Structure/Activity Analysis of TASK-3 Channel Antagonists Based on a 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine

 ,
,  ,
,  ,
, 
Abstract

1. Introduction
2. Results
2.1. Exploring the PK-THPP Binding Site in TASK-3 by Alanine Mutagenesis Screening
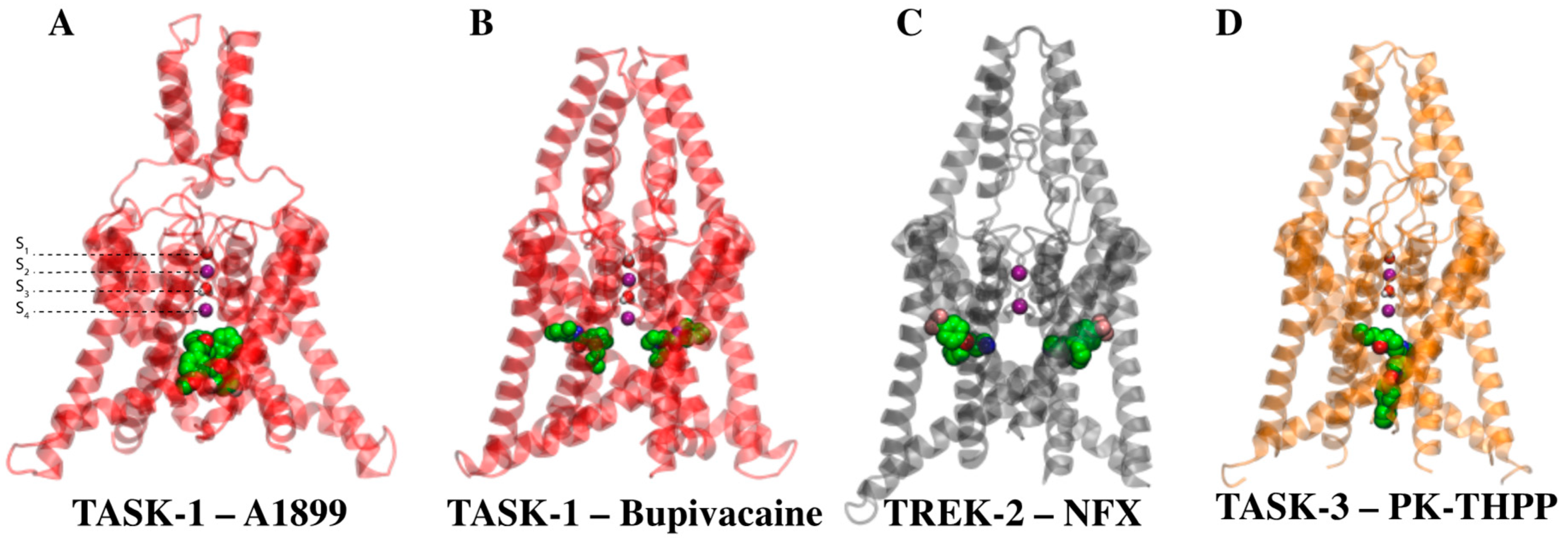
2.2. TASK-3 Modeling and Structural Characterization
2.3. Structural-Activity Relationship (SAR) of THPP Series against TASK-3 Channel
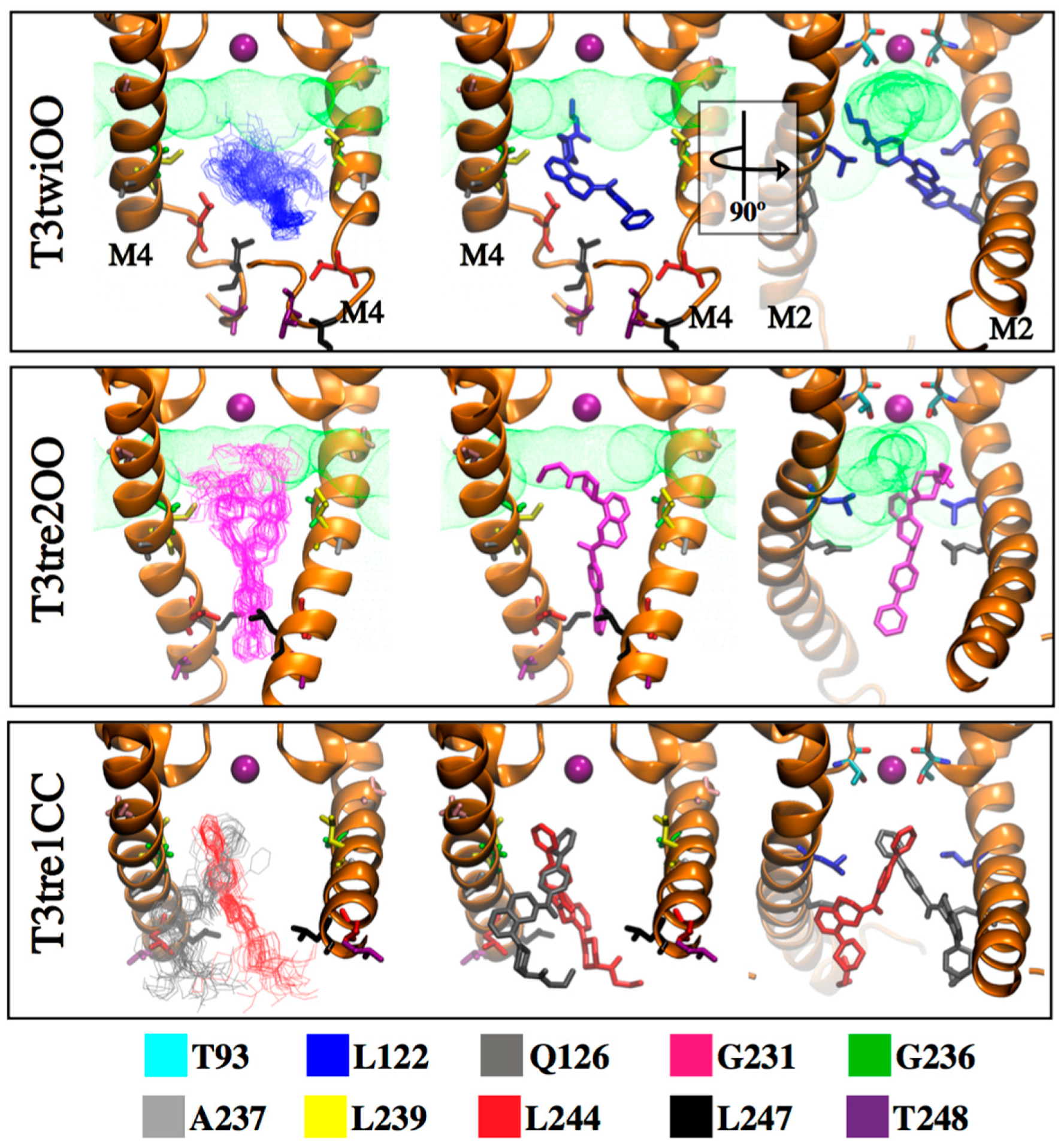
2.4. Defining the PK-THPP Binding Mode Using a Wide Conformational Sampling
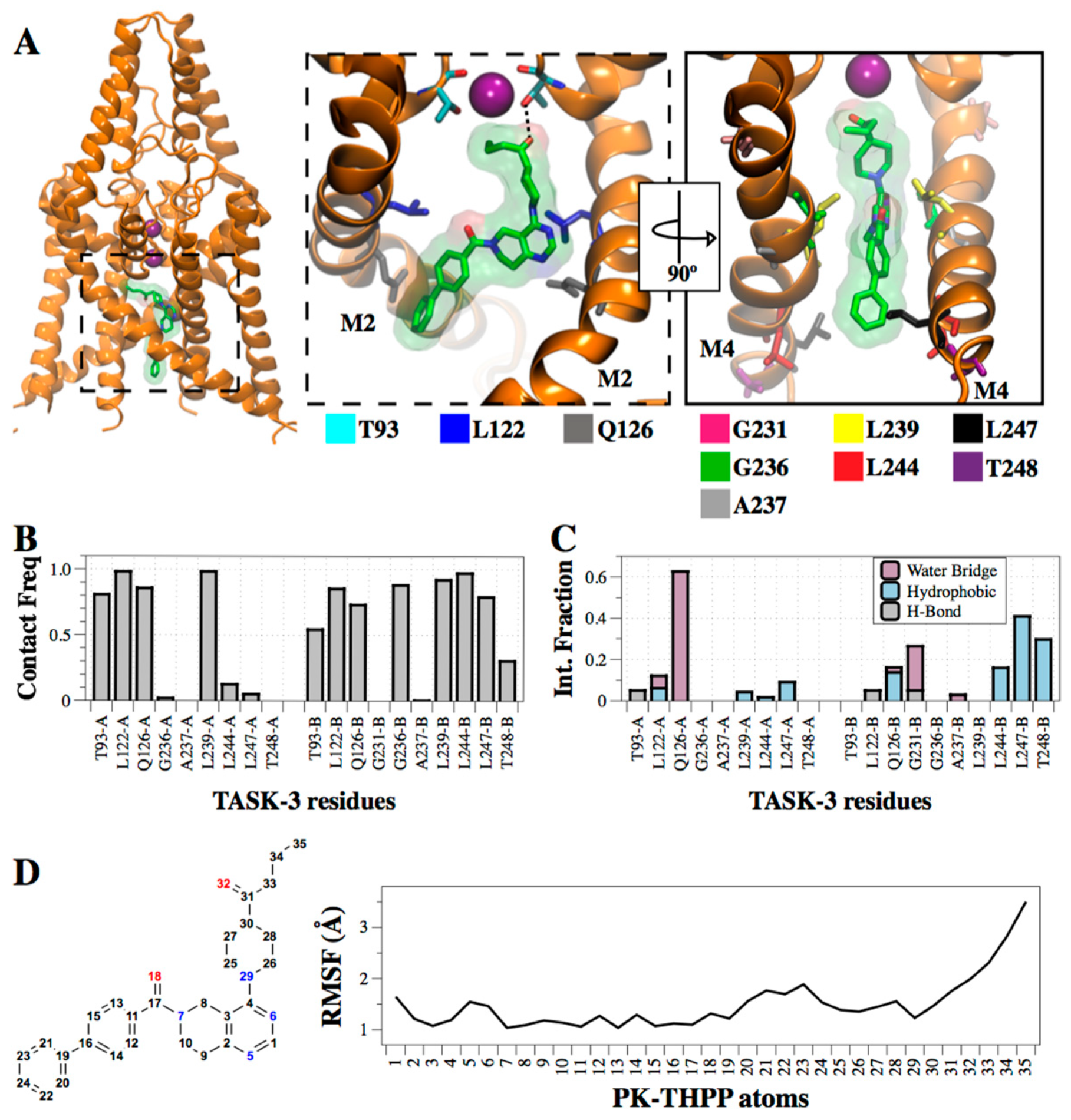
2.5. Interaction of PK-THPP with Residues of TASK-3 Binding Site by Molecular Dynamics Simulations (MDs)
3. Discussion
4. Materials and Methods
4.1. Oocyte Preparation, cRNA Synthesis, and Injection
4.2. Electrophysiology
4.3. TASK-3 Modeling
4.4. Modeling of Compounds of THPP Series
4.5. Molecular Docking
4.6. Relative Binding Free Energy calculations (Relative )
4.7. Molecular Pharmacophore Modeling
4.8. Clustering of Conformers of PK-THPP
4.9. Molecular Dynamics Simulations (MDs)
4.10. Reagents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMS | Alanine mutagenesis screening |
| K2P | Two-pore domain potassium channel |
| MDs | Molecular dynamics simulation(s) |
| MM-GBSA | The molecular mechanics-generalized Born surface area method |
| POPC | Phosphatidyl oleoylphosphatidylcholine |
| RMSD | Root-mean-square deviation |
| RMSF | Root-mean-square fluctuation |
| SASA | solvent accessible surface area |
| TASK | Tandem of pore domains in a weak inwardly rectifying K+ channel [TWIK]-related acid sensitive K+ channel |
| T3twiOO | TASK-3 from TWIK-1 in open−open fenestration state |
| T3tre1CC | TASK-3 from TREK-1 in closed−closed fenestration state |
| T1tre2OO | TASK-3 from TREK-2 in open−open fenestration state |
References
- Goldstein, S.; Bockenhauer, D.; O’Kelly, I.; Zilberberg, N. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci. 2001, 2, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Cotten, J.F. TASK-1 (KCNK3) and TASK-3 (KCNK9) Tandem Pore Potassium Channel Antagonists Stimulate Breathing in Isoflurane Anesthetized Rats. Anesth. Analg. 2013, 116, 810–816. [Google Scholar] [CrossRef]
- Rajan, S.; Wischmeyer, E.; Xin Liu, G.; Preisig-Müller, R.; Daut, J.; Karschin, A.; Derst, C. TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J. Biol. Chem. 2000, 275, 16650–16657. [Google Scholar] [CrossRef] [PubMed]
- Talley, E.M.; Lei, Q.; Sirois, J.E.; Bayliss, D.A. TASK-1, a Two-Pore Domain K+ Channel, Is Modulated by Multiple Neurotransmitters in Motoneurons. Neuron 2000, 25, 399–410. [Google Scholar] [CrossRef]
- Brohawn, S.; del Marmol, J.; MacKinnon, R. Crystal Structure of the Human K2P TRAAK, a Lipid- and Mechano-Sensitive K+ Ion Channel. Science. 2012, 335, 436–441. [Google Scholar] [CrossRef] [PubMed]
- González, W.; Zúñiga, L.; Cid, L.P.; Arévalo, B.; Niemeyer, M.I.; Sepúlveda, F. V An extracellular ion pathway plays a central role in the cooperative gating of a K2P K+ channel by extracellular pH. J. Biol. Chem. 2013, 288, 5984–5991. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, L.; Márquez, V.; González-Nilo, F.D.; Chipot, C.; Cid, L.P.; Sepúlveda, F.V.; Niemeyer, M.I. Gating of a pH-sensitive K2P potassium channel by an electrostatic effect of basic sensor residues on the selectivity filter. PLoS ONE 2011, 6, e16141. [Google Scholar] [CrossRef]
- Miller, A.; Long, S. Crystal Structure of the Human Two-Pore Domain Potassium Channel K2P1. Science 2012, 335, 432–441. [Google Scholar] [CrossRef]
- Brohawn, S.; Campbell, E.; MacKinnon, R. Domain-swapped chain connectivity and gated membrane access in a Fab-mediated crystal of the human TRAAK K+ channel. Proc. Natl. Acad. Sci. USA 2013, 110, 2129–2134. [Google Scholar] [CrossRef]
- Dong, Y.; Pike, A.; Mackenzie, A.; Mcclenaghan, C.; Aryal, P.; Dong, L.; Grieben, M.; Goubin, S.; Ruda, G.F.; Clausen, M.; et al. K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science 2015, 347, 1256–1259. [Google Scholar] [CrossRef]
- Lolicato, M.; Arrigoni, C.; Mori, T.; Sekioka, Y.; Bryant, C.; Clark, K.A.; Minor, D.L. K2P2.1 (TREK-1)–activator complexes reveal a cryptic selectivity filter binding site. Nature 2017, 547, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Aryal, P.; Abd-Wahab, F.; Bucci, G.; Sansom, M.S.P.; Tucker, S.J. A hydrophobic barrier deep within the inner pore of the TWIK-1 K2P potassium channel. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, D.; Arévalo, B.; Martínez, G.; Rinné, S.; Sepúlveda, F.V.; Decher, N.; González, W. Side fenestrations provide “anchor” for a stable binding of A1899 to the pore of TASK-1 potassium channels. Mol. Pharm. 2017, 14, 2197–2208. [Google Scholar] [CrossRef]
- Rinné, S.; Kiper, A.K.; Vowinkel, K.S.; Ramírez, D.; Schewe, M.; Bedoya, M.; Aser, D.; Gensler, I.; Netter, M.F.; Stansfeld, P.J.; et al. The molecular basis for an allosteric inhibition of K+ -flux gating in K2P channels. Elife 2019, 8, e39476. [Google Scholar] [CrossRef]
- Schewe, M.; Sun, H.; Mert, Ü.; Mackenzie, A.; Pike, A.C.W.; Schulz, F.; Constantin, C.; Vowinkel, K.S.; Conrad, L.J.; Kiper, A.K.; Gonzalez, W.; et al. A pharmacological master key mechanism that unlocks the selectivity filter gate in K+ channels. Science 2019, 363, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Coburn, C.A.; Luo, Y.; Cui, M.; Wang, J.; Soll, R.; Dong, J.; Hu, B.; Lyon, M.A.; Santarelli, V.P.; Kraus, R.L.; et al. Discovery of a pharmacologically active antagonist of the two-pore domain potassium channel K2P9.1 (TASK-3). Chem. Med. Chem. 2012, 7, 123–133. [Google Scholar] [CrossRef]
- Flaherty, D.P.; Simpson, D.S.; Miller, M.; Maki, B.E.; Zou, B.; Shi, J.; Wu, M.; McManus, O.B.; Aubé, J.; Li, M.; et al. Potent and selective inhibitors of the TASK-1 potassium channel through chemical optimization of a bis-amide scaffold. Bioorganic Med. Chem. Lett. 2014, 24, 3968–3973. [Google Scholar] [CrossRef] [PubMed]
- Streit, A.K.; Netter, M.F.; Kempf, F.; Walecki, M.; Rinné, S.; Bollepalli, M.K.; Preisig-Müller, R.; Renigunta, V.; Daut, J.; Baukrowitz, T.; et al. A specific two-pore domain potassium channel blocker defines the structure of the TASK-1 open pore. J. Biol. Chem. 2011, 286, 13977–13984. [Google Scholar] [CrossRef] [PubMed]
- Kiper, A.K.; Rinné, S.; Rolfes, C.; Ramírez, D.; Seebohm, G.; Netter, M.F.; González, W.; Decher, N. Kv1.5 blockers preferentially inhibit TASK-1 channels: TASK-1 as a target against atrial fibrillation and obstructive sleep apnea? Pflugers Arch. 2015, 467, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Chokshi, R.H.; Larsen, A.T.; Bhayana, B.; Cotten, J.F. Breathing Stimulant Compounds Inhibit TASK-3 Potassium Channel Function Likely by Binding at a Common Site in the Channel Pore. Mol. Pharmacol. 2015, 88, 926–934. [Google Scholar] [CrossRef]
- Rodríguez, Y.A.; Gutiérrez, M.; Ramírez, D.; Alzate-Morales, J.; Bernal, C.C.; Güiza, F.; Romero, A.R. Novel N -allyl/propargyl tetrahydroquinolines: Synthesis via Three-component Cationic Imino Diels—Alder Reaction, Binding Prediction, and Evaluation as Cholinesterase Inhibitors. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef]
- Rabbani, G.; Baig, M.H.; Lee, E.J.; Cho, W.-K.; Ma, J.Y.; Choi, I. Biophysical study on the interaction between eperisone hydrochloride and human serum albumin using spectroscopic, calorimetric, and molecular docking analyses. Mol. Pharm. 2017, 14, 1656–1665. [Google Scholar] [CrossRef]
- Rabbani, G.; Lee, E.J.; Ahmad, K.; Baig, M.H.; Choi, I. Binding of tolperisone hydrochloride with human serum albumin: Effects on the conformation, thermodynamics, and activity of HSA. Mol. Pharm. 2018, 15, 1445–1456. [Google Scholar] [CrossRef]
- Forrest, L.R.; Tang, C.L.; Honig, B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys. J. 2006, 91, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.D.; Lamb, M.L.; Saeh, J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef]
- Balaji, B.; Ramanathan, M. Prediction of estrogen receptor β ligands potency and selectivity by docking and MM-GBSA scoring methods using three different scaffolds. J. Enzyme Inhib. Med. Chem. 2012, 27, 832–844. [Google Scholar] [CrossRef]
- Srivastava, M.; Singh, H.; Naik, P.K. Molecular modeling evaluation of the antimalarial activity of artemisinin analogues: Molecular docking and rescoring using Prime/MM-GBSA Approach. Curr Res J Biol Sci 2010, 2, 83–102. [Google Scholar]
- Bottegoni, G.; Cavalli, A.; Recanatini, M. A comparative study on the application of hierarchical-agglomerative clustering approaches to organize outputs of reiterated docking runs. J. Chem. Inf. Model. 2006, 46, 852–862. [Google Scholar] [CrossRef]
- Bottegoni, G.; Rocchia, W.; Cavalli, A. Application of conformational clutering in protein-ligand docking. methods Mol. Biol. 2012, 819, 169–186. [Google Scholar] [CrossRef]
- Bang, H.; Kim, Y.; Kim, D. TASK-3, a new member of the Tandem pore K+ channel family*. J. Biol. Chem. 2000, 275, 9340–9346. [Google Scholar] [CrossRef]
- Bittner, S.; Budde, T.; Wiendl, H.; Meuth, S.G. From the background to the spotlight: TASK channels in pathological conditions. Brain Pathol. 2010, 20, 999–1009. [Google Scholar] [CrossRef]
- Huang, X.; Jan, L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef]
- Lolicato, M.; Riegelhaupt, P.M.; Arrigoni, C.; Clark, K.A.; Minor, D.L. Transmembrane Helix Straightening and Buckling Underlies Activation of Mechanosensitive and Thermosensitive K2P Channels. Neuron 2014, 84, 1198–1212. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinforma. 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Schrödinger, L. Desmond Molecular Dynamics System, version 3.0; Epik version 2.2; Glide, version 5.7; Impact, version 5.7; LigPrep, version 2.5; Phase, version 3.3; Prime, version 2.3. New York, NY, USA, 2011. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC ’06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Smart, O.; Neduvelil, J.; Wang, X.; Wallace, B.; Sansom, M. HOLE: A program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996, 14, 354–360. [Google Scholar] [CrossRef]
- Shelke, S.M.; Bhosale, S.H.; Dash, R.C.; Suryawanshi, M.R.; Mahadik, K.R. Exploration of new scaffolds as potential MAO-A inhibitors using pharmacophore and 3D-QSAR based in silico screening. Bioorg. Med. Chem. Lett. 2011, 21, 2419–2424. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Lindert, S.; Sinko, W.; McCammon, A. Exploring the Role of Receptor Flexibility in Structure-Based Drug Discovery. Biophys. Chem. 2015, 186, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods: I. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2012, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Adasme-Carreño, F.; Muñoz-Gutierrez, C.; Caballero, J.; Alzate-Morales, J.H. Performance of the MM/GBSA scoring using a binding site hydrogen bond network-based frame selection: The protein kinase case. Phys. Chem. Chem. Phys. 2014, 16, 14047–14058. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Rio, A.D.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Wang, W.; Kollman, P.A. Computational study of protein specificity: The molecular basis of HIV-1 protease drug resistance. Proc. Natl. Acad. Sci. 2001, 98, 14937–14942. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Hou, T.; Yu, R. Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: Mechanism for binding and drug resistance. J. Med. Chem. 2007, 50, 1177–1188. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the Differential Activity of Thrombin Inhibitors Using Docking, QSAR, Molecular Dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinforma. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Durdagi, S.; Erol, I.; Salmas, R.E.; Patterson, M.; Noskov, S.Y. First universal pharmacophore model for hERG1 K+ channel activators: ActhER. J. Mol. Graph. Model. 2017, 74, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, S.; Zhang, Y. Identification of near-native structures by clustering protein docking conformations. PROTEINS Struct. Funct. Bioinforma. 2007, 68, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Shenkin, P.S.; Mcdonald, D.Q. Cluster Analysis of Molecular Conformations. J. Comput. Chem. 1994, 15, 899–916. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| THPP Series | IC50 (µM) | ΔEMM | ΔGsol | ΔGBind | ||||
|---|---|---|---|---|---|---|---|---|
| ΔEinternal | ΔEelectrostatic | ΔEvdw | ΔGGB | ΔGSA | ||||
| Coulomb | Hbond | |||||||
| PK-THPP | 0.035 | 11.37 | −6.48 | −0.40 | −57.95 | 13.21 | -43.79 | –84.05 |
| 21 | 0.05 | 5.88 | −22.14 | 0.67 | −49.63 | 27.08 | –42.34 | –80.49 |
| 20b | 0.07 | 9.27 | −6.44 | −2.03 | −50.21 | 5.07 | –39.93 | –84.26 |
| 22 | 0.07 | 14.81 | −20.47 | −1.07 | −55.36 | 18.19 | –38.79 | –82.69 |
| 17b | 27 | 6.45 | −5.58 | −1.51 | −39.99 | 13.60 | –32.19 | –59.21 |
| # of Interactions (# by Chemical Groups) | |||
|---|---|---|---|
| Ligand Name | Hydrophobic | Polar | Hydrogen bond |
| 17b | 5 (3→ Biphenyl, 1→ Pyrimidine, 1→ Substituent) | 1 (Biphenyl) | - |
| 20b | 6 (4 → Biphenyl, 1→ Pyrimidine, 1→ Substituent) | 1 (Biphenyl) | 2 (1→Carbonyl, 1 Pyrimidine) |
| 21 | 4 (2→ Biphenyl, 1→ Tetrahydropyridine, 1→ Pyrimidine) | - | 1 (Carbonyl) |
| 22 | 2 (1→ Biphenyl, 1→Substituent) | - | 2 (1→ Carbonyl, 1 Pyrimidine) |
| 23 | 5 (3→ Biphenyl, 2→ Pyrimidine) | 1 (Substituent) | 1 (Carbonyl) |
| T3twiOO | T3tre2OO | T3tre1CC | |||
|---|---|---|---|---|---|
| No. | Pop. | No. | Pop. | No. | Pop. |
| 10 | 54 | 23 | 59 | 28 | 25 |
| 33 | 26 | ||||
| PK-THPP Pose | TASK-3 Model | ΔEMM | ΔGsol | Relative ΔGBind | ||||
|---|---|---|---|---|---|---|---|---|
| ΔEinternal | ΔEelectrostatic | ΔEvdw | ΔGGB | ΔGSA | ||||
| Coulomb | Hbond | |||||||
| 84 | T3twiOO | 25.2 | −20.0 | −1.1 | −63.7 | 18.6 | −75.1 | −115.8 |
| 177 | T3tre2OO | 11.4 | −6.5 | −0.4 | −57.9 | 13.2 | −43.8 | −84.1 |
| 270 | T3tre1CC | 6.3 | −21.8 | −1.1 | −51.9 | 34.6 | −39.7 | −73.5 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, D.; Bedoya, M.; Kiper, A.K.; Rinné, S.; Morales-Navarro, S.; Hernández-Rodríguez, E.W.; Sepúlveda, F.V.; Decher, N.; González, W. Structure/Activity Analysis of TASK-3 Channel Antagonists Based on a 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine. Int. J. Mol. Sci. 2019, 20, 2252. https://doi.org/10.3390/ijms20092252
Ramírez D, Bedoya M, Kiper AK, Rinné S, Morales-Navarro S, Hernández-Rodríguez EW, Sepúlveda FV, Decher N, González W. Structure/Activity Analysis of TASK-3 Channel Antagonists Based on a 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine. International Journal of Molecular Sciences. 2019; 20(9):2252. https://doi.org/10.3390/ijms20092252
Chicago/Turabian StyleRamírez, David, Mauricio Bedoya, Aytug K. Kiper, Susanne Rinné, Samuel Morales-Navarro, Erix W. Hernández-Rodríguez, Francisco V. Sepúlveda, Niels Decher, and Wendy González. 2019. "Structure/Activity Analysis of TASK-3 Channel Antagonists Based on a 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine" International Journal of Molecular Sciences 20, no. 9: 2252. https://doi.org/10.3390/ijms20092252
APA StyleRamírez, D., Bedoya, M., Kiper, A. K., Rinné, S., Morales-Navarro, S., Hernández-Rodríguez, E. W., Sepúlveda, F. V., Decher, N., & González, W. (2019). Structure/Activity Analysis of TASK-3 Channel Antagonists Based on a 5,6,7,8 tetrahydropyrido[4,3-d]pyrimidine. International Journal of Molecular Sciences, 20(9), 2252. https://doi.org/10.3390/ijms20092252