1. Introduction
Hypoxic pulmonary hypertension (HPH) is a well-known component of pulmonary artery hypertension (PAH), which is characterized by persistent pulmonary vasoconstriction and vascular remodeling, finally leading to heart failure. Underlying the pathogenesis, excessive pulmonary vascular smooth muscle cell (VSMC) proliferation is the convergence point. Growing evidence suggests that mitochondrial dysregulation plays a vital role in VSMC proliferation [
1], displaying mitochondria-induced suppression of apoptosis and activation of proliferating signaling. Interestingly, the functional and structural changes of remodeled arteries in HPH nearly completely reverse in normoxia after exposure to chronic hypoxia [
2,
3,
4], however it is not reversed in a murine model with endothelial Bone Morphogenetic Protein Receptor Type II (BMPRII) mutations due to mitochondrial impairment [
4]. Furthermore, Dichloroacetate, which can restore mitochondrial function, has proven its effectiveness in experimental models of cancer and PAH through the enhancement of glucose oxidation (GO) and the attenuation of mitochondria-induced hypoxia-inducible factor 1α (HIF-1α) signaling [
5].
Most cancer cells and pulmonary artery smooth muscle cells (PASMCs) undergo a metabolic shift, enhancing glycolysis and suppressing GO, leading to dysregulation of mitochondria-derived reactive oxygen species (mROS), citrate, and α-ketoglutarate and induction of the proliferation-related signaling pathways, including nuclear factor of activated T cells (NFAT), voltage-gated potassium channel 1.5 (Kv1.5), and HIF-1α [
5,
6]. In turn, downstream factors such as HIF-1α can inhibit GO via inducing the expression of glycolysis enzymes, ultimately causing a detrimental regulatory loop to enhance proliferation. The role of GO suppression and mitochondrial abnormalities in proliferative diseases is still under active investigation. A decrease in the level of mitochondrial Ca
2+ (Ca
2+m) is one of the predominant reasons underlying this impairment. As a result, the activity of Ca
2+-dependent GO enzymes, including pyruvate dehydrogenase, isocitrate dehydrogenase, and α-oxoglutarate dehydrogenase, is compromised, leading to GO inhibition [
5]. Intriguingly, hyperproliferative VSMCs have been shown to display decreased Ca
2+m influx [
7,
8,
9], which is regulated by the mitochondria-associated membranes (MAM).
MAM, a synapse-like ultrastructure between the mitochondria and endoplasmic reticulum (ER), is known to transfer ER Ca
2+ to the mitochondria through the residing inositol 1,4,5-trisphosphate receptor (IP
3R)-glucose regulated protein 75 (GRP75)-voltage-dependent anion-selective channel 1 (VDAC1) complex. Considering that the Ca
2+m uniporter has a low Ca
2+ affinity, Ca
2+m influx mainly relies on the proximal microdomains. In addition to Ca
2+m regulation, MAM is also involved in steroid synthesis, phospholipid metabolism, proliferation, autophagy, and mitochondrial biology [
10]. Lines of constitutive proteins, kinases, and signaling pathways (such as pAkt-inositol 1,4,5-trisphosphate receptor type 3 (IP
3R3), pAkt-phosphofurin acidic cluster sorting protein 2 [PACS2]) have been discovered at the MAM surface, which maintain close spatial proximity and biological functions [
11]. Recent studies have revealed that MAM formation and functions are enhanced in several conditions, such as senescence, Alzheimer’s disease, gangliosidosis, obesity, and insulin resistance [
12]. It has also been shown that enhanced MAM formation increases the apoptosis of PASMCs and ameliorates HPH [
8], which it is induced by silencing Nogo-B via increase in ER Ca
2+ flux to the mitochondria. Likewise, blocking ER stress, an upstream factor of Nogo-B, has been shown to be a potential therapeutic strategy for HPH in a murine model [
9].
Nogo-B receptor (NgBR) resides in the ER and stimulates its ligand, Nogo-B, to regulate migration and proliferation in endothelial cells and VSMCs [
13]. NgBR is required for cerebral vasculature development [
14] and vascular angiogenesis [
15]. It has been shown that the reduction of the NgBR level attenuates the vascular endothelial growth factor (VEGF)-induced phosphorylation of Akt, the phenotype of chemotaxis, and morphogenesis in endothelial cells, while transfection with active Akt plasmid reverses this effect. Follow-up studies suggest that the cytoplasmic domain of NgBR resembles hydrophobic pockets to bind prenylated Ras and promotes its accumulation and activation at the membrane. Recently, it was reported that the decrease of NgBR expression promoted PASMC proliferation through regulation of reactive oxygen species (ROS) production in an intrauterine pulmonary hypertension (IPH) model [
16]. However, the underlying mechanism remains unclear.
This study aims to explore the role of NgBR in MAM and subsequent mitochondrial function in VSMCs. Here, we analyzed the alteration in NgBR expression in vivo and in vitro under hypoxia-renormoxia and elucidated its function in MAM formation and MAM-associated pAkt signaling. Our results provide a better understanding of the role of NgBR in mitochondrial dysfunction-related diseases.
3. Discussion
Mitochondrial dysfunction is a well-known contributor of VSMC proliferation and vascular remodeling, but the mechanism by which it occurs remains unclear. In this study, we demonstrated that NgBR expression is suppressed in vivo and in vitro under hypoxia. Downregulation of NgBR promotes the proliferation of hypoxic VSMCs through mitochondrial impairment, which is caused by a decreased level of Ca2+m. Disruption of MAM and enhanced pAkt/IP3R3 signaling lead to attenuation of mitochondrial Ca2+ influx. These results indicate that NgBR plays an important role in mitochondrial impairment from excessive VSMC proliferation.
PAH is a fatal disease characterized by persistent vascular contraction, excessive proliferation of VSMCs, and aberrant secretion of endothelial cells. Using a specific spontaneous regression model of HPH, we investigated the role of NgBR in VSMC proliferation. Contrary to the increased level of Nogo-B in HPH, we observed that exposure to hypoxia suppressed NgBR level in the remodeled pulmonary arteries, while NgBR expression was increased after normoxic recovery. The pathogenesis of “recovery” has received particular interest. Not only is the proinflammatory and pro-proliferative environment reversed during the recovery [
21], but there is also a change in the characteristic phenotype of VSMCs towards a proapoptotic phenotype [
22]. This observation enhances our understanding on the relationship between NgBR and VSMC proliferation. In our in vitro model, VSMCs proliferation was not reversed in reoxygenation. This may be explained by the time of renormoxia exposure not being sufficient to reflect the restoration of excessive proliferation, as our observations suggested that wall thickness in remodeled arteries was nearly reversed to that in normoxia until two-months recovery later. Here, we discovered that exposure to hypoxia suppressed NgBR expression, and downregulation of NgBR mimicked hypoxia-induced proliferation in vitro. NgBR OE attenuated this effect.
ROS play a critical role in VSMC proliferation and endothelial dysfunction, and negatively regulates NgBR expression. Elevation of ROS has been shown to inhibit NgBR expression in normal PASMCs exhibiting hyperproliferation, while scavenging ROS restored the reduced NgBR expression in PASMCs in a model of IPH [
16]. This negative feedback regulation also pertains to IPH-vascular endothelial cells. The similarity in compromised NgBR expression between IPH and HPH suggests possible common upstream pathways, given that hypoxic PASMCs show sustained increase of ROS production. Other PH models may display downregulated NgBR expression.
We next investigated the role of NgBR in mitochondrial function. Mitochondrial hyperpolarization has been well described in the proliferating PASMCs of PH patients and animal models [
5]. Likewise, NgBR KD VSMCs show mitochondrial hyperpolarization with reduced Ca
2+m influx. mROS generation is a byproduct of unpaired electron transfer in complex I and complex III during oxidative phosphorylation. Despite controversial conclusions regarding the elevation or attenuation of mROS production in PH-PASMCs, it is clear that mROS plays a principal role in the development of mitochondrial abnormalities through affecting redox-sensitive proteins, such as HIF-1α and Kv 1.5. Our data revealed that mROS production was decreased in NgBR KD VSMCs. We also detected a decline of Ca
2+m in NgBR KD cells. Attenuation of Ca
2+m influx has a causal role in decreased dehydrogenases activities in the Krebs cycle, thereby inhibiting mitochondrial respiration. NgBR OE enhanced mitochondrial respiration through an increase in Ca
2+m. Unexpectedly, basal respiration was not reduced while NgBR was downregulated. Basal respiration includes two consumptions: mitochondrial oxidative phosphorylation and proton leak. Recent work suggests that downregulation of NgBR contributes to mitochondrial dysfunction due to a breakdown of the glycosylation pathway on part of oxidative phosphorylation system subunits [
23]. This may lead to a perturbation of the electron transference in respiratory chain, and subsequently abnormalities of proton leak. Thus, that may cover up the reduction of mitochondrial oxidative phosphorylation. The above results suggest that downregulation of NgBR disturbs mitochondrial function.
Next, we found that downregulation of NgBR led to the activation of downstream HIF-1α under normoxia. Integration of abnormalities in the mitochondria activates downstream effectors, such as HIF-1α, which is induced at both transcriptional and protein levels in PASMCs and pulmonary arterial endothelial cells obtained from patients with pulmonary hypertension and animal models. Heterozygous HIF-1α
+/– mice subjected to chronic hypoxic exposure remarkably lack elevated pulmonary arterial pressure and right ventricular hypertrophy [
24]. Reduced mROS relieves the inhibition of HIF-prolyl-hydroxylases and alleviates p53 activity, which inhibits HIF-1 activation at both transcription and protein levels. Concomitantly, HIF-1α itself inhibits mitochondrial biology via decreasing mitochondrial biogenesis and inducing the expression of glycolytic enzymes, in the same way that pyruvate dehydrogenase (PDH) kinase phosphorylates and inhibits PDH, ultimately causing a detrimental regulatory loop.
We then explored a causal factor for mitochondrial impairment and showed that NgBR mediates communications between the ER and mitochondria. Recently, ER stress and mitochondrial dysfunction have been implicated in the progress of various cardiovascular diseases, including cardiac hypertrophy, ischemic heart disease, pulmonary hypertension, and atherosclerosis [
5,
25]. Several intriguing studies link these two fundamental cellular processes and provide novel strategies on restoring mitochondrial function [
9,
12]. MAM, a specialized microdomain of the ER membrane in proximity to the mitochondria, specially conducts ER Ca
2+ transmission to the mitochondria. Grp75 is required for this ultrastructure integrity, and induces mitochondrial dysfunction under pathological status [
26,
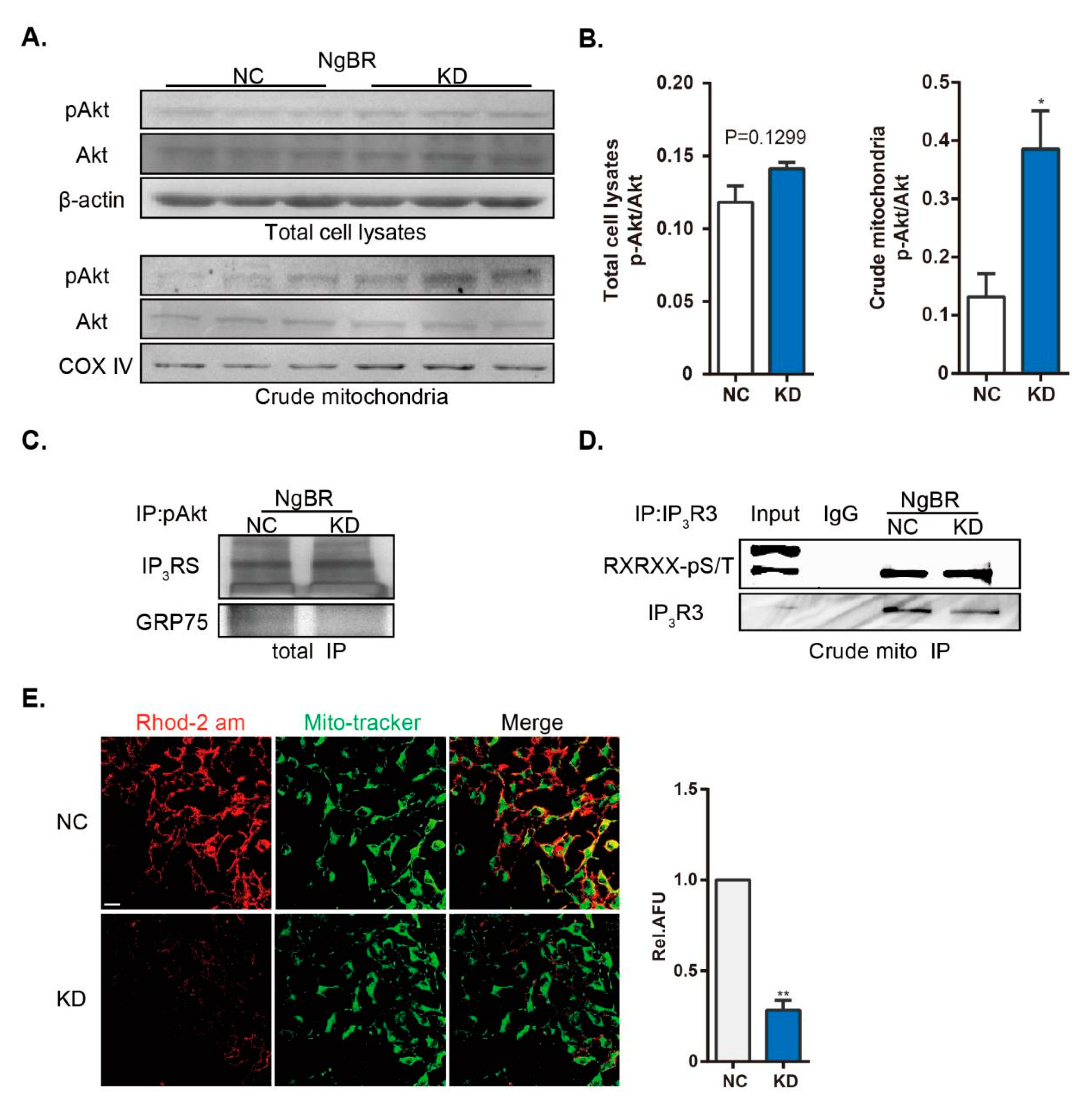
27]. We found that the interaction between Grp75 and pAkt was downregulated in NgBR knock-down cells, this indicates a reduction of MAM. Our data here link the two constitutive cellular events via NgBR, and show that downregulation of NgBR reduces ER-mitochondria interaction under normoxia, causing reduced Ca
2+m.
Apart from the mechanistic regulation of ER-mitochondria distance, we reported a potential regulatory role of NgBR in pAkt signaling at the MAM surface. Akt, a pivotal regulator of localized proteins at the MAM surface, targets several substrates, such as IP
3Rs, hexokinase 2, and PACS2, followed by regulation of mitochondrial biology [
11]. Intriguingly, we found that downregulation of NgBR promoted pAkt/IP
3R3 signaling in MAM. Depletion of NgBR inhibits VEGF-stimulated phosphorylation of Akt in human umbilical vein endothelial cells, contributing to diminished cell migration and morphogenesis. A reverse effect by constitutive Akt activation demonstrates that Akt is a downstream target of NgBR. However, we found no discernible changes in pAkt/Akt level following NgBR downregulation in VSMCs, which is in agreement with a recent report [
16]. Surprisingly, downregulated NgBR expression enhanced phosphorylation of Akt in the crude mitochondrial extracts, thereby increasing the level of phosphorylated IP
3R3 and further reducing the transfer of ER Ca
2+ into the mitochondria. The mechanism of NgBR-induced subcellular Akt activation remains unclear. Recently, NgBR was reported to recruit non-caveolae-rich lipid raft subdomains associated with H-Ras localization at the cell membrane [
28]. Accumulation of H-Ras at the membrane is a canonical upstream activator of the phosphatidylinositol-4,5-bisphosphate 3-kinase-Akt pathway leading to tumorigenesis and tumor resistance. In addition to plasma membrane localization, Ras proteins also activate Akt in other cellular compartments, such as the ER and Golgi [
29,
30]. As expected, several membrane Ras family proteins have been recognized in isolates from both the brain and liver MAM by depth proteomic analyses [
31]. Therefore, it may explain the effect of subcellular pAkt activation. However, the mechanism by which NgBR downregulation activates MAM-associated pAkt signaling, by multiple steps or direct binding, needs to be explored in future studies.
There are certain limitations in our study. First, we used the VSMC cell line A10 instead of primary PASMCs for studying the role of NgBR in MAM and VSMC proliferation. However, the amount of subcellular fraction that can be obtained from primary PASMCs is limited. Hence, we utilized the vascular smooth muscle-derived cell line to acquire enough subcellular extracts, and established a hypoxia-induced proliferative model that displayed downregulated NgBR expression, in keeping with the results of HPH. Furthermore, MAM has a well-defined role in related proliferative/apoptotic conditions, such as PH-PASMCs, cancer, and hypoxia-reoxygenation injury [
32,
33]. In the present study, we demonstrate that downregulation of NgBR triggers pseudohypoxic disruption of MAM.
In summary, we report a novel role of downregulated NgBR expression in mitochondrial impairment and VSMC proliferation through suppressing MAM formation and promoting MAM-associated pAkt signaling. A better understanding about the mechanisms of NgBR is required, which may have potential implications in the treatment of vascular remodeling and mitochondrial dysfunction-related diseases.
4. Materials and Methods
4.1. Chemicals
Anti-pAkt1/2/3 (Ser473; sc-7985), IP3R3 (sc-7277), and NgBR (sc-138044) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against proliferating cell nuclear antigen (PCNA; BM0104), VDAC1 (PB0478), GRP75 (PB0668), and IP3R1 (PB0223) were obtained from Boster (Pleasanton, CA, USA). Antibodies against fatty acid-CoA ligase 4 (FACL-4; ab155282), cytochrome c oxidase complex IV (COX IV; ab 14744), and NgBR (ab168351) were acquired from Abcam (Cambridge, Cambs, UK). Antibodies against IP3Rs (#3763), phospho-(Ser/Thr) Akt substrate (#9611), and cleaved caspase-3 (#9664) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-Akt (60203-2-Ig) was purchased from proteintech (Wuhan, Hubei, China). Anti-IP3R3 (610312) was obtained from BD Biosciences (Franklin Lakes, NJ, USA). Anti-HIF-1α (NB100-134) was obtained from Novus Biologicals (Littleton, CO, USA). Anti-β-actin (bsm-33036M) was obtained from Bioss (Woburn, MA, USA). Horseradish peroxidase (HRP)-labeled goat anti-rabbit/mouse IgG (H+L) was purchased from Boster. Rhod-2 AM (R1244), tetramethylrhodamine methyl ester (TMRM) reagent (I34361), and MitoSOX™ Red (M36008) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Protein A/G magnetic beads were purchased from Bio-Rad Laboratories (Hercules, CA, USA). 5-Ethynyl-2′-deoxyuridine (EdU) detection kits (C10310-3) were purchased from RiboBio (Guangzhou, Guangdong, China). Mitochondrial separation reagent (C3601) was obtained from Beyotime (Beijing, China).
4.2. Animals
Animal experiments were approved by the Laboratory Animal Welfare and Ethics Committee of the Army Medical University (Chongqing, SCXK 2012-0011, 01082012, China). Adult male rats (180–200 g) were randomly divided into 6 groups (H45d, R30d, R60d, N45d, N75d, and N105d) and fed under normoxia (21% O
2) or hypobaric hypoxia (10.0% O
2) for the indicated days. Prior to lung isolation, the mean pulmonary artery pressure (mPAP) was measured as previously described [
34]. The right ventricle (RV) free wall and the left ventricle plus septum (LV + S) were separated, and the weight ratio of RV/(LV + S) was calculated as an index of RV hypertrophy. Rats were anesthetized with urethane and perfused with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). The lung tissues were fixed with 4% PFA overnight at 4 °C and then embedded in optimal cutting temperature (OCT) compound. Following this, 5-μm-thick sections were made for hematoxylin and eosin staining. Images were captured using an Olympus IX-70 inverted microscope (Olympus, Tokyo, Japan). The percent medial wall thickness was analyzed as previously described [
34]. Distal pulmonary arteries (50–300 μm) were identified and measured at the two ends of the shortest external diameter of the distal pulmonary arteries, and the average was taken ([2 × wall thickness/external diameter] × 100).
4.3. Cell Culture
The VSMC cell line A10 was purchased from the American Type Culture Collection and cultured in high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Experimental groups (H48 and R24) were exposed to hypoxia (4% O2) for 48 h, followed by exposure to normoxia (21% O2) for 24 h. Control groups were maintained under normoxia (21% O2) for 48 and 72 h. Hypoxia was established by using a mixture of ultra-high purity gases (5% CO2, 4% O2, and 91% N2) in a 37 °C hypoxia workstation (InvivO2; Baker Ruskinn, Sanford, ME, USA). Most experiments involving hypoxic groups, including Rhod-2 AM staining and cells collection for crude mitochondrial extracts were performed in the hypoxia workstation.
4.4. Transient Transfection with Plasmids
Short hairpin RNA (shRNA) plasmids targeting NgBR were designed with the following sequences: (sense) 5′-CGGCAGTATGCAGCTTGTGAA-3′, (antisense) 5′-TTCACAAGCTGCATACTGCCG-3′, which was provided by GeneChem (Shanghai, China), as well as an NgBR overexpression (OE) plasmid. A non-target plasmid was generated as a control. Plasmid transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
4.5. Subcellular Fraction
Cells (1–2 × 10
7) were harvested, trypsinized, washed once with cold PBS, and centrifuged at 235×
g for 10 min. Cell pellets were resuspended in mitochondrial separation reagent (Beyotime) on ice for 15 min. The suspension was homogenized with a total of 16 strokes using a pre-cooled glass homogenizer and centrifuged at 750×
g for 10 min. The pellet contained nuclei and intact cells. The acquired supernatant was further centrifuged at 11,000×
g for 10 min. All centrifugation procedures were performed at 4°C. Supernatants (crude mitochondrial fraction) were collected [
35].
4.6. Immunoprecipitation
Total cell lysates were obtained by re-suspending cell pellets in immunoprecipitation (IP) buffer (20 mM Tris (pH 7.5), 150 mM NaCl, and 1% Triton X-100) supplemented with a protease inhibitor tablet (Roche, Mannheim, Germany) and phosphatase inhibitor (Boster). Crude mitochondrial extracts were obtained as mentioned above. Specialized antibodies, including pAkt1/2/3 (sc-7985) and IP
3R3 (C-20) antibodies (Santa Cruz Biotechnology) and control antibodies, including goat and rabbit IgG (Beyotime), were incubated with protein A/G magnetic beads at room temperature for 15 min, prior to incubation with lysates overnight at 4 °C. Then, 1× sodium dodecyl sulfate buffer was added and the precipitated immune complex was boiled at 65 °C for 25 min, following a previously described protocol [
11].
4.7. Western Blotting
Equal amounts of protein samples were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories). After blocking with skimmed milk powder at room temperature for 1 h, primary antibodies were added and incubated overnight at 4 °C. The bands were washed with Tris-buffered saline with Tween 20 and incubated with HRP-labeled secondary antibodies at room temperature for 1 h. The immunoreactive bands were visualized using NBT/BCIP (Thermo Fisher Scientific). β-Actin and COX IV were used as the loading controls for total lysates and crude mitochondrial extracts.
4.8. Electron Microscopy
Detailed assessment of the morphology of mitochondria and ER was performed using a scanning transmission electron microscope (Libra 200 FEG, Zeiss, Oberkochen, Germany; 200 kV). Morphological calculations were performed according to a previously established method using ImageJ software [
8]. Cells were fixed with Karnovsky fixative, pelleted, and post-fixed with 1% osmium tetroxide. Cells were then serially dehydrated in ethanol, rinsed in propylene oxide, and incubated in a mixture of propylene oxide and resin prior to embedding. Samples were sliced, mounted on metal gratings, and imaged with the magnification of 50,000×. The shortest line between the outer mitochondrial membrane and ER was drawn to represent the minimum distance between the two organelles and measured using ImageJ as previously reported [
8].
4.9. EdU Detection
EdU assay was used to assess cell proliferation and was performed according to the manufacturer’s instructions. Images were taken using an Olympus FV1000 confocal laser scanning microscope. Proliferating cells were detected and calculated as previously reported [
36]. The number of EdU-positive cells was counted in random optical fields and the ratio of EdU-positive cells to Hoechst-stained cells was calculated.
4.10. Immunofluorescence Staining
The right lung tissues were embedded in OCT compound and snap-frozen with liquid nitrogen. The tissues were cut into 15-μm sections. Cells were washed twice with PBS and fixed with 4% PFA for 30 min at room temperature. The tissue sections were permeated with 0.1% Triton-X-PBS and subsequently blocked with normal serum for 1 h at room temperature. Diluted antibodies (NgBR and α-SMA) were added and maintained overnight at 4 °C. Cells and tissue sections were washed and incubated with secondary antibodies for 1 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Beyotime). Confocal images were captured with an Olympus FV1000 microscope using AIM4.2 software. For analysis of NgBR expression in pulmonary arteries, NgBR fluorescence was excited at 543 nm and emission was 560–660 nm. VSMC marker α-SMA exciation was performed at 488 nm and emission was 505–525 nm. Then NgBR fluorescence was calculated using AIM4.2 software. For analysis of the MAM formation [
12], cells were transfected with plasmids expressing mCherry fused with a targeting sequence of subunit IV of COX (from GeneChem). Protein disulfide isomerase (PDI; Abcam) was used as an ER marker. Pictures were acquired with the magnification of 1000×. Then the mCherry positive cells were used to calculate the Pearson’s correlation coefficient, which indicates the strength of mitochondria-ER interaction.
4.11. Rhod-2 AM Staining
Rhod-2 AM (Gibco, Invitrogen, Carlsbad, CA) staining was used to measure Ca2+m, and was performed according to the commercially available protocol. Cells were incubated with dihydrorhod-2 AM (4 μM) at 37 °C in the presence of 0.003% (w/v) pluronic acid for 40 min. After 0.5 h of de-esterification, cells were washed and incubated with mitochondrial green marker at 37 °C for 15 min. Then cells were washed and mounted. Rhod-2 fluorescence was excited at 543 nm and emission was 560–660 nm. MitoTracker Green exciation was performed at 488 nm and emission was 505–525 nm.
4.12. mROS Detection
MitoSOX™ Red mitochondrial superoxide indicator was used to quantify ROS production. Cells were treated with MitoSOX™ working solution (5 μM) and incubated for 15 min at 37 °C. Then, cells were gently washed three times with PBS, and incubated with mitochondrial green marker at 37 °C for 15 min. Images were taken using an Olympus FV1000 microscope. MitoSOX™ Red excitation was performed at 543 nm and emission was 560–660 nm. MitoTracker Green exciation was performed at 488 nm and emission was 505–525 nm.
4.13. Mitochondrial Inner Membrane Potential
Mitochondrial inner membrane potential (ΔΨ) was measured by treating cells with 10 nM TMRM (Invitrogen) for 30 min at 37 °C. Then, cells were gently washed three times with PBS, and incubated with mitochondrial green marker at 37 °C for 15 min. Images were taken using an Olympus FV1000 microscipe. TMRM excitation was performed at 543 nm and emission was 560–660 nm. MitoTracker Green exciation was performed at 488 nm and emission was 505–525 nm.
4.14. Mitochondrial Respiration
The oxygen consumption rate (OCR) was measured with an XF-96 extracellular flux analyzer (Seahorse Bioscience, Billerica, MA). Cells were seeded in 96-well plates at a density of 8,000 cells per well. After 24 h of transfection with shNgBR or NgBR OE plasmid, the plates were incubated at 37 °C in 5% CO
2 for 48 h. OCR was measured under basal conditions and after the sequential addition of 25 µL oligomycin, 25 µL carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and 25 µL antimycin A/rotenone to 175 µL medium. The final working concentrations reached to 2.4 µM (oligomycin), 0.3 µM (FCCP), and 0.6 µM (antimycin A/rotenone). The maximal respiration, basal respiration, and spare capacity were determined as previously described [
37]. Briefly, basal OCR was calculated as OCR before oligomycin minus OCR after antimycin A/rotenone. Maximal OCR was calculated as the OCR after FCCP minus non-mitochondrial OCR. Spare capacity was calculated as the difference between maximal OCR after FCCP minus basal OCR. To allow comparison between different experiments, data were calculated as the fold of control group. Data are presented as the mean ± standard error of the mean (SEM).
4.15. Statistical Analysis
Individual experiments were performed at least three times. All data are presented as the mean ± SEM. The statistical analysis and graph were performed with SPSS 16.0 and GraphPad Prism6 software. Normality test was performed, and then statistical differences between groups were evaluated by one-way analysis of variance followed by Bonferroni’s test for multiple comparisons. For experiments comparing two groups containing one-way variance, a two-sided, unpaired Student’s t-test was performed to evaluate the differences. p < 0.05 was statistically significant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






