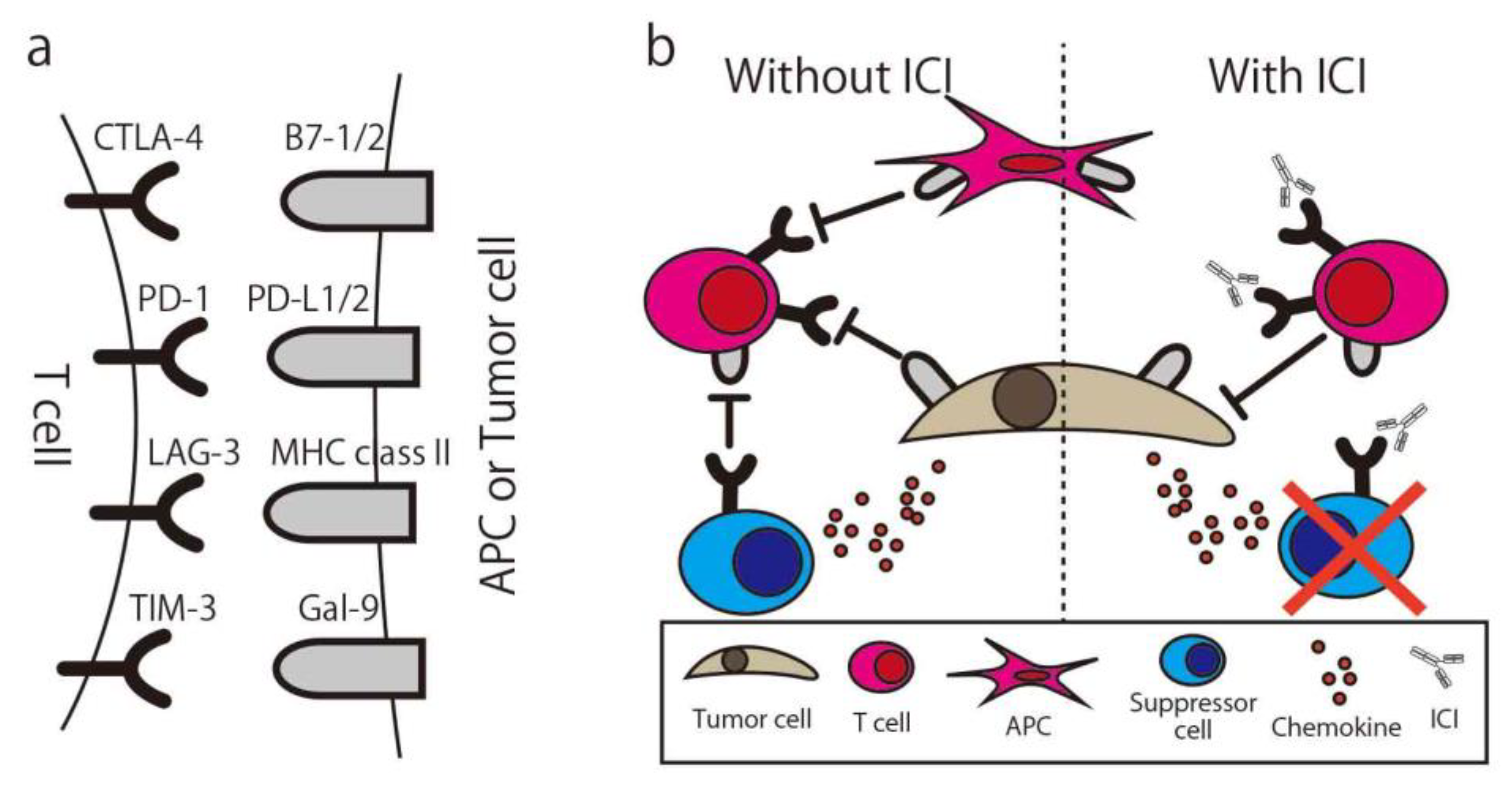
2.3. Immune Checkpoint Inhibitor
3. Challenges for the Future
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ICI | Immune checkpoint inhibitor |
| CAR | Chimeric antigen receptor |
| DC | Dendritic cell |
| TAA | Tumor associated antigen |
| APC | antigen presenting cell |
| GS-CSF | granulocyte-macrophage colony-stimulating factor |
| MHC | major histocompatibility complex |
| IL | interleukin |
| HER | human epidermal growth factor receptor |
| GD2 | Disialoganglioside |
| NK cell | Natural killer cell |
| TCR | T-cell receptor |
| Treg | regulatory T-cells |
| CTLA-4 | Cytotoxic T-Lymphocyte Associated Protein 4 |
| PD-1 | Programmed cell death 1 |
| TIM-3 | T-cell immunoglobulin and mucin domain-containing protein 3 |
| TIL | Tumor infiltrated T-cell |
| IFN-γ | interferon gamma |
References
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Rosen, G.; Caparros, B.; Huvos, A.G.; Kosloff, C.; Nirenberg, A.; Cacavio, A.; Marcove, R.C.; Lane, J.M.; Mehta, B.; Urban, C. Preoperative chemotherapy for osteogenic sarcoma: Selection of postoperative adjuvant chemotherapy based on the response of the primary tumor to preoperative chemotherapy. Cancer 1982, 49, 1221–1230. [Google Scholar] [CrossRef]
- Durfee, R.A.; Mohammed, M.; Luu, H.H. Review of Osteosarcoma and Current Management. Rheumatol. Ther. 2016, 3, 221–243. [Google Scholar] [CrossRef]
- Misaghi, A.; Goldin, A.; Awad, M.; Kulidjian, A.A. Osteosarcoma: A comprehensive review. Sicot J. 2018, 4, 12. [Google Scholar] [CrossRef]
- Italiano, A.; Mir, O.; Mathoulin-Pelissier, S.; Penel, N.; Piperno-Neumann, S.; Bompas, E.; Chevreau, C.; Duffaud, F.; Entz-Werlé, N.; Saada, E.; et al. Cabozantinib in patients with advanced Ewing sarcoma or osteosarcoma (CABONE): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 446–455. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Shaikh, A.B.; Li, F.; Li, M.; He, B.; He, X.; Chen, G.; Guo, B.; Li, D.; Jiang, F.; Dang, L.; et al. Present Advances and Future Perspectives of Molecular Targeted Therapy for Osteosarcoma. Int. J. Mol. Sci. 2016, 17, 506. [Google Scholar] [CrossRef]
- Hiddemann, W.; Roessner, A.; Wörmann, B.; Mellin, W.; Klockenkemper, B.; Bösing, T.; Büchner, T.; Grundmann, E. Tumor heterogeneity in osteosarcoma as identified by flow cytometry. Cancer 1987, 59, 324–328. [Google Scholar] [CrossRef]
- Wang, D.; Niu, X.; Wang, Z.; Song, C.-L.; Huang, Z.; Chen, K.-N.; Duan, J.; Bai, H.; Xu, J.; Zhao, J.; et al. Multiregion Sequencing Reveals the Genetic Heterogeneity and Evolutionary History of Osteosarcoma and Matched Pulmonary Metastases. Cancer Res. 2019, 79, 7–20. [Google Scholar] [CrossRef]
- Li, B.; Chan, H.L.; Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 2019, 26, 3009–3025. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- van Erp, A.E.M.; Versleijen-Jonkers, Y.M.H.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Gorris, M.A.J.; van Dam, L.S.; Mentzel, T.; Weidema, M.E.; Savci-Heijink, C.D.; Desar, I.M.E.; et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8(+) lymphocytes in primary sarcomas is subtype dependent. Oncotarget 2017, 8, 71371–71384. [Google Scholar] [CrossRef] [PubMed]
- Kienle, G.S. Fever in Cancer Treatment: Coley’s Therapy and Epidemiologic Observations. Glob. Adv. Health Med. 2012, 1, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Burnet, M. Cancer; a biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu Rev. Immunol 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; p. 1548. [Google Scholar]
- Paul, W.E. Immunity; Johns Hopkins University Press: Baltimore, MA, USA, 2015. [Google Scholar]
- Hanna, M.G., Jr.; Peters, L.C. Specific immunotherapy of established visceral micrometastases by BCG-tumor cell vaccine alone or as an adjunct to surgery. Cancer 1978, 42, 2613–2625. [Google Scholar] [CrossRef]
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.-Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475. [Google Scholar] [CrossRef]
- Weir, C.; Oksa, A.; Millar, J.; Alexander, M.; Kynoch, N.; Walton-Weitz, Z.; Mackenzie-Wood, P.; Tam, F.; Richards, H.; Naylor, R.; et al. The Safety of an Adjuvanted Autologous Cancer Vaccine Platform in Canine Cancer Patients. Vet. Sci. 2018, 5, 87. [Google Scholar] [CrossRef]
- Sondak, V.K.; Sabel, M.S.; Mulé, J.J. Allogeneic and autologous melanoma vaccines: Where have we been and where are we going? Clin. Cancer Res. 2006, 12, 2337s–2341s. [Google Scholar] [CrossRef]
- Simons, J.W.; Carducci, M.A.; Mikhak, B.; Lim, M.; Biedrzycki, B.; Borellini, F.; Clift, S.M.; Hege, K.M.; Ando, D.G.; Piantadosi, S.; et al. Phase I/II trial of an allogeneic cellular immunotherapy in hormone-naïve prostate cancer. Clin. Cancer Res. 2006, 12, 3394–3401. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Asquith, J.M.; Leatherman, J.M.; Kobrin, B.J.; Petrik, S.; Laiko, M.; Levi, J.; Daphtary, M.M.; Biedrzycki, B.; Wolff, A.C.; et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: A chemotherapy dose-ranging factorial study of safety and immune activation. J. Clin. Oncol. 2009, 27, 5911–5918. [Google Scholar] [CrossRef] [PubMed]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [CrossRef]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Tsukahara, T.; Nabeta, Y.; Kawaguchi, S.; Ikeda, H.; Sato, Y.; Shimozawa, K.; Ida, K.; Asanuma, H.; Hirohashi, Y.; Torigoe, T.; et al. Identification of human autologous cytotoxic T-lymphocyte-defined osteosarcoma gene that encodes a transcriptional regulator, papillomavirus binding factor. Cancer Res. 2004, 64, 5442–5448. [Google Scholar] [CrossRef]
- Tsuda, N.; Murayama, K.; Ishida, H.; Matsunaga, K.; Komiya, S.; Itoh, K.; Yamada, A. Expression of a newly defined tumor-rejection antigen SART3 in musculoskeletal tumors and induction of HLA class I-restricted cytotoxic T lymphocytes by SART3-derived peptides. J. Orthop Res. 2001, 19, 346–351. [Google Scholar] [CrossRef]
- Srivastava, A.; Fuchs, B.; Zhang, K.; Ruan, M.; Halder, C.; Mahlum, E.; Weber, K.; Bolander, M.E.; Sarkar, G. High WT1 expression is associated with very poor survival of patients with osteogenic sarcoma metastasis. Clin. Cancer Res. 2006, 12, 4237–4243. [Google Scholar] [CrossRef]
- Maheswaran, S.; Englert, C.; Bennett, P.; Heinrich, G.; Haber, D.A. The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev. 1995, 9, 2143–2156. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Tsuboi, A.; Nakata, J.; Nishida, S.; Hosen, N.; Kumanogoh, A.; Oji, Y.; Sugiyama, H. Wilms’ Tumor Gene 1 (WT1) Peptide Vaccine Therapy for Hematological Malignancies: From CTL Epitope Identification to Recent Progress in Clinical Studies Including a Cure-Oriented Strategy. Oncol. Res. Treat. 2017, 40, 682–690. [Google Scholar] [CrossRef]
- Suehara, Y.; Kubota, D.; Kikuta, K.; Kaneko, K.; Kawai, A.; Kondo, T. Discovery of biomarkers for osteosarcoma by proteomics approaches. Sarcoma 2012, 2012, 425636. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Laschi, M.; Geminiani, M.; Santucci, A. Proteomics of osteosarcoma. Expert Rev. Proteom. 2014, 11, 331–343. [Google Scholar] [CrossRef]
- Kawano, M.; Nishida, H.; Nakamoto, Y.; Tsumura, H.; Tsuchiya, H. Cryoimmunologic antitumor effects enhanced by dendritic cells in osteosarcoma. Clin. Orthop Relat Res. 2010, 468, 1373–1383. [Google Scholar] [CrossRef][Green Version]
- Timmerman, J.M.; Levy, R. Dendritic cell vaccines for cancer immunotherapy. Annu. Rev. Med. 1999, 50, 507–529. [Google Scholar] [CrossRef]
- Hsu, F.J.; Benike, C.; Fagnoni, F.; Liles, T.M.; Czerwinski, D.; Taidi, B.; Engleman, E.G.; Levy, R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat. Med. 1996, 2, 52–58. [Google Scholar] [CrossRef]
- Reichardt, V.L.; Okada, C.Y.; Stockerl-Goldstein, K.E.; Bogen, B.; Levy, R. Rationale for adjuvant idiotypic vaccination after high-dose therapy for multiple myeloma. Biol. Blood Marrow Transpl. 1997, 3, 157–163. [Google Scholar]
- Chauvin, C.; Philippeau, J.-M.; Hémont, C.; Hubert, F.-X.; Wittrant, Y.; Lamoureux, F.; Trinité, B.; Heymann, D.; Rédini, F.; Josien, R. Killer Dendritic Cells Link Innate and Adaptive Immunity against Established Osteosarcoma in Rats. Cancer Res. 2008, 68, 9433–9440. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Nishida, H.; Tanzawa, Y.; Takeuchi, A.; Hayashi, K.; Yamamoto, N.; Mizukoshi, E.; Nakamoto, Y.; Kaneko, S.; Tsuchiya, H. Phase 1/2 study of immunotherapy with dendritic cells pulsed with autologous tumor lysate in patients with refractory bone and soft tissue sarcoma. Cancer 2017, 123, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Himoudi, N.; Wallace, R.; Parsley, K.L.; Gilmour, K.; Barrie, A.U.; Howe, K.; Dong, R.; Sebire, N.J.; Michalski, A.; Thrasher, A.J.; et al. Lack of T-cell responses following autologous tumour lysate pulsed dendritic cell vaccination, in patients with relapsed osteosarcoma. Clin. Transl. Oncol. 2012, 14, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, L.C.; Bonnyay, D.P.; Charalambous, A.; Darguste, D.I.; Fujii, S.; Soares, H.; Brimnes, M.K.; Moltedo, B.; Moran, T.M.; Steinman, R.M. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 2004, 199, 815–824. [Google Scholar] [CrossRef]
- Mulé, J.J.; Shu, S.; Schwarz, S.L.; Rosenberg, S.A. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science 1984, 225, 1487–1489. [Google Scholar] [CrossRef]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Scheuermann, R.H.; Racila, E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Sergina, N.V.; Moasser, M.M. The HER family and cancer: Emerging molecular mechanisms and therapeutic targets. Trends Mol. Med. 2007, 13, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Maximiano, S.; Magalhães, P.; Guerreiro, M.P.; Morgado, M. Trastuzumab in the Treatment of Breast Cancer. BioDrugs 2016, 30, 75–86. [Google Scholar] [CrossRef]
- Ebb, D.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Ratnayake, M.; Savoldo, B.; Perlaky, L.; Dotti, G.; Wels, W.S.; Bhattacharjee, M.B.; Gilbertson, R.J.; Shine, H.D.; Weiss, H.L.; et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007, 67, 5957–5964. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef]
- Huang, G.; Yu, L.; Cooper, L.J.; Hollomon, M.; Huls, H.; Kleinerman, E.S. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012, 72, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Park, H.; Greene, J.; Pao, J.; Mulvey, E.; Zhou, S.X.; Albert, C.M.; Moy, F.; Sachdev, D.; Yee, D.; et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS ONE 2015, 10, e0133152. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef] [PubMed]
- Born, W.; Miles, C.; White, J.; O’Brien, R.; Freed, J.H.; Marrack, P.; Kappler, J.; Kubo, R.T. Peptide sequences of T-cell receptor delta and gamma chains are identical to predicted X and gamma proteins. Nature 1987, 330, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.-h.; Meyer, C.; Bonneville, M. γδ T Cells: First Line of Defense and Beyond. Annu. Rev. Immunol. 2014, 32, 121–155. [Google Scholar] [CrossRef]
- Zhao, Y.; Niu, C.; Cui, J. Gamma-delta (γδ) T cells: Friend or foe in cancer development? J. Transl. Med. 2018, 16, 3. [Google Scholar] [CrossRef]
- Dar, A.A.; Patil, R.S.; Chiplunkar, S.V. Insights into the Relationship between Toll Like Receptors and Gamma Delta T Cell Responses. Front. Immunol. 2014, 5, 366. [Google Scholar] [CrossRef]
- Kato, Y.; Tanaka, Y.; Miyagawa, F.; Yamashita, S.; Minato, N. Targeting of tumor cells for human gammadelta T cells by nonpeptide antigens. J. Immunol. 2001, 167, 5092–5098. [Google Scholar] [CrossRef]
- Kondo, M.; Izumi, T.; Fujieda, N.; Kondo, A.; Morishita, T.; Matsushita, H.; Kakimi, K. Expansion of human peripheral blood γδ T cells using zoledronate. J. Vis. Exp. 2011. [Google Scholar] [CrossRef]
- Dieli, F.; Vermijlen, D.; Fulfaro, F.; Caccamo, N.; Meraviglia, S.; Cicero, G.; Roberts, A.; Buccheri, S.; D’Asaro, M.; Gebbia, N.; et al. Targeting human {gamma}delta} T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007, 67, 7450–7457. [Google Scholar] [CrossRef]
- Tanaka, Y.; Murata-Hirai, K.; Iwasaki, M.; Matsumoto, K.; Hayashi, K.; Kumagai, A.; Nada, M.H.; Wang, H.; Kobayashi, H.; Kamitakahara, H.; et al. Expansion of human γδ T cells for adoptive immunotherapy using a bisphosphonate prodrug. Cancer Sci. 2018, 109, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Nicol, A.J.; Tokuyama, H.; Mattarollo, S.R.; Hagi, T.; Suzuki, K.; Yokokawa, K.; Nieda, M. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br. J. Cancer 2011, 105, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Tanaka, Y.; Yagi, J.; Minato, N.; Tanabe, K. Phase I/II study of adoptive transfer of γδ T cells in combination with zoledronic acid and IL-2 to patients with advanced renal cell carcinoma. Cancer Immunol. Immunother. 2011, 60, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Matsushita, H.; Hoshikawa, M.; Hasegawa, K.; Kokudo, N.; Kakimi, K. Adjuvant combination therapy with gemcitabine and autologous γδ T-cell transfer in patients with curatively resected pancreatic cancer. Cytotherapy 2017, 19, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Muraro, M.; Mereuta, O.M.; Carraro, F.; Madon, E.; Fagioli, F. Osteosarcoma cell line growth inhibition by zoledronate-stimulated effector cells. Cell Immunol. 2007, 249, 63–72. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Z.; Wang, Z.; Li, S.; Li, B.; Sun, L.; Li, H.; Lin, P.; Wang, S.; Teng, W.; Zhou, X.; et al. Decitabine Enhances Vγ9Vδ2 T Cell-Mediated Cytotoxic Effects on Osteosarcoma Cells via the NKG2DL-NKG2D Axis. Front. Immunol. 2018, 9, 1239. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Ye, C.; Lin, P.; Li, B.; Zhang, W.; Sun, L.; Wang, Z.; Xue, D.; Teng, W.; et al. Valproic Acid Combined with Zoledronate Enhance gammadelta T Cell-Mediated Cytotoxicity against Osteosarcoma Cells via the Accumulation of Mevalonate Pathway Intermediates. Front. Immunol. 2018, 9, 377. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Callahan, M.K.; Wolchok, J.D.; Allison, J.P. Anti-CTLA-4 antibody therapy: Immune monitoring during clinical development of a novel immunotherapy. Semin. Oncol. 2010, 37, 473–484. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Wedekind, M.F.; Wagner, L.M.; Cripe, T.P. Immunotherapy for osteosarcoma: Where do we go from here? Pediatr Blood Cancer 2018, 65, e27227. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.K.; Cote, G.M.; Choy, E.; Yang, P.; Harmon, D.; Schwab, J.; Nielsen, G.P.; Chebib, I.; Ferrone, S.; Wang, X.; et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol. Res. 2014, 2, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Okamoto, M.; Sasaki, J.; Kuroda, C.; Ishida, H.; Ueda, K.; Ideta, H.; Kamanaka, T.; Sobajima, A.; Takizawa, T.; et al. Anti-PD-1 antibody decreases tumour-infiltrating regulatory T cells. BMC Cancer 2020, 20, 25. [Google Scholar] [CrossRef] [PubMed]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef]
- Yoshida, K.; Okamoto, M.; Sasaki, J.; Kuroda, C.; Ishida, H.; Ueda, K.; Okano, S.; Ideta, H.; Kamanaka, T.; Sobajima, A.; et al. Clinical outcome of osteosarcoma and its correlation with programmed death-ligand 1 and T cell activation markers. Oncotargets Ther. 2019, 12, 2513–2518. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet. Oncol. 2017, 18, e143–e152. [Google Scholar] [CrossRef]
- Mulkey, F.; Theoret, M.R.; Keegan, P.; Pazdur, R.; Sridhara, R. Comparison of iRECIST versus RECIST V.1.1 in patients treated with an anti-PD-1 or PD-L1 antibody: Pooled FDA analysis. J. Immunother. Cancer 2020, 8, e000146. [Google Scholar] [CrossRef] [PubMed]
- Houdek, Š.; Büchler, T.; Kindlová, E. [Comparison of RECIST 1.1 and iRECIST for Response Evaluation in Solid Tumours]. Klin Onkol 2017, 30, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Borcoman, E.; Nandikolla, A.; Long, G.; Goel, S.; Tourneau, C.L. Patterns of Response and Progression to Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book 2018, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef]
- Nishino, M.; Giobbie-Hurder, A.; Gargano, M.; Suda, M.; Ramaiya, N.H.; Hodi, F.S. Developing a common language for tumor response to immunotherapy: Immune-related response criteria using unidimensional measurements. Clin. Cancer Res. 2013, 19, 3936–3943. [Google Scholar] [CrossRef]
- Sakamoto, A.; Iwamoto, Y. Current status and perspectives regarding the treatment of osteo-sarcoma: Chemotherapy. Rev. Recent Clin. Trials 2008, 3, 228–231. [Google Scholar] [CrossRef]
- Wu, C.-C.; Beird, H.C.; Andrew Livingston, J.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.-L.; Ju, Z.; et al. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef]
- Ladle, B.H.; Phillips, M.J.; Yu, C.; Gamper, C.J. Immune modulatory effects of chemotherapy increase the effectiveness of anti-PD1 immunotherapy in a poorly immunogenic murine model of osteosarcoma. J. Immunol. 2017, 198, 15. [Google Scholar]
- Malas, S.; Harrasser, M.; Lacy, K.E.; Karagiannis, S.N. Antibody therapies for melanoma: New and emerging opportunities to activate immunity (Review). Oncol. Rep. 2014, 32, 875–886. [Google Scholar] [CrossRef]
- You, W.; Liu, M.; Miao, J.D.; Liao, Y.Q.; Song, Y.B.; Cai, D.K.; Gao, Y.; Peng, H. A Network Meta-analysis Comparing the Efficacy and Safety of Anti-PD-1 with Anti-PD-L1 in Non-small Cell Lung Cancer. J. Cancer 2018, 9, 1200–1206. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, A.; Gridelli, C. “Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer”: Is there a substantial difference or not? J. Thorac. Dis. 2018, 10, S4065–S4068. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Behera, M.; Owonikoko, T.K.; Kamphorst, A.O.; Pakkala, S.; Belani, C.P.; Khuri, F.R.; Ahmed, R.; Ramalingam, S.S. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer: A systematic analysis of the literature. Cancer 2018, 124, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Dodagatta-Marri, E.; Meyer, D.S.; Reeves, M.Q.; Paniagua, R.; To, M.D.; Binnewies, M.; Broz, M.L.; Mori, H.; Wu, D.; Adoumie, M.; et al. α-PD-1 therapy elevates Treg/Th balance and increases tumor cell pSmad3 that are both targeted by α-TGFβ antibody to promote durable rejection and immunity in squamous cell carcinomas. J. Immunother. Cancer 2019, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Piperno-Neumann, S.; Le Deley, M.-C.; Rédini, F.; Pacquement, H.; Marec-Bérard, P.; Petit, P.; Brisse, H.; Lervat, C.; Gentet, J.-C.; Entz-Werlé, N.; et al. Zoledronate in combination with chemotherapy and surgery to treat osteosarcoma (OS2006): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 1070–1080. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Innate | Adaptive | |
|---|---|---|
| Specificity | Non-specific | Specific |
| Response | Rapid | Slow |
| Memory | No | Yes |
| Main Players | NK cell Macrophage Granulocyte | T-cell B cell |
| Trial ID | Type of Immunotherapy | Target Disease | Techniques | Phase | Status |
|---|---|---|---|---|---|
| NCT01241162 | DC vaccine | Osteosarcoma, other cancer | DC vaccine, Decitabine | 1 | Completed without result |
| NCT01803152 | DC vaccine | Osteosarcoma, other sarcoma | DC vaccine, Gemcitabine | 1 | Active, not recruiting |
| NCT02107963 | CAR T-cell | Osteosarcoma, other cancer | GD2-CAR, AP1903, Cyclophosphamide | 1 | Completed without result |
| NCT01953900 | CAR T-cell | Osteosarcoma, Neuroblastoma | GD2-CAR, VZV vaccine, Fludarabine, Cyclophosphamide | 1 | Active, not recruiting |
| NCT03635632 | CAR T-cell | Osteosarcoma, other cancer | C7R-GD2-CAR, Fludarabine, Cyclophosphamide | 1 | Recruiting |
| NCT03356782 | CAR T-cell | Osteosarcoma, other sarcoma | Each sarcoma specific CAR-T-cell | 1/2 | Recruiting |
| NCT03628209 | Anti-PD-1 antibody | Osteosarcoma | Nivolumab, Azacitidine, surgery | 1/2 | Recruiting |
| NCT03277924 | Anti-PD-1 antibody | Osteosarcoma, other sarcoma | Nivolumab, Sunitinib | 1/2 | Recruiting |
| NCT04294511 | Anti-PD-1 antibody | Osteosarcoma | Camrelizumab, Neoadjuvant chemotherapy | 2 | Recruiting |
| NCT03359018 | Anti-PD-1 antibody | Osteosarcoma | SHR-1210, Apatinib | 2 | Completed |
| NCT04351308 | Anti-PD-1 antibody | Osteosarcoma | Camrelizumab, MAPI, Apatinib | 2 | Recruiting |
| NCT03013127 | Anti-PD-1 antibody | Osteosarcoma | Pembrolizumab | 2 | Active, not recruiting |
| NCT04044378 | Anti-PD-1 antibody | Osteosarcoma | Camrelizumab, Famitinib, Isosfamide | 2 | Withdrawn (Toxicity) |
| NCT03676985 | Anti-PD-L1 antibody | Osteosarcoma | ZKAB001 | 1/2 | Recruiting |
| NCT04359550 | Anti-PD-L1 antibody | Osteosarcoma | ZKAB001 | 3 | Not yet recruiting |
| NCT03006848 | Anti-PD-L1 antibody | Osteosarcoma | Avelumab | 2 | Active, not recruiting |
| NCT02500797 | Anti-PD-1 a/o L1 antibody | Osteosarcoma, other cancer | Nivolumab, Ipilimumab | 2 | Active, not recruiting |
| NCT02982486 | Anti-PD-1 a/o L1 antibody | Osteosarcoma, other cancer | Nivolumab, Ipilimumab | 2 | Not yet recruiting |
| NCT02815995 | Anti-PD-1/L1 antibody | Osteosarcoma, other sarcoma | Durvalumab, Tremelimumab | 2 | Active, not recruiting |
| NCT04074564 | DC vaccine, Anti-PD-1 antibody | Osteosarcoma, other sarcoma | MASCT-I, anti-PD-1 antibody, Apatinib | 1 | Not yet recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshida, K.; Okamoto, M.; Aoki, K.; Takahashi, J.; Saito, N. A Review of T-Cell Related Therapy for Osteosarcoma. Int. J. Mol. Sci. 2020, 21, 4877. https://doi.org/10.3390/ijms21144877
Yoshida K, Okamoto M, Aoki K, Takahashi J, Saito N. A Review of T-Cell Related Therapy for Osteosarcoma. International Journal of Molecular Sciences. 2020; 21(14):4877. https://doi.org/10.3390/ijms21144877
Chicago/Turabian StyleYoshida, Kazushige, Masanori Okamoto, Kaoru Aoki, Jun Takahashi, and Naoto Saito. 2020. "A Review of T-Cell Related Therapy for Osteosarcoma" International Journal of Molecular Sciences 21, no. 14: 4877. https://doi.org/10.3390/ijms21144877
APA StyleYoshida, K., Okamoto, M., Aoki, K., Takahashi, J., & Saito, N. (2020). A Review of T-Cell Related Therapy for Osteosarcoma. International Journal of Molecular Sciences, 21(14), 4877. https://doi.org/10.3390/ijms21144877