A Thermostable Aspergillus fumigatus GH7 Endoglucanase Over-Expressed in Pichia pastoris Stimulates Lignocellulosic Biomass Hydrolysis

 ,
,
Abstract
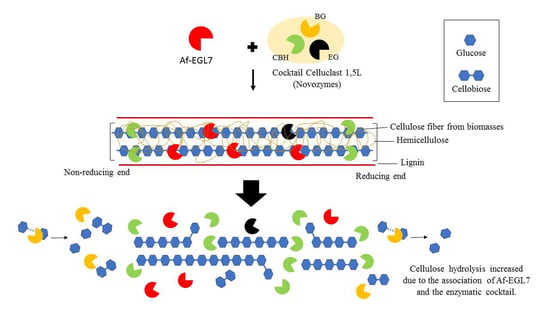
:
1. Introduction
2. Results and Discussion
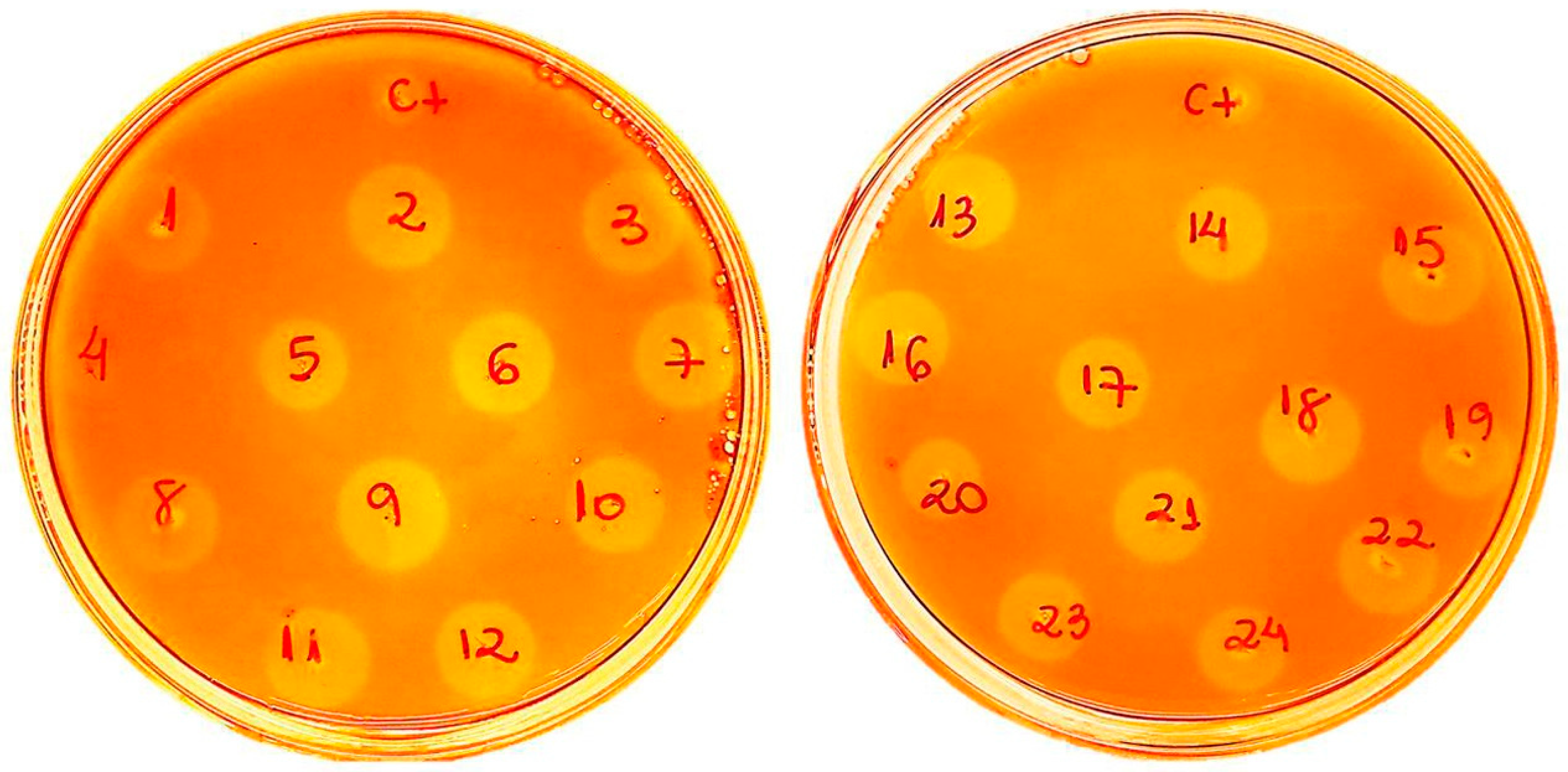
2.1. Af-EGL7 Cloning and Expression
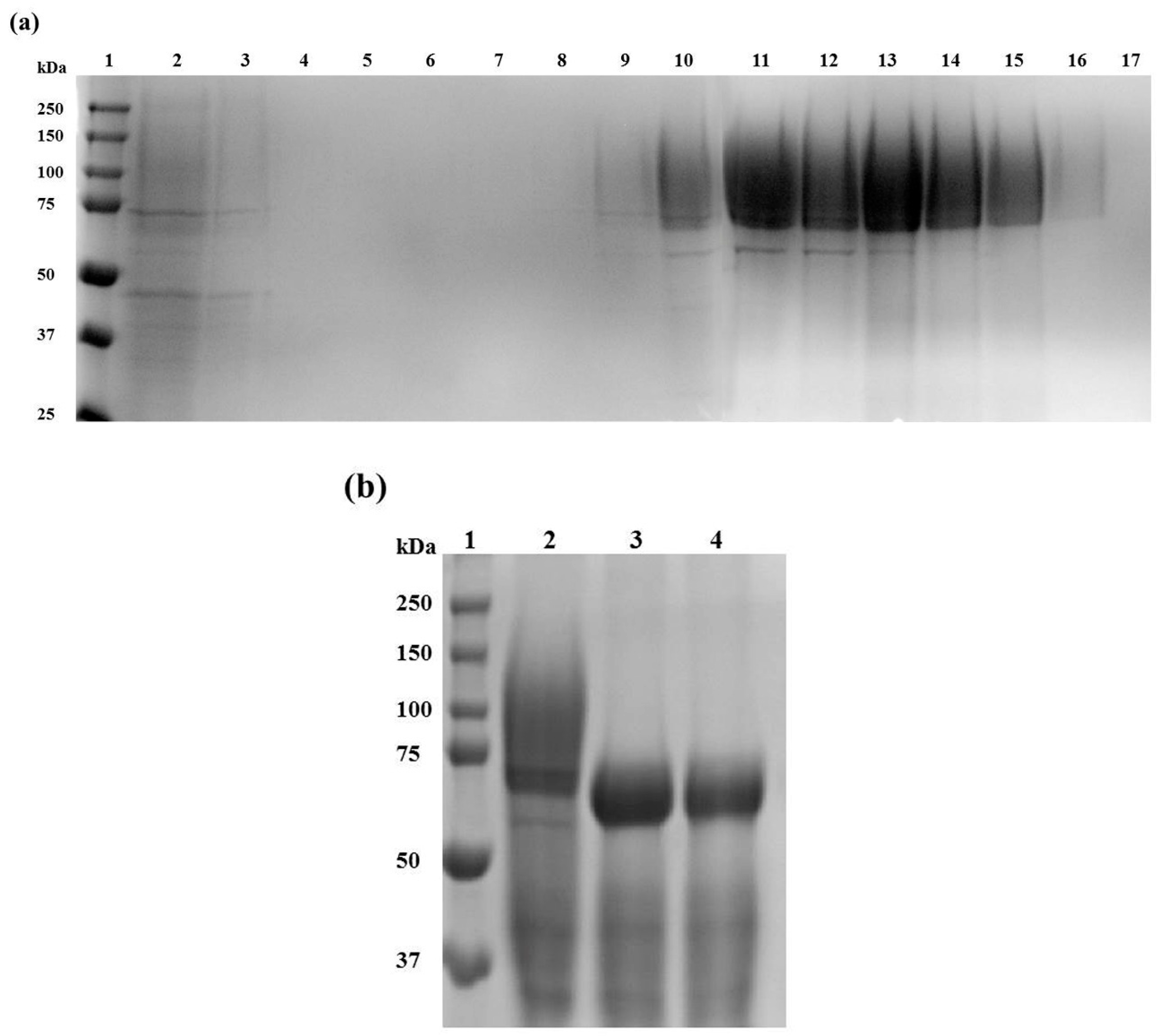
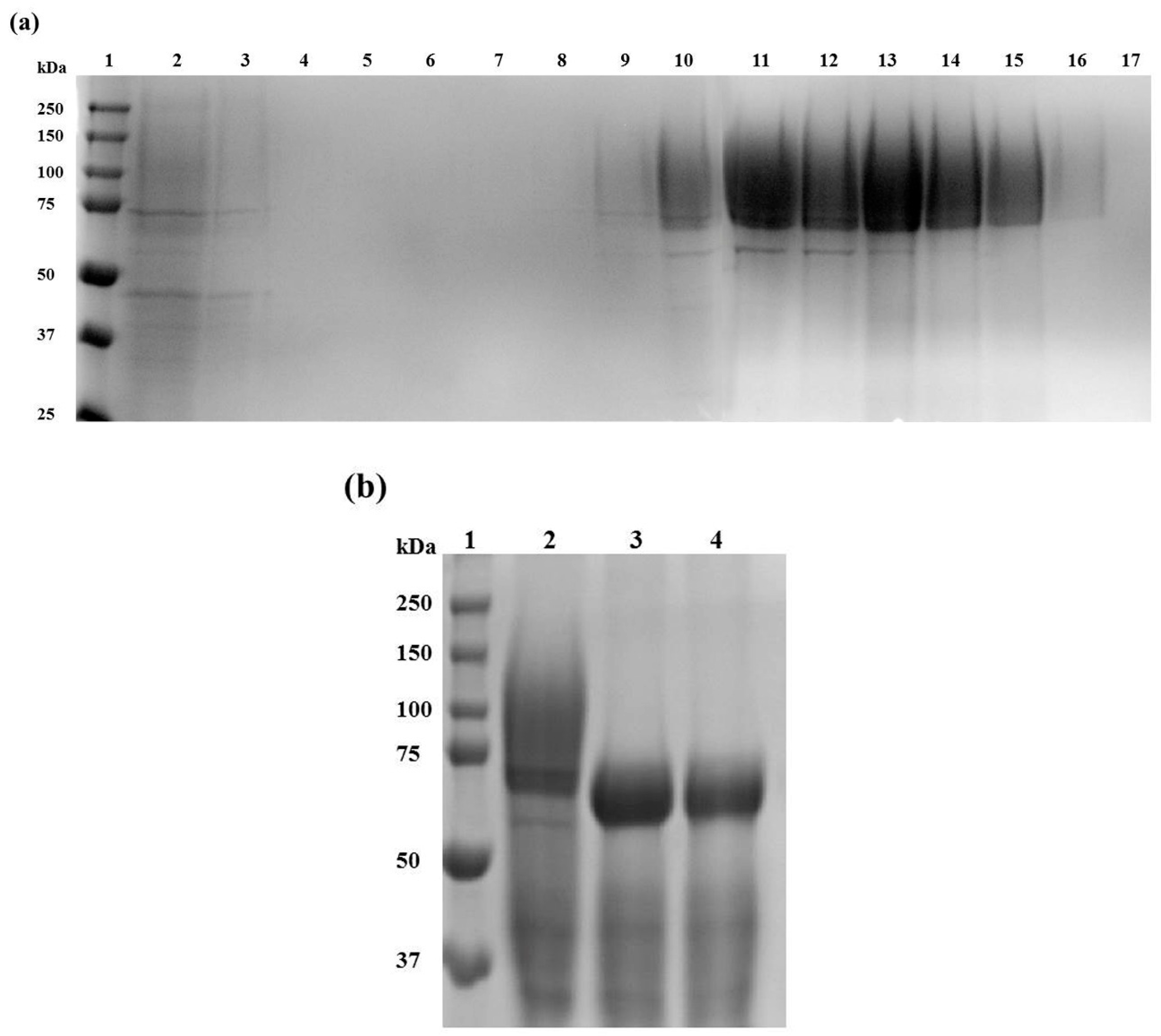
2.2. Af-EGL7 Expression and Purification
2.3. Biochemical Characterization
2.3.1. Enzyme Activity
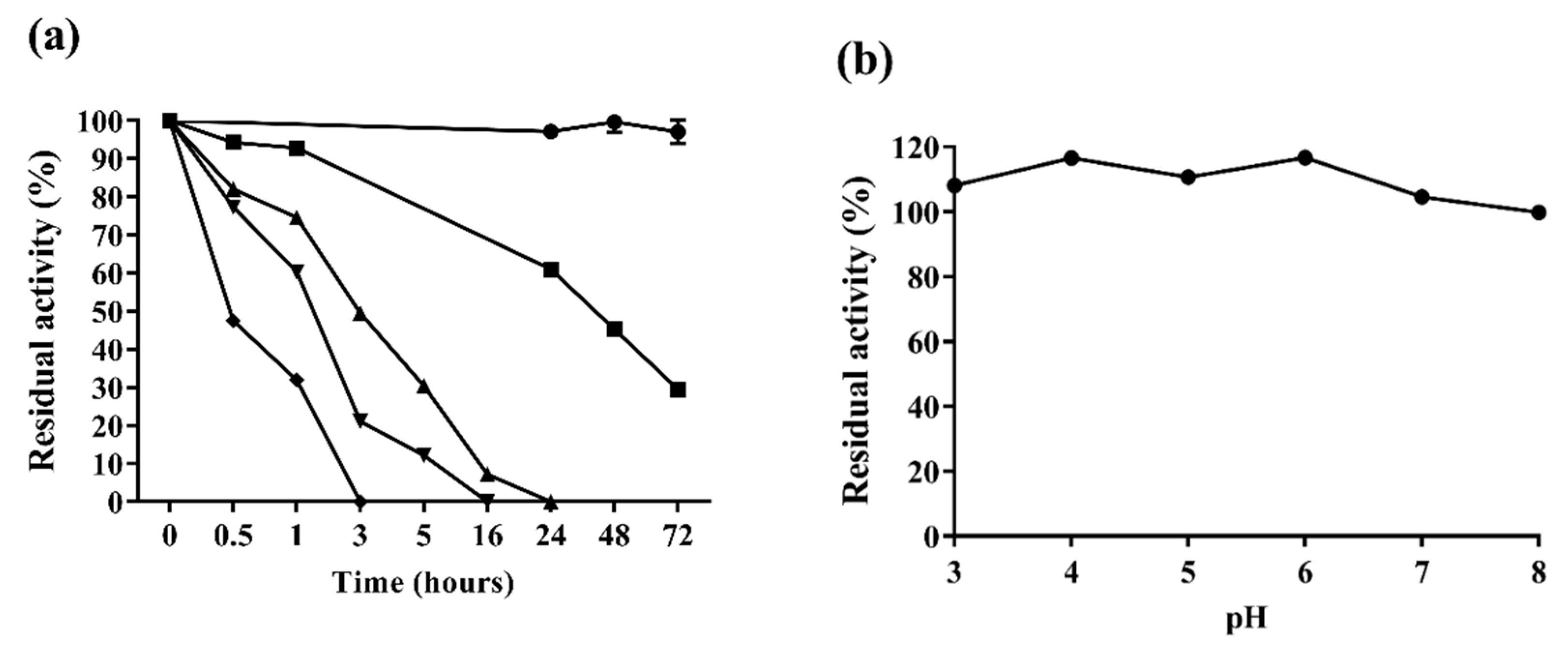
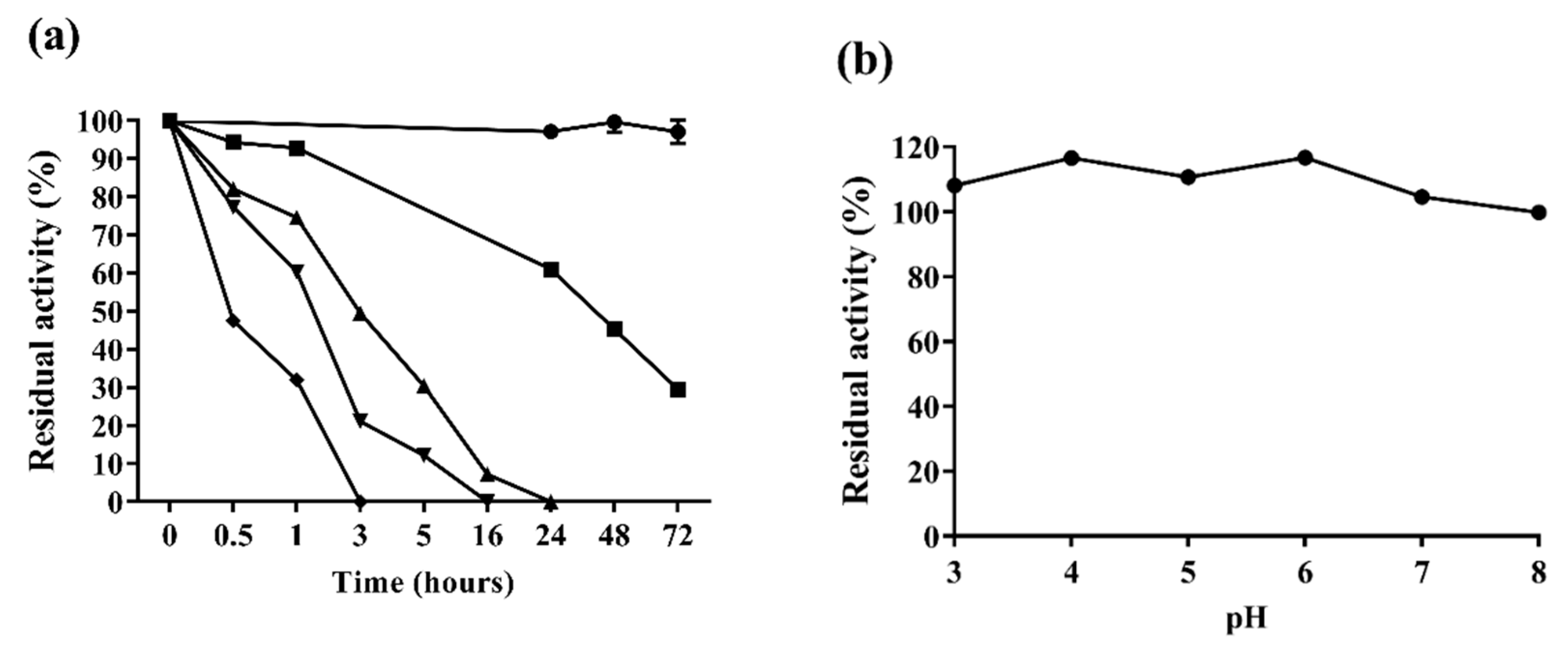
2.3.2. Enzyme Stability
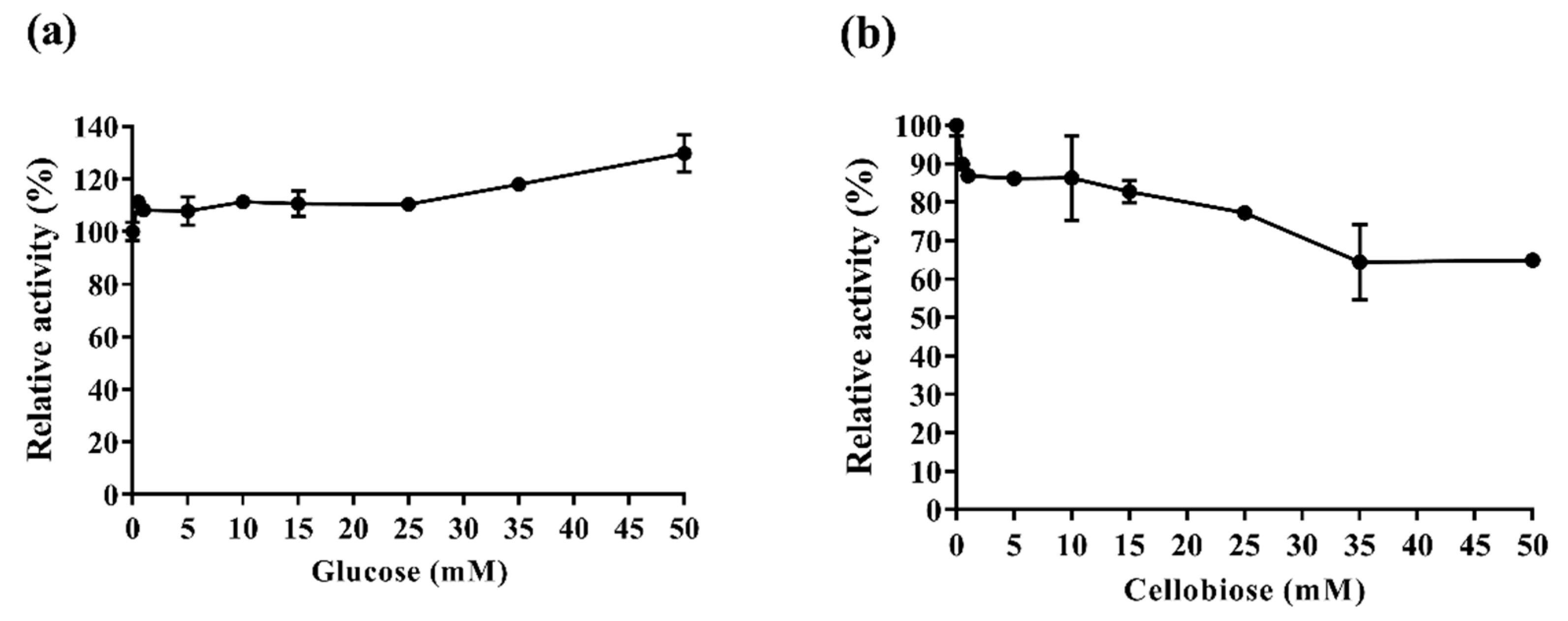
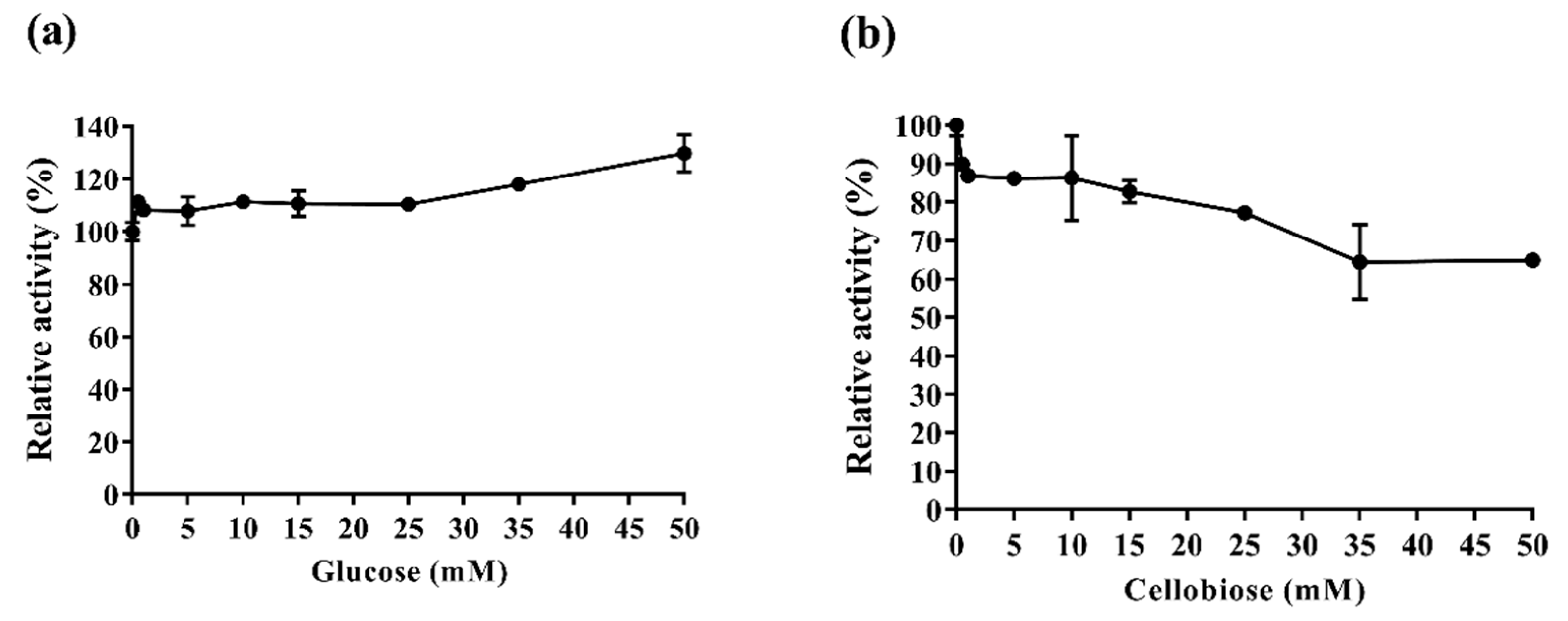
2.3.3. Af-EGL7 Tolerance to Product Inhibition
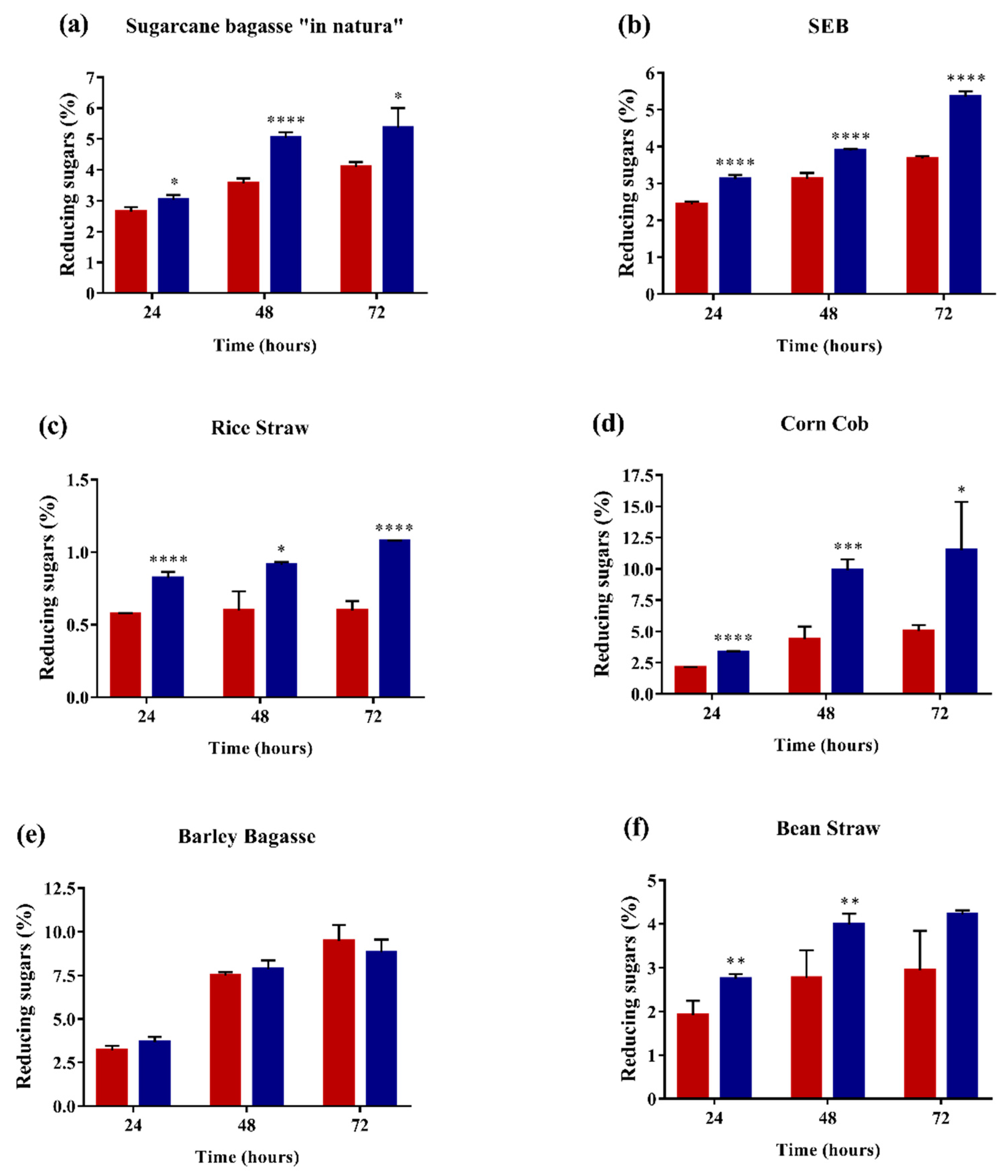
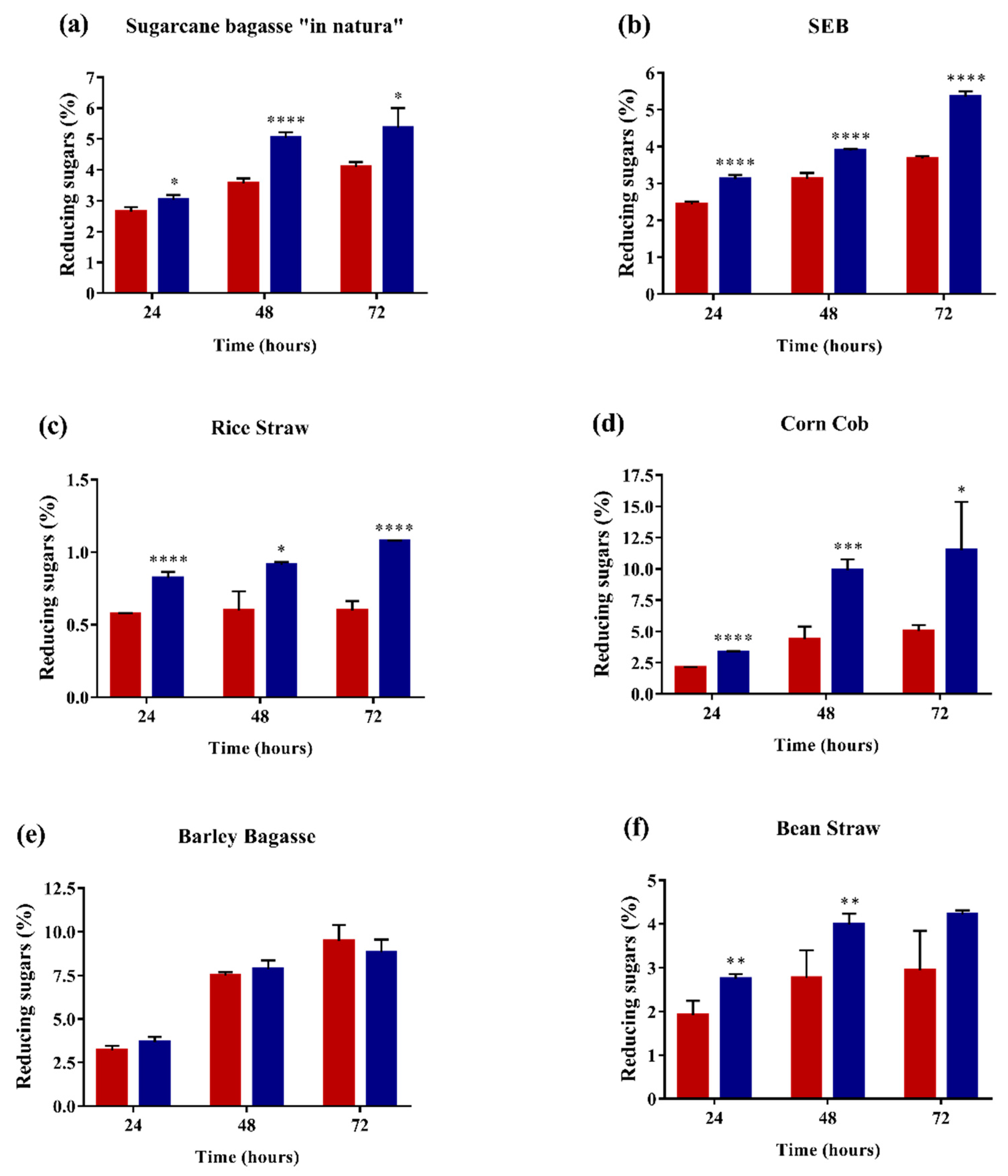
2.4. Enzyme Performance in Agroindustrial Residue Degradation
3. Materials and Methods
3.1. Strains, Culture Conditions and Vector
3.2. RNA Extraction, cDNA Synthesis, and Gene Amplification
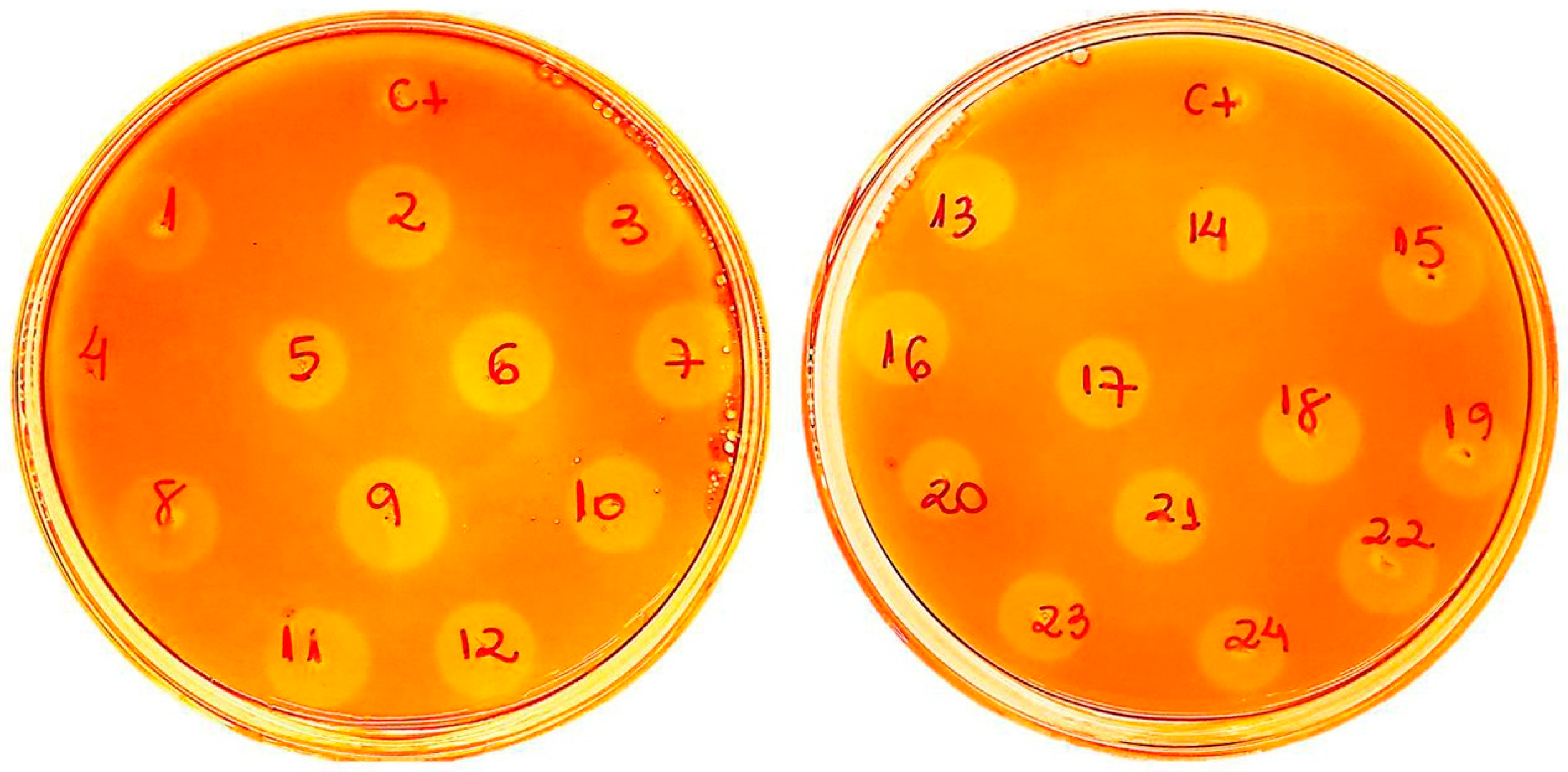
3.3. Cloning, Transformation of P. pastoris, and Screening of Recombinant Transformants
3.4. Recombinant Af-EGL7 Heterologous Expression in P. pastoris
3.5. Recombinant Af-EGL7 Purification
3.6. Endoglucanase Activity Assay
3.7. Af-EGL7 Deglycosylation by Endo H
3.8. Af-EGL7 Stability Assays
3.9. Determination of Kinetic Parameters
3.10. Glucose and Cellobiose Effect on Af-EGL7 Activity
3.11. Lignocellulosic Biomass saccharification
3.12. Statistical Analysis
3.13. Reproducibility of the Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jain, K.K.; Kumar, S.; Bhardwaj, K.N.; Kuhad, R.C. Functional Expression of a Thermostable Endoglucanase from Thermoascus aurantiacus RCKK in Pichia pastoris X-33 and Its Characterization. Mol. Biotechnol. 2018, 60, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.P.; de Albuquerque, F.S.; dos Santos, C.W.V.; Franco, M.; Caetano, L.C.; Pereira, H.J.V. Production, purification, characterization and application of a new halotolerant and thermostable endoglucanase of Botrytis ricini URM 5627. Bioresour. Technol. 2018, 270, 263–269. [Google Scholar] [CrossRef]
- Ramos, J.L.; Duque, E. Twenty-first-century chemical odyssey: Fuels versus commodities and cell factories versus chemical plants. Microb. Biotechnol. 2019, 12, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, A.V.; De Gouvêa, P.F.; Gerolamo, L.E.; Yonamine, D.K.; De Lourdes, L.; Balico, D.L.; Uyemura, S.A. Functional characterization of GH7 endo-1,4-β-glucanase from Aspergillus fumigatus and its potential industrial application. Protein Expr. Purif. 2018, 150, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Li, W.; Han, W.; Wang, Q.; Bi, P.; Han, C.; Zhu, L. Characterization of a novel thermostable GH7 endoglucanase from Chaetomium thermophilum capable of xylan hydrolysis. Int. J. Biol. Macromol. 2018, 117, 342–349. [Google Scholar] [CrossRef]
- Rigoldi, F.; Donini, S.; Redaelli, A.; Parisini, E.; Gautieri, A. Review: Engineering of thermostable enzymes for industrial applications. APL Bioeng. 2018, 2. [Google Scholar] [CrossRef]
- de Gouvêa, P.F.; Bernardi, A.V.; Gerolamo, L.E.; Santos, E.S.; Riano-Pachon, D.; Uyemura, S.A.; Dinamarco, T.M. Transcriptome and secretome analysis of Aspergillus fumigatus in the presence of sugarcane bagasse. BMC Genomics 2018, 19, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Aich, S.; Singh, R.K.; Kundu, P.; Pandey, S.P.; Datta, S. Genome-wide characterization of cellulases from the hemi-biotrophic plant pathogen, Bipolaris sorokiniana, reveals the presence of a highly stable GH7 endoglucanase. Biotechnol. Biofuels 2017, 10, 1–14. [Google Scholar] [CrossRef]
- Liu, Y.; Dun, B.; Shi, P.; Ma, R.; Luo, H.; Bai, Y.; Xie, X.; Yao, B. A novel GH7 Endo-β-1,4-glucanase from Neosartorya fischeri P1 with good thermostability, broad substrate specificity and potential application in the brewing industry. PLoS ONE 2015, 10, e0137485. [Google Scholar] [CrossRef]
- Berto, G.L.; Velasco, J.; Tasso Cabos Ribeiro, C.; Zanphorlin, L.M.; Noronha Domingues, M.; Tyago Murakami, M.; Polikarpov, I.; de Oliveira, L.C.; Ferraz, A.; Segato, F. Functional characterization and comparative analysis of two heterologous endoglucanases from diverging subfamilies of glycosyl hydrolase family 45. Enzyme Microb. Technol. 2019, 120, 23–35. [Google Scholar] [CrossRef]
- Karnaouri, A.C.; Topakas, E.; Christakopoulos, P. Cloning, expression, and characterization of a thermostable GH7 endoglucanase from Myceliophthora thermophila capable of high-consistency enzymatic liquefaction. Appl. Microbiol. Biotechnol. 2014, 98, 231–242. [Google Scholar] [CrossRef]
- Pellegrini, V.O.A.; Serpa, V.I.; Godoy, A.S.; Camilo, C.M.; Bernardes, A.; Rezende, C.A.; Junior, N.P.; Franco Cairo, J.P.L.; Squina, F.M.; Polikarpov, I. Recombinant Trichoderma harzianum endoglucanase I (Cel7B) is a highly acidic and promiscuous carbohydrate-active enzyme. Appl. Microbiol. Biotechnol. 2015, 99, 9591–9604. [Google Scholar] [CrossRef]
- Moroz, O.V.; Maranta, M.; Shaghasi, T.; Harris, P.V.; Wilson, K.S.; Davies, G.J. The three-dimensional structure of the cellobiohydrolase Cel7A from Aspergillus fumigatus at 1.5Å resolution. Acta Crystallogr. Sect. FStructural Biol. Commun. 2015, 71, 114–120. [Google Scholar] [CrossRef]
- Miao, J.; Li, T.; Ma, L.; Liu, D.; Shen, Q.; Wang, M.; Huang, Q. Effects of amino acids on the lignocellulose degradation by Aspergillus fumigatus Z5: Insights into performance, transcriptional, and proteomic profiles. Biotechnol. Biofuels 2019, 12, 1–19. [Google Scholar] [CrossRef]
- Chahed, H.; Boumaiza, M.; Ezzine, A.; Marzouki, M.N. Heterologous expression and biochemical characterization of a novel thermostable Sclerotinia sclerotiorum GH45 endoglucanase in Pichia pastoris. Int. J. Biol. Macromol. 2018, 106, 629–635. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Ranaei Siadat, S.O.; Motallebi, M.; Zamani, M.R.; Barshan Tashnizi, M.; Moshtaghi, S. Characterization and high level expression of acidic endoglucanase in Pichia pastoris. Appl. Biochem. Biotechnol. 2014, 172, 2253–2265. [Google Scholar] [CrossRef]
- Quan, J.; Tian, J. Circular polymerase extension cloning for high-throughput cloning of complex and combinatorial DNA libraries. Nat. Protoc. 2011, 6, 242–251. [Google Scholar] [CrossRef]
- David, B.Y. The colorimetric determination of the serum proteins. J. Biol. Chem. 1929, 82, 545–550. [Google Scholar]
- Hamilton, S.R.; Gerngross, T.U. Glycosylation engineering in yeast: The advent of fully humanized yeast. Curr. Opin. Biotechnol. 2007, 18, 387–392. [Google Scholar] [CrossRef]
- Amore, A.; Knott, B.C.; Supekar, N.T.; Shajahan, A.; Azadi, P.; Zhao, P.; Wells, L.; Linger, J.G.; Hobdey, S.E.; Vander Wall, T.A.; et al. Distinct roles of N- and O-glycans in cellulase activity and stability. Proc. Natl. Acad. Sci. USA 2017, 114, 13667–13672. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Gao, G.; Li, Y.; Yang, L.; Liang, Y.; Jin, H.; Han, W.; Feng, Y.; Zhang, Z. Cloning, expression, and characterization of a thermophilic endoglucanase, AcCel12B from Acidothermus cellulolyticus 11B. Int. J. Mol. Sci. 2015, 16, 25080–25095. [Google Scholar] [CrossRef] [PubMed]
- Narra, M.; Dixit, G.; Divecha, J.; Kumar, K.; Madamwar, D.; Shah, A.R. Production, purification and characterization of a novel GH 12 family endoglucanase from Aspergillus terreus and its application in enzymatic degradation of delignified rice straw. Int. Biodeterior. Biodegrad. 2014, 88, 150–161. [Google Scholar] [CrossRef]
- Valenzuela, S.V.; Valls, C.; Schink, V.; Sánchez, D.; Roncero, M.B.; Diaz, P.; Martínez, J.; Pastor, F.I.J. Differential activity of lytic polysaccharide monooxygenases on celluloses of different crystallinity. Effectiveness in the sustainable production of cellulose nanofibrils. Carbohydr. Polym. 2019, 207, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.V.; Zubieta, M.P.; Franco Cairo, J.P.L.; Calzado, F.; Paes Leme, A.F.; Squina, F.M.; Prade, R.A.; De Lima Damásio, A.R. Mapping N-linked glycosylation of carbohydrate-active enzymes in the secretome of Aspergillus nidulans grown on lignocellulose. Biotechnol. Biofuels 2016, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, L.; Liu, Z.; Yuan, S. Gene cloning, heterologous expression and characterization of a Coprinopsis cinerea endo-β-1,3(4)-glucanase. Fungal Biol. 2017, 121, 61–68. [Google Scholar] [CrossRef]
- Patel, A.K.; Singhania, R.R.; Sim, S.J.; Pandey, A. Thermostable cellulases: Current status and perspectives. Bioresour. Technol. 2019, 279, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Luo, H.; Shi, P.; Huang, H.; Bai, Y.; Yao, B. A highly-active endo-1,3-1,4-β-glucanase from thermophilic Talaromyces emersonii CBS394.64 with application potential in the brewing and feed industries. Process Biochem. 2014, 49, 1448–1456. [Google Scholar] [CrossRef]
- Mohd Azhar, S.H.; Abdulla, R.; Jambo, S.A.; Marbawi, H.; Gansau, J.A.; Mohd Faik, A.A.; Rodrigues, K.F. Yeasts in sustainable bioethanol production: A review. Biochem. Biophys. Reports. 2017, 10, 52–61. [Google Scholar] [CrossRef]
- Azizi, M.; Hemmat, J.; Seifati, S.M.; Torktaz, I.; Karimi, S. Characterization of a thermostable endoglucanase produced by Isoptericola variabilis sp. IDAH9. Brazilian J. Microbiol. 2015, 46, 1225–1234. [Google Scholar] [CrossRef]
- Brodeur, G.; Yau, E.; Badal, K.; Collier, J.; Ramachandran, K.B.; Ramakrishnan, S. Chemical and physicochemical pretreatment of lignocellulosic biomass: A review. Enzyme Res. 2011, 2011, 1–17. [Google Scholar] [CrossRef]
- Kumar, P.; Barrett, D.M.; Delwiche, M.J.; Stroeve, P. Methods for pretreatment of lignocellulosic biomass for efficient hydrolysis and biofuel production. Ind. Eng. Chem. Res. 2009, 48, 3713–3729. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, P.; Yuan, T.; Huang, H.; Li, Z.; Meng, K.; Yang, P.; Yao, B. Purification, gene cloning and characterization of an acidic β-1,4-glucanase from Phialophora sp. G5 with potential applications in the brewing and feed industries. J. Biosci. Bioeng. 2012, 114, 379–384. [Google Scholar] [CrossRef]
- Luo, H.; Yang, J.; Li, J.; Shi, P.; Huang, H.; Bai, Y.; Fan, Y.; Yao, B. Molecular cloning and characterization of the novel acidic xylanase XYLD from Bispora sp. MEY-1 that is homologous to family 30 glycosyl hydrolases. Appl. Microbiol. Biotechnol. 2010, 86, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Meleiro, L.P.; Carli, S.; Fonseca-Maldonado, R.; da Silva Torricillas, M.; Zimbardi, A.L.R.L.; Ward, R.J.; Jorge, J.A.; Furriel, R.P.M. Overexpression of a Cellobiose-Glucose-Halotolerant Endoglucanase from Scytalidium thermophilum. Appl. Biochem. Biotechnol. 2017, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Pang, Q.; Zhao, L.; Fan, S.; Shi, H. Thermoanaerobacterium thermosaccharolyticum β-glucosidase: A glucose-tolerant enzyme with high specific activity for cellobiose. Biotechnol. Biofuels 2012, 5, 1–10. [Google Scholar] [CrossRef]
- Andrić, P.; Meyer, A.S.; Jensen, P.A.; Dam-Johansen, K. Reactor design for minimizing product inhibition during enzymatic lignocellulose hydrolysis: I. Significance and mechanism of cellobiose and glucose inhibition on cellulolytic enzymes. Biotechnol. Adv. 2010, 28, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Day, D.F. Composition of sugarcane, energy cane, and sweet sorghum suitable for ethanol production at Louisiana sugar mills. J. Ind. Microbiol. Biotechnol. 2011, 38, 803–807. [Google Scholar] [CrossRef]
- Prassad, S.; Singh, A.; Joshi, H.C. Ethanol as an alternative fuel from agricultural, industrial and urban residues. Res. Conserv. Recycl. 2007, 50, 1–39. [Google Scholar] [CrossRef]
- Mello, L.R.P.F.; Mali, S. Use of malt bagasse to produce biodegradable baked foams made from cassava starch. Ind. Crops Prod. 2014, 55, 187–193. [Google Scholar] [CrossRef]
- Brum, S.S.; de Oliveira, L.C.A.; Bianchi, M.L.; Guerreiro, M.C.; de Oliveira, L.K.; Carvalho, K.T.G. Síntese de acetato de celulose a partir da palha de feijão utilizando N-bromossuccinimida (NBS) como catalisador. Polímeros 2012, 22, 447–452. [Google Scholar] [CrossRef]
- Miller, G.L. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | KM (mg·mL−1) | Vmax (µmol·min−1·mg−1) | kcat (s−1) | kcat/KM (mL·mg−1·s−1) |
|---|---|---|---|---|
| CM-Cellulose | 24.5 ± 0.6 | 6193 ± 140 | 5037 ± 114 | 205.9 ± 0.8 |
| β-glucan | 5.6 ± 0.3 | 13722 ± 519 | 11161 ± 422 | 1992 ± 37 |
| Xyloglucan | 8.6 ± 0.3 | 9082 ± 293 | 7386 ± 238 | 859.9 ± 0.2 |
| Cu2+ Concentration (mM) | Af-EGL7 Relative Activity (%) |
|---|---|
| 0.0 | 100.0 ± 1.7 |
| 0.1 | 107.2 ± 1.4 |
| 0.5 | 115.5 ± 0.5 |
| 1.0 | 117.4 ± 0.7 |
| 5.0 | 85.9 ± 1.5 |
| 10.0 | 49.8 ± 1.5 |
| 15.0 | 21.7 ± 0.3 |
| Source Organism | Expression System | Substrate | Vmax (U·mg−1) | KM (mg·mL−1) | kcat (s−1) | kcat/KM (mL·mg−1·s−1) | Thermal Stability | pH Stability |
|---|---|---|---|---|---|---|---|---|
| Aspergillus fumigatus (this work) | Pichia pastoris | β-Glucan | 13722 ± 519 | 5.6 ± 0.3 | 11161 ± 422 | 1992 ± 37 | 100% after 72 h at 55 °C; 30% after 1 h at 90 °C | No loss after 72 h in the pH range 3.0–8.0 |
| Xyloglucan | 9082 ± 293 | 8.6 ± 0.3 | 7386 ± 238 | 859.9 ± 0.2 | ||||
| CMC-Na | 6193 ± 140 | 24.5 ± 0.6 | 5037 ± 114 | 205.9 ± 0.8 | ||||
| Aspergillus fumigatus [4] | Escherichia coli | β-Glucan | 191.9 ± 0.0007 | 113.9 ± 0.005 | 159.9 | 1.4 | 85% after 48 h at 50 °C; 40% after 1 h at 55 °C | Start to decrease after 24 h at pH 6.0 |
| CMC-Na | 51.9 ± 0.007 | 209.7 ± 0.1 | 43.3 | 0.2 | ||||
| Neosartorya fischeri [9] | Pichia pastoris | β-Glucan | 5000 ± 186 | 4.5 ± 0.2 | - | - | 16.1% after 1h at 70 °C | No loss after 1 h in the pH range 3.0–8.0 |
| Talaromyces emersonii [27] | Pichia pastoris | β-Glucan | 17951 ± 69 | 4.0 ± 1.5 | - | 3156 ± 24 | More than 50% after 2 min at 80 °C | More than 70% and 50% after 1 h in the pH range 1.0–11.0 and at pH 12.0, respectively |
| CMC-Na | 2257 ± 79 | 20.8 ± 2.1 | - | 78.9 ± 3.8 | ||||
| Myceliophthora thermophile [11] | Pichia pastoris | CMC-Na | 622.5 ± 86.4 | 24.0 ± 0.5 | - | 0.313667 | t1/2 = 9.96 h (70 °C); t1/2 = 6.5 h (80 °C) | No loss after 24 h in the pH range 3.0–11.0 |
| Trichoderma harzianum [12] | Aspergillus niger | Xyloglucan | 0.22 ± 0.095 (μM·s−1) | 1.98 ± 0.47 | 0.45 | - | 100% after two months of incubation at 55 °C and pH 5.0 | - |
| Chaetomium thermophilum [5] | Pichia pastoris | CMC-Na | 59.6 ± 8.2 | 79.2 ± 5.8 | 2.11 × 10−3 | 0.02673 | 61.3% after 60 min at 80 °C; Almost all activity lost after 100 min at 90 °C | - |
| β-Glucan | 12.6 ± 0.6 (µg·min−1·mL−1) | 9.8 ± 0.6 | 0.7 × 10−3 | 0.07849 | ||||
| Bispora sp. MEY-1 [33] | Pichia pastoris | β-glucan | 6737 | 9.16 | - | - | 100% after 1 h at 60 °C; More than 30% after 1 h at 70 °C | More than 85% after 1 h in the pH range 1.0–8.0 |
| CMC-Na | 3460 | 287 | - | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vianna Bernardi, A.; Kimie Yonamine, D.; Akira Uyemura, S.; Magnani Dinamarco, T. A Thermostable Aspergillus fumigatus GH7 Endoglucanase Over-Expressed in Pichia pastoris Stimulates Lignocellulosic Biomass Hydrolysis. Int. J. Mol. Sci. 2019, 20, 2261. https://doi.org/10.3390/ijms20092261
Vianna Bernardi A, Kimie Yonamine D, Akira Uyemura S, Magnani Dinamarco T. A Thermostable Aspergillus fumigatus GH7 Endoglucanase Over-Expressed in Pichia pastoris Stimulates Lignocellulosic Biomass Hydrolysis. International Journal of Molecular Sciences. 2019; 20(9):2261. https://doi.org/10.3390/ijms20092261
Chicago/Turabian StyleVianna Bernardi, Aline, Deborah Kimie Yonamine, Sergio Akira Uyemura, and Taisa Magnani Dinamarco. 2019. "A Thermostable Aspergillus fumigatus GH7 Endoglucanase Over-Expressed in Pichia pastoris Stimulates Lignocellulosic Biomass Hydrolysis" International Journal of Molecular Sciences 20, no. 9: 2261. https://doi.org/10.3390/ijms20092261
APA StyleVianna Bernardi, A., Kimie Yonamine, D., Akira Uyemura, S., & Magnani Dinamarco, T. (2019). A Thermostable Aspergillus fumigatus GH7 Endoglucanase Over-Expressed in Pichia pastoris Stimulates Lignocellulosic Biomass Hydrolysis. International Journal of Molecular Sciences, 20(9), 2261. https://doi.org/10.3390/ijms20092261