Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
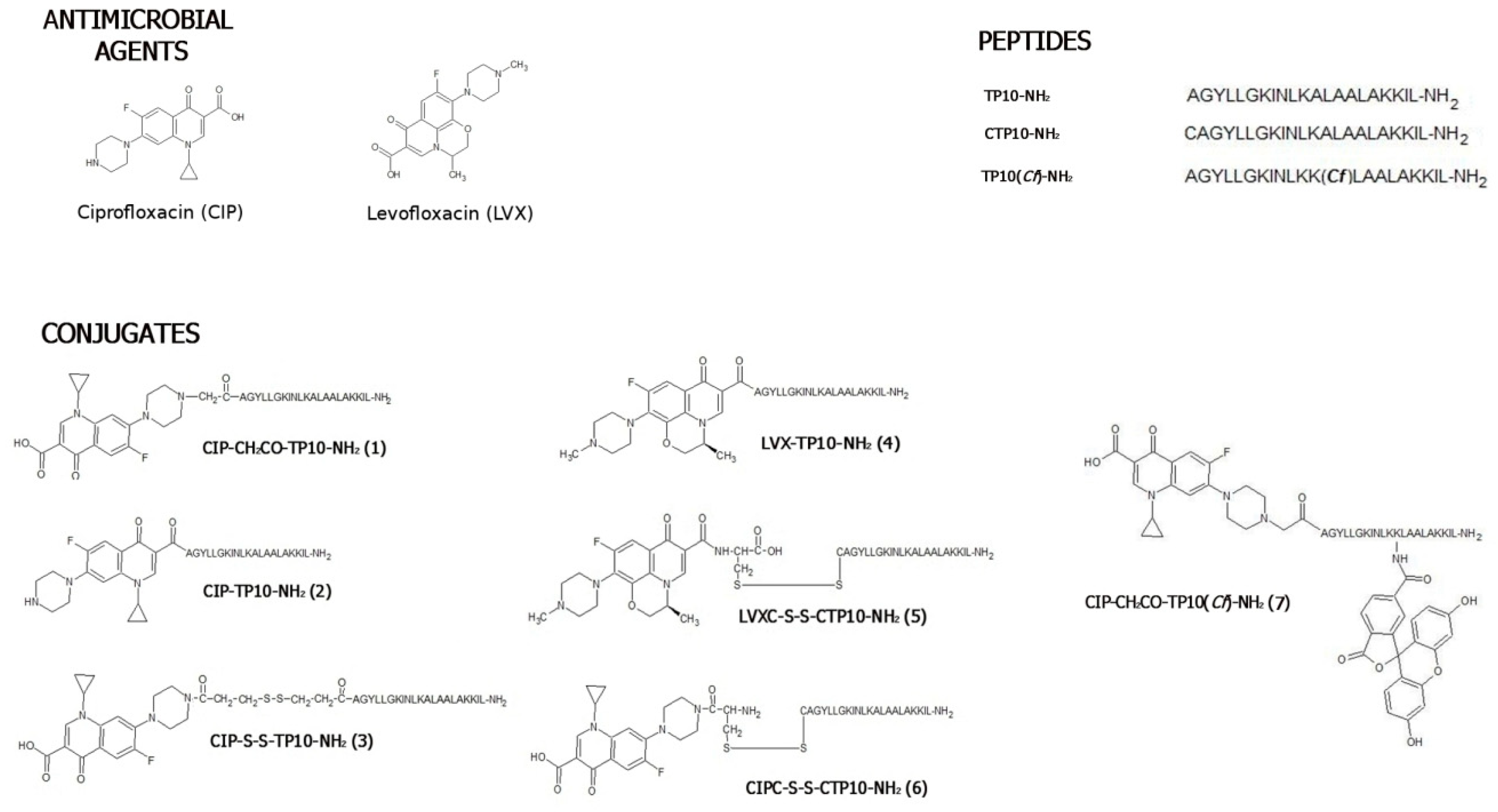
2.1. Chemical Synthesis and Stability Studies
2.1.1. Synthesis
2.1.2. Stability
2.2. Antibacterial Activity
2.2.1. Bacteriostatic Activity
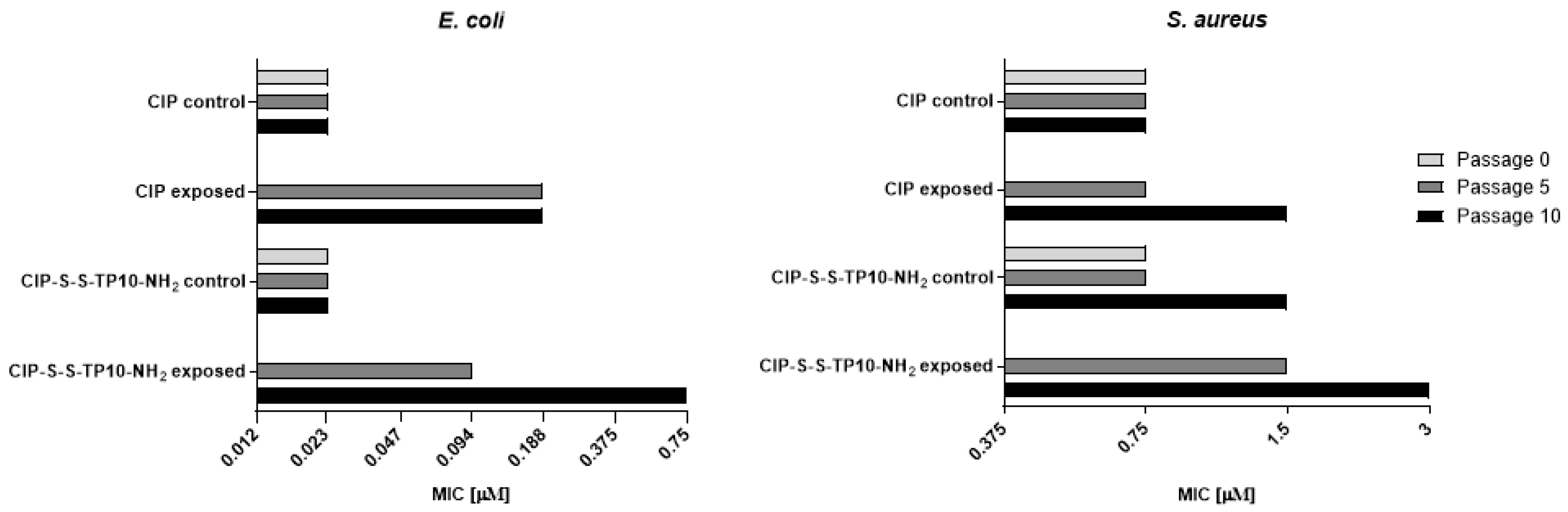
2.2.2. Long-Term Resistance Development
2.3. Bactericidal Effect
2.3.1. Minimal Bactericidal Concentration
2.3.2. Kinetics of Bactericidal Effect
2.4. Antifungal Activity
2.5. Molecular Mechanism of Antifungal Activity
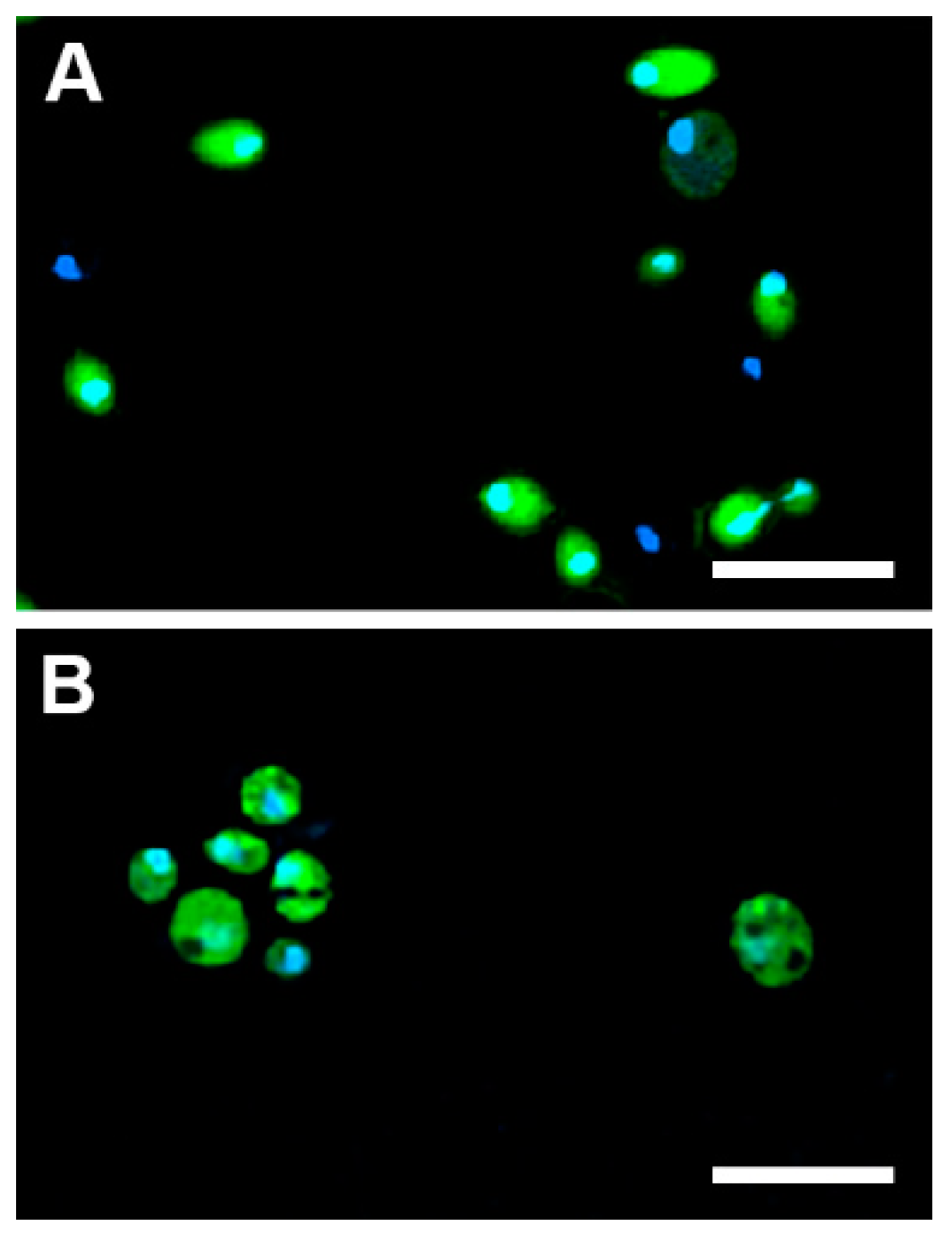
2.5.1. Cellular Uptake
2.5.2. Inhibition of Yeast DNA Topoisomerase II
2.6. Mammalian Toxicity
2.6.1. Cytotoxicity
2.6.2. Hemolysis
2.6.3. ROS Generation
2.7. Selectivity in Relation to Mammalian Cells
2.7.1. Antibacterial
2.7.2. Antifungal
2.8. Summary
3. Materials and Methods
3.1. Solid-Phase Peptide Synthesis (SPPS)
3.2. Synthesis of Levofloxacin and Ciprofloxacin-Based Conjugates
3.2.1. Synthesis of LVX-Cys(Npys)
3.2.2. Synthesis of Cys(Npys)-CIP
3.3. Synthesis of Fluorescently Labelled Compounds
3.4. Microorganism Strains and Growth Conditions
3.5. Stability Testing
3.6. Antibacterial Activity Assay
3.7. Determination of Resistance Induction Potential
3.8. Antifungal Activity Assay
3.9. Cytotoxicity Assay
3.10. Measurement of ROS Generation
3.11. Determination of the Haemolytic Potential
3.12. Monitoring of the Cellular Uptake
3.13. Inhibition of DNA Relaxation Mediated by DNA Topoisomerase II
3.14. Bacterial Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMP | antimicrobial peptide |
| BcSI | Bactericidal Selectivity Index |
| BSI | Bacteriostatic Selectivity Index |
| CCA | α-cyano-4-hydroxycinnamic acid matrix |
| Cf | 5(6)-carboxyfluorescein |
| CIP | ciprofloksacin |
| CPP | cell-penetrating peptide |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DCM | dichloromethane |
| DHB | 2,5-dihydroxybenzoic acid matrix |
| DIC | Differential Interference Contrast |
| DIPCI | N,N′-diisopropylocarbodiimide |
| DIPEA | N,N-diisopropylethylamine |
| DMF | N,N-dimethylformamide |
| EtBr | ethidium bromide |
| HATU | (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4 ,5-b]pyridinium 3-oxide hexafluorophosphate |
| HEK293 | human embryonic kidney cells |
| HeLa | human cervical cancer cells |
| HepG2 | human liver cancer cells |
| HL-60 | human myeloid leukemia cells |
| HOAt | 1-hydroksy-7-azabenzotriazol |
| HOBt | 1-hydroxybenzotriazole |
| IH50 | Minimum Hemolytic Concentration |
| LLC-PK1 | old male pig kidney cells |
| LVX | levofloxacin |
| LVX-NHS | levofloxacin-N-hydroxysuccinimidyl ester |
| MBC | Minimal Bactericidal Concentration |
| MDR | multidrug-resistance |
| MHA | Mueller Hinton Agar |
| MHB | Mueller Hinton Broth |
| MIC | Minimal Inhibitory Concentration |
| MSI | Mycostatic Selectivity Index |
| Mtt | 4-methyltrityl |
| NMM | N-methylmorpholine |
| Npys | Npys |
| PBS | phosphate-buffered saline |
| ROS | reactive oxygen species |
| TAT | trans-activator protein |
| TBTU | N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uroniumtetrafluoroborate |
| TFA | trifluoroacetic acid |
References
- Ezelarab, H.A.A.; Abbas, S.H.; Hassan, H.A.; Abuo-Rahma, G.E.D.A. Recent updates of fluoroquinolones as antibacterial agents. Arch. Pharm. 2018, 351, e1800141. [Google Scholar] [CrossRef] [PubMed]
- WHO Model List of Essential Medicines (19th List); World Health Organization: Geneva, Switzerland, 2015.
- Hooper, D.C. Mechanisms of Action of Antimicrobials: Focus on Fluoroquinolones. Clin. Infect. Dis. 2001, 32, S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, L.S.; Sutton, S.B.; Webber, M.A.; Piddock, L.J.V. Fluoroquinolone resistance: Mechanisms, impact on bacteria, and role in evolutionary success. Trends Microbiol. 2014, 22, 438–445. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.U.; Hearn, B.; Mack, D.; Wender, P.; Rothbard, J.; Kirisits, M.J.; Mui, E.; Wernimont, S.; Roberts, C.W.; Muench, S.P.; et al. Delivery of antimicrobials into parasites. Proc. Natl. Acad. Sci. USA 2003, 100, 14281–14286. [Google Scholar] [CrossRef]
- Cohen, D.T.; Zhang, C.; Fadzen, C.M.; Mijalis, A.J.; Hie, L.; Johnson, K.D.; Shriver, Z.; Plante, O.; Miller, S.J.; Buchwald, S.L.; et al. A chemoselective strategy for late-stage functionalisation of complex small molecules with polypeptides and proteins. Nat. Chem. 2019, 11, 78–85. [Google Scholar] [CrossRef]
- Purkayastha, N.; Capone, S.; Beck, A.K.; Seebach, D.; Leeds, J.; Thompson, K.; Moser, H.E. Antibacterial Activity of Enrofloxacin and Ciprofloxacin Derivatives of β-Octaarginine. Chem. Biodivers. 2015, 12, 179–193. [Google Scholar] [CrossRef]
- Ptaszyńska, N.; Olkiewicz, K.; Okońska, J.; Gucwa, K.; Łęgowska, A.; Gitlin-Domagalska, A.; Dębowski, D.; Lica, J.; Heldt, M.; Milewski, S.; et al. Peptide Conjugates of Lactoferricin Analogues and Antimicrobials—Design, Chemical Synthesis, and Evaluation of Antimicrobial Activity and Mammalian Cytotoxicity. Peptides 2019, 117, 170079. [Google Scholar] [CrossRef]
- Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Łȩgowska, A.; Okońska, J.; Ruczyński, J.; Gitlin-Domagalska, A.; Dȩbowski, D.; Milewski, S.; Rolka, K. Antibiotic-Based Conjugates Containing Antimicrobial HLopt2 Peptide: Design, Synthesis, Antimicrobial and Cytotoxic Activities. ACS Chem. Biol. 2019, 14, 2233–2242. [Google Scholar] [CrossRef]
- Stewart, K.M.; Horton, K.L.; Kelley, S.O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008, 6, 2242–2255. [Google Scholar] [CrossRef]
- Guo, Z.; Peng, H.; Kang, J.; Sun, D. Cell-penetrating peptides: Possible transduction mechanisms and therapeutic applications (review). Biomed. Rep. 2016, 4, 528–534. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesised human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Usmani, S.S.; Singh, S.; Chaudhary, K.; Raghava, G.P.S.; Gautam, A. CPPsite 2.0: A repository of experimentally validated cell-penetrating peptides. Nucleic Acids Res. 2016, 44, D1098–D1103. [Google Scholar] [CrossRef] [PubMed]
- Pooga, M.; Hällbrink, M.; Zorko, M.; Langel, U. Cell penetration by transportan. FASEB J. 1998, 12, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hällbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta 2000, 1467, 165–176. [Google Scholar] [CrossRef]
- Nekhotiaeva, N.; Elmquist, A.; Rajarao, G.K.; Hällbrink, M.; Langel, U.; Good, L. Cell entry and antimicrobial properties of eukaryotic cell-penetrating peptides. FASEB J. 2004, 18, 394–396. [Google Scholar] [CrossRef] [PubMed]
- Horváti, K.; Bacsa, B.; Mlinkó, T.; Szabó, N.; Hudecz, F.; Zsila, F.; Bősze, S. Comparative analysis of internalisation, haemolytic, cytotoxic and antibacterial effect of membrane-active cationic peptides: Aspects of experimental setup. Amino Acids 2017, 49, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Bárány-Wallje, E.; Gaur, J.; Lundberg, P.; Langel, Ü.; Gräslund, A. Differential membrane perturbation caused by the cell penetrating peptide Tp10 depending on attached cargo. FEBS Lett. 2007, 581, 2389–2393. [Google Scholar] [CrossRef]
- Izabela, R.; Jarosław, R.; Magdalena, A.; Piotr, R.; Ivan, K. Transportan 10 improves the anticancer activity of cisplatin. Naunyn-Schmiedeberg’s. Arch. Pharmacol. 2016, 389, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Gou, Y.; Zhao, Q.; Li, S.; Zhang, W.; Song, J.; Mou, L.; Li, J.; Wang, K.; Zhang, B.; et al. Antimicrobial activities and action mechanism studies of transportan 10 and its analogues against multidrug-resistant bacteria. J. Pept. Sci. 2015, 21, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 1998, 42, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.; Abdelkhalek, A.; Seleem, M.N. Evaluation of short synthetic antimicrobial peptides for treatment of drug-resistant and intracellular Staphylococcus aureus. Sci. Rep. 2016, 6, 29707. [Google Scholar] [CrossRef] [PubMed]
- Franz, R.; Kelly, S.L.; Lamb, D.C.; Kelly, D.E.; Ruhnke, M.; Morschhäuser, J. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob. Agents Chemother. 1998, 42, 3065–3072. [Google Scholar] [CrossRef]
- Shen, L.L.; Pernet, A.G. Mechanism of inhibition of DNA gyrase by analogues of nalidixic acid: The target of the drugs is DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 307–311. [Google Scholar] [CrossRef]
- Tunitskaya, V.L.; Khomutov, A.R.; Kochetkov, S.N.; Kotovskaya, S.K.; Charushin, V.N. Inhibition of DNA Gyrase by Levofloxacin and Related Fluorine-Containing Heterocyclic Compounds. Acta Naturae 2011, 3, 94–99. [Google Scholar] [CrossRef]
- Moreau, N.J.; Robaux, H.; Baron, L.; Tabary, X. Inhibitory effects of quinolones on pro- and eucaryotic DNA topoisomerases I and II. Antimicrob. Agents Chemother. 1990, 34, 1955–1960. [Google Scholar] [CrossRef]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of α-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef]
- Song, J.; Kai, M.; Zhang, W.; Zhang, J.; Liu, L.; Zhang, B.; Liu, X.; Wang, R. Cellular uptake of transportan 10 and its analogs in live cells: Selectivity and structure-activity relationship studies. Peptides 2011, 32, 1934–1941. [Google Scholar] [CrossRef]
- Aguiar, L.; Biosca, A.; Lantero, E.; Gut, J.; Vale, N.; Rosenthal, P.J.; Nogueira, F.; Andreu, D.; Fernàndez-Busquets, X.; Gomes, P. Coupling the antimalarial cell penetrating peptide TP10 to classical antimalarial drugs primaquine and chloroquine produces strongly hemolytic conjugates. Molecules 2019, 24, 4559. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, E.; Camm, J. Cardiotoxicity of fluoroquinolones. J. Antimicrob. Chemother. 2002, 49, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Adikwu, E.; Brambaifa, N. Ciprofloxacin Cardiotoxicity and Hepatotoxicity in Humans and Animals. Pharmacol. Pharm. 2012, 3, 207–213. [Google Scholar] [CrossRef]
- Qutrio Baloch, Z.; Raza, M.A.; Abbas, S.A.; Bukhari, S. Ciprofloxacin-induced Hepatotoxicity in a Healthy Young Adult. Cureus 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Łęgowska, A.; Dębowski, D.; Łukajtis, R.; Wysocka, M.; Czaplewski, C.; Lesner, A.; Rolka, K. Implication of the disulfide bridge in trypsin inhibitor SFTI-1 in its interaction with serine proteinases. Bioorg. Med. Chem. 2010, 18, 8188–8193. [Google Scholar] [CrossRef]
- Lica, J.J.; Grabe, G.J.; Heldt, M.; Misiak, M.; Bloch, P.; Serocki, M.; Switalska, M.; Wietrzyk, J.; Baginski, M.; Hellmann, A.; et al. Cell Density-Dependent Cytological Stage Profile and Its Application for a Screen of Cytostatic Agents Active Toward Leukemic Stem Cells. Stem Cells Dev. 2018, 27, 488–513. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.C.; Nelson, C.E.; Yu, S.S.; Beavers, K.R.; Kim, A.J.; Li, H.; Nelson, H.M.; Giorgio, T.D.; Duvall, C.L. Ex Vivo Red Blood Cell Hemolysis Assay for the Evaluation of pH-responsive Endosomolytic Agents for Cytosolic Delivery of Biomacromolecular Drugs. J. Vis. Exp. 2013, 73, e50166. [Google Scholar] [CrossRef]
- Saar, K.; Lindgren, M.; Hansen, M.; Eiríksdóttir, E.; Jiang, Y.; Rosenthal-Aizman, K.; Sassian, M.; Langel, Ü. Cell-penetrating peptides: A comparative membrane toxicity study. Anal. Biochem. 2005, 345, 55–65. [Google Scholar] [CrossRef]
- Yandek, L.E.; Pokorny, A.; Florén, A.; Knoelke, K.; Langel, Ü.; Almeida, P.F.F. Mechanism of the cell-penetrating peptide transportan 10 permeation of lipid bilayers. Biophys. J. 2007, 92, 2434–2444. [Google Scholar] [CrossRef]
- Ye, S.P.; Hsueh, E.J.; Yu, M.S.; Wu, H.; Wang, Y.C. Oral ciprofloxacin as antibacterial prophylaxis after allogeneic bone marrow transplantation: A reappraisal. Bone Marrow Transplant. 1999, 24, 1207–1211. [Google Scholar]
- Wang, C.H.; Chang, F.Y.; Chao, T.Y.; Kao, W.Y.; Ho, C.L.; Chen, Y.C.; Dai, M.S.; Chang, P.Y.; Wu, Y.Y.; Lin, J.C. Characteristics comparisons of bacteremia in allogeneic and autologous hematopoietic stem cell-transplant recipients with levofloxacin prophylaxis and influence on resistant bacteria emergence. J. Microbiol. Immunol. Infect. 2018, 51, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Doan, V.P.; Yeh, J.C.; Gulbis, A.M.; Aitken, S.L.; Ariza-Heredia, E.; Ahmed, S. Levofloxacin versus Cefpodoxime for Antibacterial Prophylaxis in Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 1637–1641. [Google Scholar] [CrossRef] [PubMed]
- De Pauw, B.E.; Donnelly, J.P.; De Witte, T.; Novakova, I.R.; Schattenberg, A. Options and limitations of long-term oral ciprofloxacin as antibacterial prophylaxis in allogeneic bone marrow transplant recipients. Bone Marrow Transplant. 1990, 5, 179–182. [Google Scholar]
- Trenschel, R.; Peceny, R.; Runde, V.; Elmaagacli, A.; Dermoumi, H.; Von Heinegg, E.H.; Müller, K.D.; Schaefer, U.W.; Beelen, D.W. Fungal colonization and invasive fungal infections following allogeneic BMT using metronidazole, ciprofloxacin and fluconazole or ciprofloxacin and fluconzole as intestinal decontamination. Bone Marrow Transplant. 2000, 26, 993–997. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC [μM] | |||||||
|---|---|---|---|---|---|---|---|---|
| Gram (+) | Gram (-) | |||||||
| S. aureus ATCC 29213 | S. epidermidis ATCC 12228 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | |||||
| MIC90 | MIC50 | MIC90 | MIC50 | MIC90 | MIC50 | MIC90 | MIC50 | |
| CIP | 0.2 | 0.2 | 1.6 | 0.8 | 0.0125 | <0.0125 | 0.4 | 0.4 |
| LVX | 0.4 | 0.4 | 1.6 | 0.8 | 0.025 | 0.025 | 0.8 | 0.8 |
| TP10-NH2 | 3.125 | 3.125 | 1.6 | 1.6 | 1.6 | 1.6 | >100 | >100 |
| 1 | 12.5 | 12.5 | 12.5 | 6.25 | 25 | 25 | >100 | >100 |
| 2 | 0.8 | 0.8 | 1.6 | 1.6 | 1.6 | 1.6 | 3.25 | 3.25 |
| 3 | 0.8 | 0.8 | 0.8 | 0.8 | 0.025 | 0.0125 | 1.6 | 0.8 |
| 4 | 1.6 | 1.6 | 1.6 | 1.6 | 50 | 50 | 100 | 100 |
| 5 | 6.25 | 6.25 | 3.125 | 3.125 | 3.125 | 3.125 | 25 | 12.5 |
| 6 | 3.125 | 3.125 | 6.25 | 6.25 | 6.25 | 6.25 | >100 | >100 |
| Compound | MIC [μM] | |||||||
|---|---|---|---|---|---|---|---|---|
| C. albicans SC 5314 | C. albicans Δopt1Δopt5Δptr2Δptr22 | C. albicans Gu4 | C. albicans Gu5 | C. glabrata DSM 11226 | C. krusei DSM 6128 | |||
| MIC90 | MIC50 | MIC90 | MIC50 | MIC50 | MIC50 | MIC50 | MIC50 | |
| CIP | >200 | >200 | >200 | >200 | >200 | >200 | >200 | >200 |
| LVX | >200 | >200 | >200 | >200 | >200 | >200 | >200 | >200 |
| TP10-NH2 | 100 | 100 | 100 | 100 | 100 | 100 | >200 | >200 |
| 1 | 100 | 50 | 200 | 100 | 50 | 50 | >200 | 50 |
| 2 | 12.5 | 12.5 | 12.5 | 6.25 | 25 | 25 | >200 | 50 |
| 3 | 25 | 6.25 | 25 | 12.5 | 6.25 | 12.5 | >200 | 25 |
| 4 | 100 | 50 | 100 | 100 | 25 | 25 | >200 | 100 |
| 5 | 25 | 12.5 | 25 | 12.5 | 12.5 | 12.5 | >200 | 50 |
| 6 | 50 | 50 | 50 | 50 | 25 | 25 | >200 | >200 |
| FLU | 100 | 50 | 100 | 50 | 50 | >200 (1600) | 100 | 50 |
| Compound | IC50 ± SD [μM] | ||
|---|---|---|---|
| HEK 293 | Hep G2 | LLC-PK1 | |
| CIP | >200 | >200 | >200 |
| LVX | >200 | >200 | >200 |
| TP10-NH2 | 25.08 ± 0.86 | 32.12 ± 1.65 | 22.73 ± 5.65 |
| CTP10-NH2 | 19.47 ± 2.93 | 59.97 ±5.75 | 51.60 ± 8.66 |
| 1 | 10.81 ± 1.87 | 21.70 ± 2.18 | 17.79 ±1.93 |
| 2 | 10.42 ± 1.39 | 11.46 ± 2.03 | 27.06 ± 1.82 |
| 3 | 13.11 ± 1.22 | 19.03 ± 2.08 | 16.83 ± 2.62 |
| 4 | 22.54 ± 9.70 | 72.58 ± 12.95 | 52.92 ± 8.15 |
| 5 | 41.34 ± 1.10 | 101.5 ± 4.90 | 104.70 ± 15.62 |
| 6 | 20.84 ± 10.91 | 37.71 ± 2.84 | 21.33 ± 1.33 |
| Antimicrobial Activity | ||||||
|---|---|---|---|---|---|---|
| Antibacterial Selectivity | Antifungal Selectivity | |||||
| Compound | HEK 293 LLC-PK1 for E. coli | HEK 293 LLC-PK1 for Gram + | HEK 293 LLC-PK1 for C. albicans | HEK 293 LLC-PK1 for C. krusei | HEK 293 LLC-PK1 for ΔC. albicans | HEK 293 LLC-PK1 for C. glabrata |
| TP10-NH2 | ++ | ++ | - | - | - | - |
| 1 | - | + | - | - | - | - |
| 2 | ++ | ++ | - | - | + | - |
| 3 | ++ | ++ | - | - | - | - |
| 4 | ++ | ++ | - | - | - | - |
| 5 | ++ | ++ | + | + | + | - |
| 6 | - | + | - | + | - | - |
| Mammalian Toxicity | ||||||
| ROS | Hemolysis Selectivity | |||||
| HL-60 | HEK 293 | HL-60 | HEK 293 | |||
| TP10-NH2 | ++ | ++ | - | - | ||
| CTP10-NH2 | N/D | N/D | + | ++ | ||
| 2 | ++ | ++ | + | + | ||
| 3 | + | ++ | N/D | - | ||
| 4 | + | ++ | N/D | - | ||
| 5 | ++ | ++ | - | + | ||
| 6 | - | ++ | + | + | ||
| Molecular Interactions | ||||||
| Topoisomerase II Inhibition | Cellular Uptake | |||||
| Bacterial | Fungal | |||||
| Gram - | Gram + | C. albicans | C. glabrata | |||
| Wall | Nucleus | |||||
| TP10-NH2 | N/D | N/D | N/D | + | - | |
| 3 | + | ++ | + | N/D | N/D | |
| 4 | ++ | ++ | + | N/D | N/D | |
| 5 | + | ++ | + | N/D | N/D | |
| 7 | N/D | N/D | N/D | + | - | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ptaszyńska, N.; Gucwa, K.; Olkiewicz, K.; Heldt, M.; Serocki, M.; Stupak, A.; Martynow, D.; Dębowski, D.; Gitlin-Domagalska, A.; Lica, J.; et al. Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 4696. https://doi.org/10.3390/ijms21134696
Ptaszyńska N, Gucwa K, Olkiewicz K, Heldt M, Serocki M, Stupak A, Martynow D, Dębowski D, Gitlin-Domagalska A, Lica J, et al. Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity. International Journal of Molecular Sciences. 2020; 21(13):4696. https://doi.org/10.3390/ijms21134696
Chicago/Turabian StylePtaszyńska, Natalia, Katarzyna Gucwa, Katarzyna Olkiewicz, Mateusz Heldt, Marcin Serocki, Anna Stupak, Dorota Martynow, Dawid Dębowski, Agata Gitlin-Domagalska, Jan Lica, and et al. 2020. "Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity" International Journal of Molecular Sciences 21, no. 13: 4696. https://doi.org/10.3390/ijms21134696
APA StylePtaszyńska, N., Gucwa, K., Olkiewicz, K., Heldt, M., Serocki, M., Stupak, A., Martynow, D., Dębowski, D., Gitlin-Domagalska, A., Lica, J., Łęgowska, A., Milewski, S., & Rolka, K. (2020). Conjugates of Ciprofloxacin and Levofloxacin with Cell-Penetrating Peptide Exhibit Antifungal Activity and Mammalian Cytotoxicity. International Journal of Molecular Sciences, 21(13), 4696. https://doi.org/10.3390/ijms21134696