The Pathway to Cancer Cachexia: MicroRNA-Regulated Networks in Muscle Wasting Based on Integrative Meta-Analysis

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
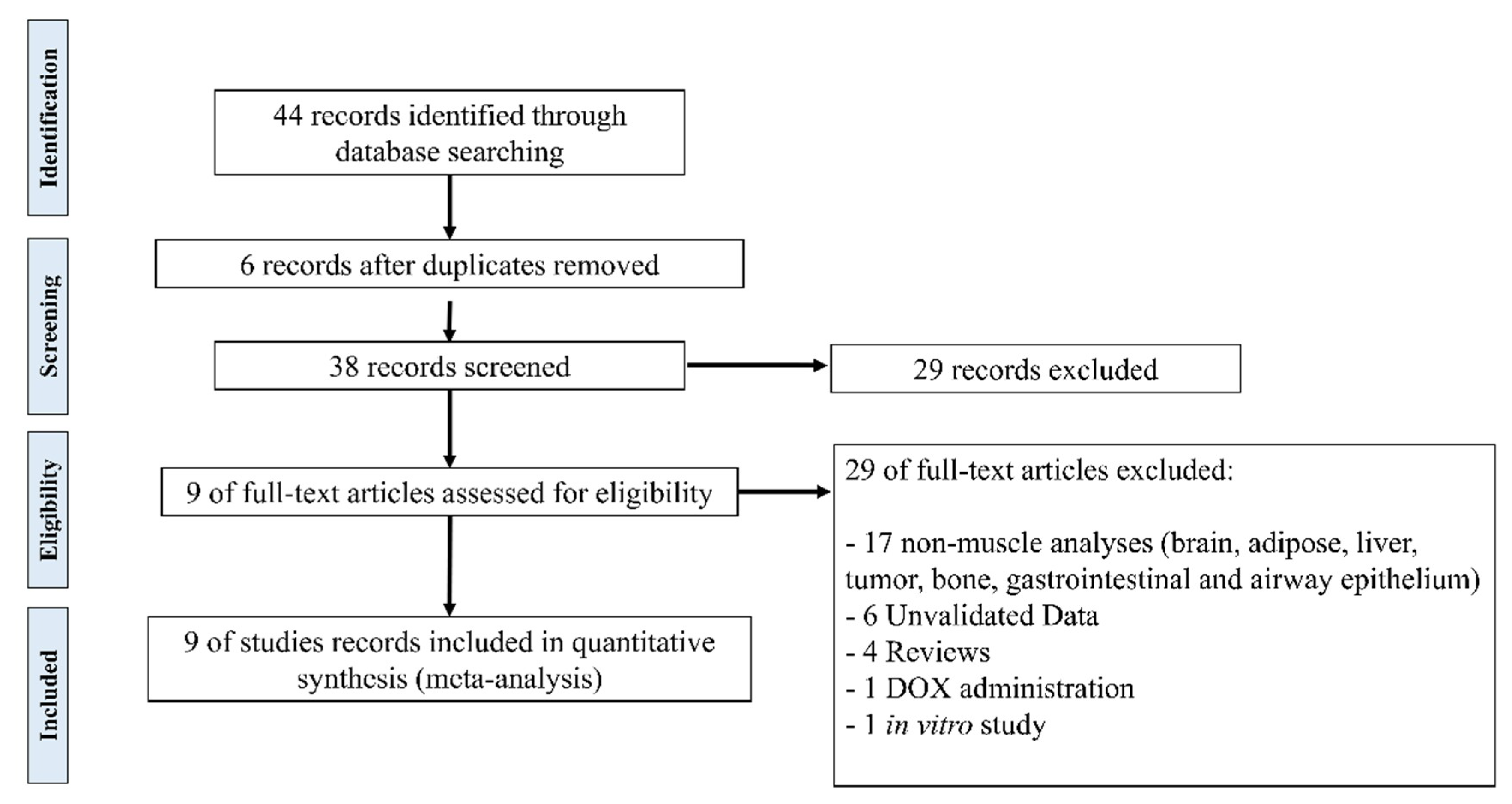
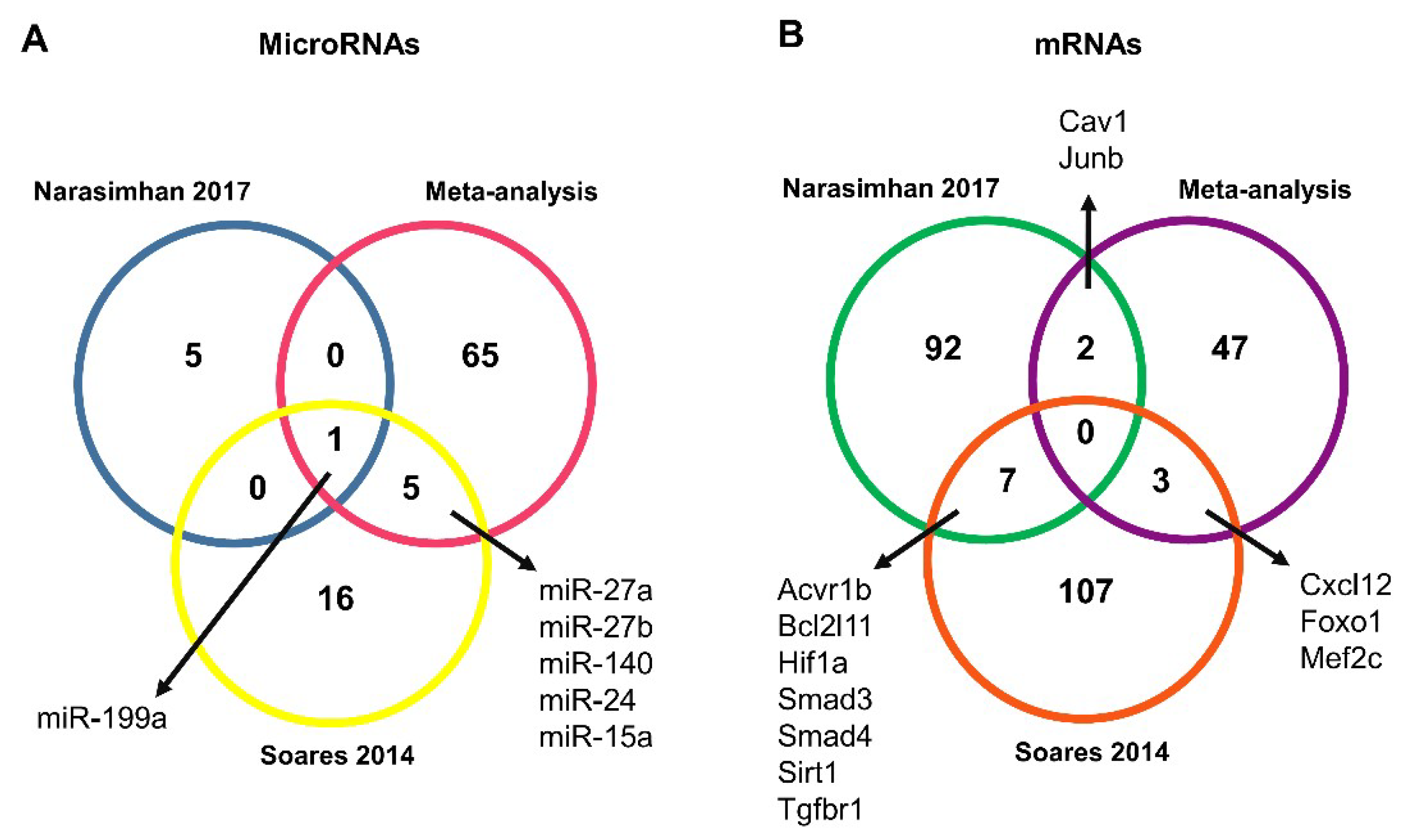
2.1. Study Selection and Characteristics
2.2. Validated Data Selection of Differentially Expressed Genes in Cancer Cachexia
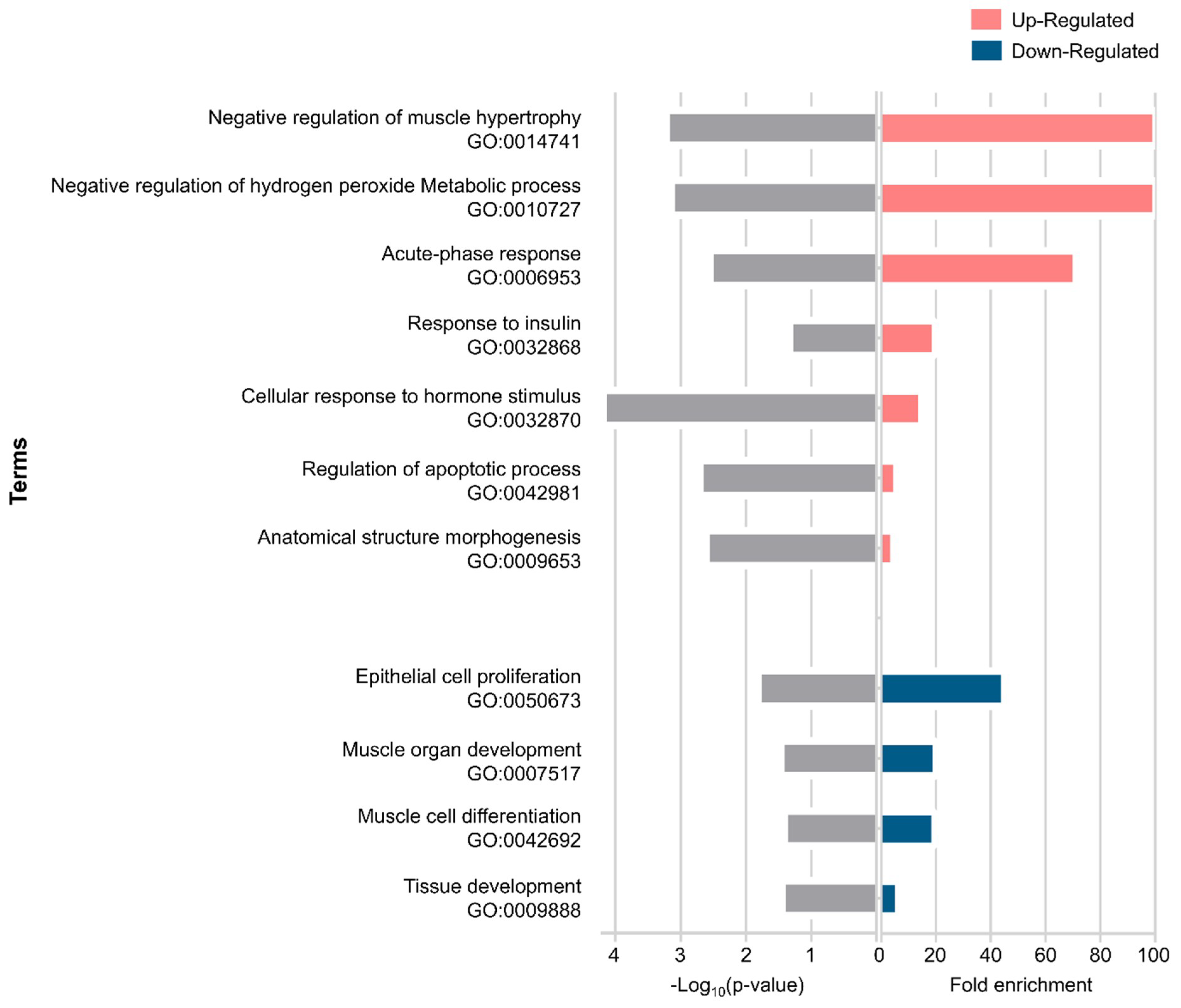
2.3. Gene Ontology Enrichment Analysis of Differentially Expressed Genes in Muscle Wasting in Cancer Cachexia
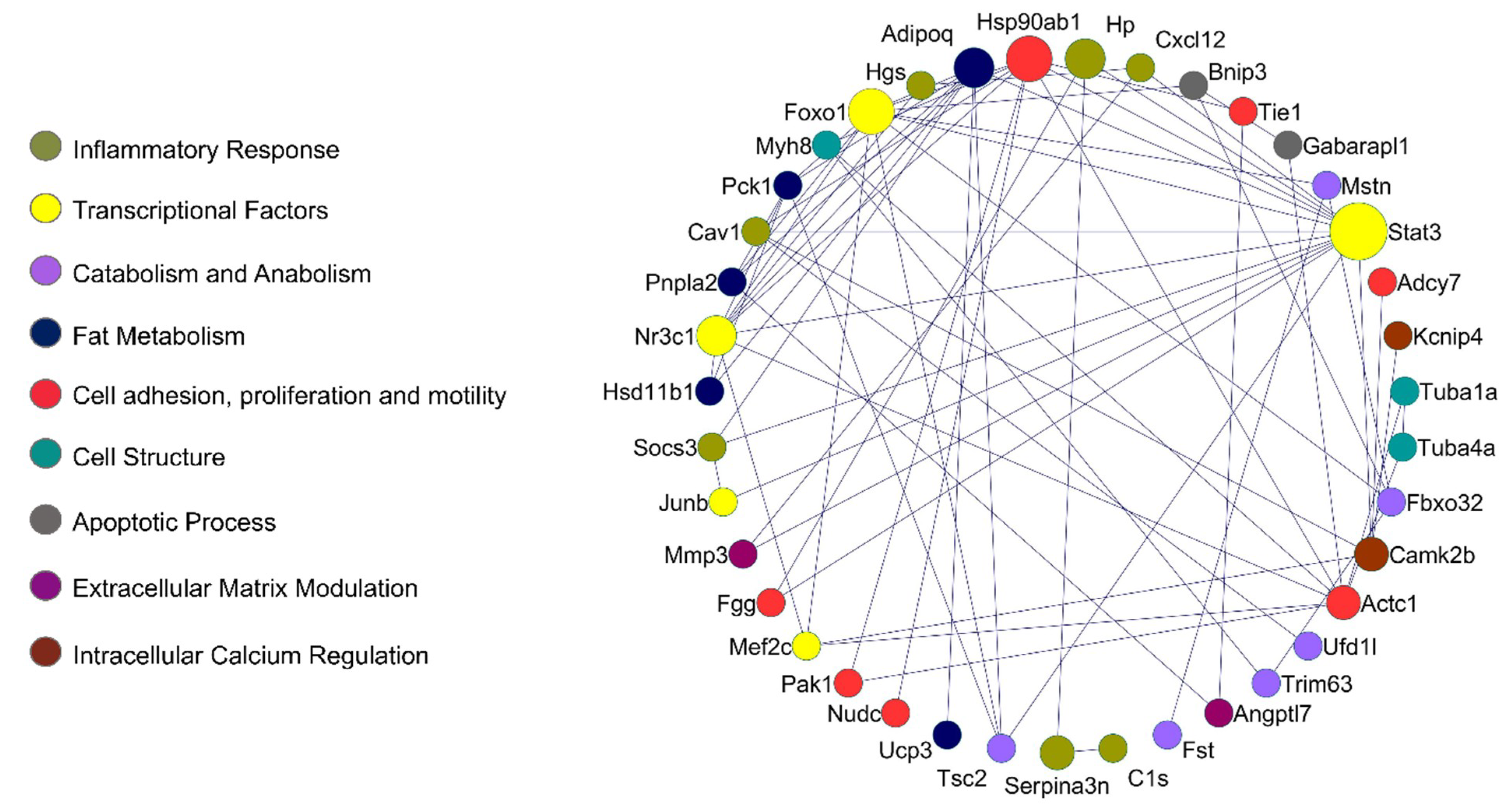
2.4. Protein–Protein Interaction Network in Muscle Wasting in Cancer Cachexia
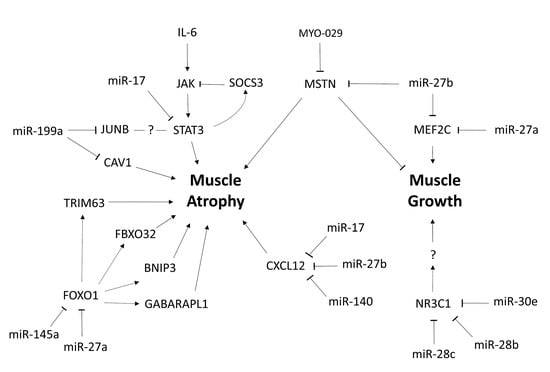
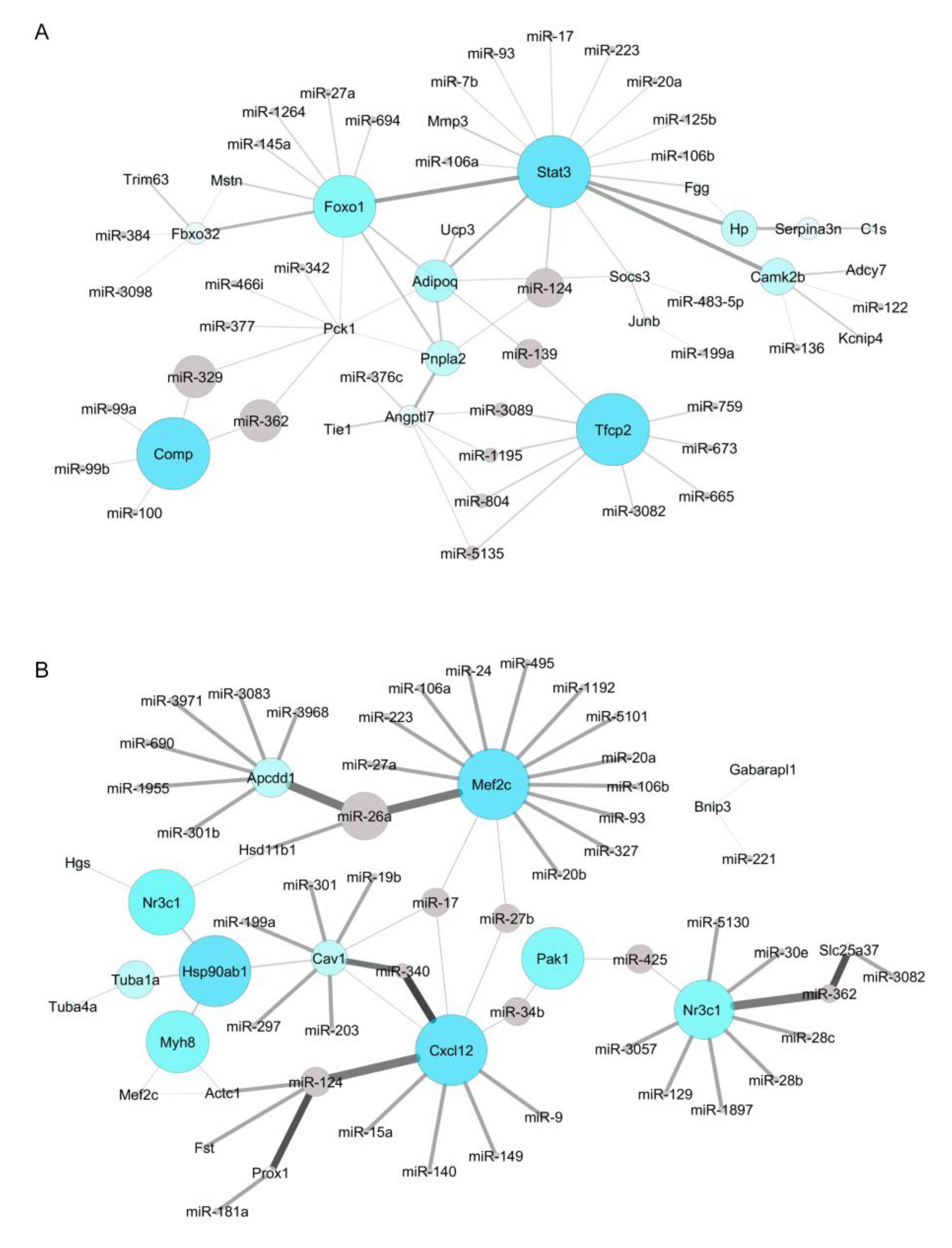
2.5. Identification of New Potential microRNA-Regulated Networks in Muscle Wasting in Cancer Cachexia
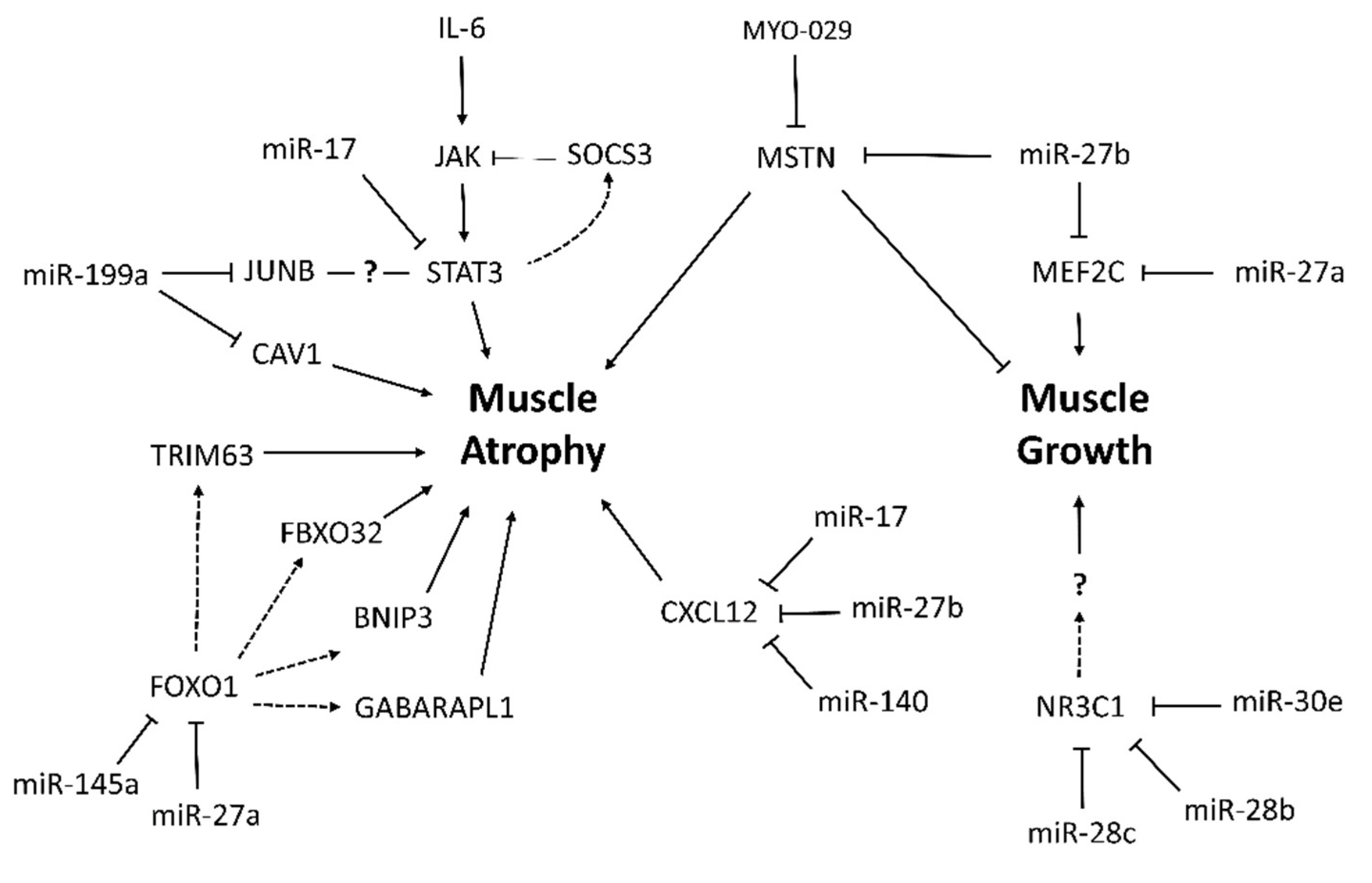
2.6. Identification of Potential Target Agents for the Treatment of Muscle Wasting in Cancer Cachexia
3. Discussion
4. Methods
4.1. Meta-Analysis of Global Gene Expression Data in Muscle Wasting in Cancer Cachexia
4.2. Meta-Analysis of Global microRNA Expression Data in Muscle Wasting in Cancer Cachexia
4.3. Identification of Muscle microRNAs as Potential Modulators of Deregulated Genes in Cancer Cachexia
4.4. Identification of Candidate Drug Targets Based on microRNA-Regulated Networks in Cancer Cachexia
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Holecek, M. Muscle wasting in animal models of severe illness. Int. J. Exp. Pathol. 2012, 93, 157–171. [Google Scholar] [CrossRef]
- Hauser, C.A.; Stockler, M.R.; Tattersall, M.H.N. Prognostic factors in patients with recently diagnosed incurable cancer: A systematic review. Support. Care Cancer 2006, 14, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Meguid, M.M.; Inui, A.; Muscaritoli, M.; Rossi-Fanelli, F. Therapy insight: Cancer anorexia-cachexia syndrome--when all you can eat is yourself. Nat. Clin. Pract. Oncol. 2005, 2, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.H. Cancer cachexia: Developing multimodal therapy for a multidimensional problem. Eur. J. Cancer 2008, 44, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Cachexia in cancer patients. Nat. Rev. Cancer 2002, 3, 883–889. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Morley, J.E.; Thomas, D.R.; Wilson, M.-M.G. Cachexia: Pathophysiology and clinical relevance. Am. J. Clin. Nutr. 2006, 83, 735–743. [Google Scholar] [CrossRef]
- Penafuerte, C.A.; Gagnon, B.; Sirois, J.; Murphy, J.; Macdonald, N.; Tremblay, M.L. Identification of neutrophil-derived proteases and angiotensin II as biomarkers of cancer cachexia. Br. J. Cancer 2016, 114, 680–687. [Google Scholar] [CrossRef]
- Kuroda, K.; Nakashima, J.; Kanao, K.; Kikuchi, E.; Miyajima, A.; Horiguchi, Y. Interleukin 6 is associated with cachexia in patients with prostate cancer. Urology 2007, 69, 113–117. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Wang, C.-J.; Chao, Y.-J.; Chen, H.-Y.; Wang, H.-C.; Tung, H.-L.; Lin, J.-T.; Shan, Y.-S. Elevated Serum Interleukin-8 Level Correlates with Cancer-Related Cachexia and Sarcopenia: An Indicator for Pancreatic Cancer Outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef] [PubMed]
- Kandarian, S.C.; Nosacka, R.L.; Delitto, A.E.; Judge, A.R.; Judge, S.M.; Ganey, J.D.; Moreira, J.D.; Jackman, R.W. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 1109–1120. [Google Scholar] [CrossRef]
- Moldawer, L.L.; Georgieff, M.; Lundholm, K. Interleukin 1, tumour necrosis factor-alpha (cachectin) and the pathogenesis of cancer cachexia. Clin. Physiol. 1987, 7, 263–274. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J. The role of cytokines in cancer cachexia. Med. Res. Rev. 1999, 19, 223–248. [Google Scholar] [CrossRef]
- Ait-Ali, D.; Turquier, V.; Grumolato, L.; Yon, L.; Jourdain, M.; Alexandre, D.; Eiden, L.E.; Vaudry, H.; Anouar, Y. The proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1 stimulate neuropeptide gene transcription and secretion in adrenochromaffin cells via activation of extracellularly regulated kinase 1/2 and p38 protein kinases, and activator pro. Mol. Endocrinol. 2004, 18, 1721–1739. [Google Scholar] [CrossRef][Green Version]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, Y.; Wu, Y.; Wang, L.; Wang, X.; Du, J. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J. Biol. Chem. 2013, 288, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef]
- Cao, P.R.; Kim, H.J.; Lecker, S.H. Ubiquitin-protein ligases in muscle wasting. Int. J. Biochem. Cell Biol. 2005, 37, 2088–2097. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef]
- Samuels, S.E.; Knowles, A.L.; Tilignac, T.; Debiton, E.; Madelmont, J.C.; Attaix, D. Higher skeletal muscle protein synthesis and lower breakdown after chemotherapy in cachectic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R133–R139. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, E.J.; Giresi, P.G.; Koncarevic, A.; Kandarian, S.C. Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J. Physiol. 2003, 551, 33–48. [Google Scholar] [CrossRef]
- Giresi, P.G. Identification of a molecular signature of sarcopenia. Physiol. Genom. 2005, 21, 253–263. [Google Scholar] [CrossRef]
- Stevenson, E.J.; Koncarevic, A.; Giresi, P.G.; Jackman, R.W.; Kandarian, S.C. Transcriptional profile of a myotube starvation model of atrophy. J. Appl. Physiol. 2005, 98, 1396–1406. [Google Scholar] [CrossRef]
- Hasselgren, P.O. Glucocorticoids and muscle catabolism. Curr. Opin. Clin. Nutr. Metab. Care 1999, 2, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Hyatt, J.-P.K.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007, 21, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Stephens, N.A.; Gallagher, I.J.; Rooyackers, O.; Skipworth, R.J.; Tan, B.H.; Marstrand, T.; Ross, J.A.; Guttridge, D.C.; Lundell, L.; Fearon, K.C.; et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome Med. 2010, 2, 1. [Google Scholar] [CrossRef]
- Gallagher, I.J.; Stephens, N.A.; MacDonald, A.J.; Skipworth, R.J.E.; Husi, H.; Greig, C.A.; Ross, J.A.; Timmons, J.A.; Fearon, K.C.H. Suppression of skeletal muscle turnover in cancer cachexia: Evidence from the transcriptome in sequential human muscle biopsies. Clin. Cancer Res. 2012, 18, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Güller, I.; Russell, A.P. MicroRNAs in skeletal muscle: Their role and regulation in development, disease and function. J. Physiol. 2010, 588, 4075–4087. [Google Scholar] [CrossRef]
- Eisenberg, I.; Alexander, M.S.; Kunkel, L.M. miRNAS in normal and diseased skeletal muscle. J. Cell. Mol. Med. 2009, 13, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar] [CrossRef]
- van de Worp, W.R.P.H.; Theys, J.; van Helvoort, A.; Langen, R.C.J. Regulation of muscle atrophy by microRNAs: “AtromiRs” as potential target in cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, A.; Ghosh, S.; Stretch, C.; Greiner, R.; Bathe, O.F.; Baracos, V.; Damaraju, S. Small RNAome profiling from human skeletal muscle: Novel miRNAs and their targets associated with cancer cachexia. J. Cachexia Sarcopenia Muscle 2017, 8, 405–416. [Google Scholar] [CrossRef]
- Soares, R.J.; Cagnin, S.; Chemello, F.; Silvestrin, M.; Musaro, A.; De Pitta, C.; Lanfranchi, G.; Sandri, M. Involvement of microRNAs in the regulation of muscle wasting during catabolic conditions. J. Biol. Chem. 2014, 289, 21909–21925. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-C.; Kulp, S.K.; Lai, I.-L.; Hsu, E.-C.; He, W.A.; Frankhouser, D.E.; Yan, P.S.; Mo, X.; Bloomston, M.; Lesinski, G.B.; et al. Preclinical Investigation of the Novel Histone Deacetylase Inhibitor AR-42 in the Treatment of Cancer-Induced Cachexia. J. Natl. Cancer Inst. 2015, 107, djv274. [Google Scholar] [CrossRef]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.B.; et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Kunzevitzky, N.; Guttridge, D.C.; Khuri, S.; Koniaris, L.G.; Zimmers, T.A. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS ONE 2011, 6, e22538. [Google Scholar] [CrossRef]
- Gilabert, M.; Calvo, E.; Airoldi, A.; Hamidi, T.; Moutardier, V.; Turrini, O.; Iovanna, J. Pancreatic cancer-induced cachexia is Jak2-dependent in mice. J. Cell. Physiol. 2014, 229, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.M.Y.; Fung, D.C.Y.; Corley, S.M.; McGill, M.C.; Bentley, N.L.; Tan, T.C.; Wilkins, M.R.; Polly, P. Cardiac and skeletal muscles show molecularly distinct responses to cancer cachexia. Physiol. Genom. 2015, 47, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Fontes-Oliveira, C.C.; Busquets, S.; Fuster, G.; Ametller, E.; Figueras, M.; Olivan, M.; Toledo, M.; López-Soriano, F.J.; Qu, X.; Demuth, J.; et al. A differential pattern of gene expression in skeletal muscle of tumor-bearing rats reveals dysregulation of excitation–contraction coupling together with additional muscle alterations. Muscle Nerve 2014, 49, 233–248. [Google Scholar] [CrossRef]
- Penna, F.; Busquets, S.; Argilés, J.M. Experimental cancer cachexia: Evolving strategies for getting closer to the human scenario. Semin. Cell Dev. Biol. 2016, 54, 20–27. [Google Scholar] [CrossRef]
- Martinelli, G.B.; Olivari, D.; Re Cecconi, A.D.; Talamini, L.; Ottoboni, L.; Lecker, S.H.; Stretch, C.; Baracos, V.E.; Bathe, O.F.; Resovi, A.; et al. Activation of the SDF1/CXCR4 pathway retards muscle atrophy during cancer cachexia. Oncogene 2016, 35, 6212–6222. [Google Scholar] [CrossRef]
- Matsumoto, S.; Takebayashi, K.; Aso, Y. The effect of spironolactone on circulating adipocytokines in patients with type 2 diabetes mellitus complicated by diabetic nephropathy. Metabolism 2006, 55, 1645–1652. [Google Scholar] [CrossRef]
- Hiuge, A.; Tenenbaum, A.; Maeda, N.; Benderly, M.; Kumada, M.; Fisman, E.Z.; Tanne, D.; Matas, Z.; Hibuse, T.; Fujita, K.; et al. Effects of peroxisome proliferator-activated receptor ligands, bezafibrate and fenofibrate, on adiponectin level. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.M.; Duong, M.; Masson, D.; Buisson, M.; Duvillard, L.; Bour, J.B.; Brindisi, M.C.; Galland, F.; Guiguet, M.; Gambert, P.; et al. Serum adiponectin and metabolic parameters in HIV-1-infected patients after substitution of nevirapine for protease inhibitors. Eur. J. Clin. Investig. 2004, 34, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.L.; Lee, J.H.; Cribbs, L.L.; Perez-Reyes, E.; Hanck, D.A. Mibefradil block of cloned T-type calcium channels. J. Pharmacol. Exp. Ther. 2000, 295, 302–308. [Google Scholar]
- Liao, P.; Yu, D.; Li, G.; Yong, T.F.; Soon, J.L.; Chua, Y.L.; Soong, T.W. A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J. Biol. Chem. 2007, 282, 35133–35142. [Google Scholar] [CrossRef]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef]
- Mohamed, H.A.; Girgis, N.M.R.; Wilcken, R.; Bauer, M.R.; Tinsley, H.N.; Gary, B.D.; Piazza, G.A.; Boeckler, F.M.; Abadi, A.H. Synthesis and molecular modeling of novel tetrahydro-β-carboline derivatives with phosphodiesterase 5 inhibitory and anticancer properties. J. Med. Chem. 2011, 54, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Koo, C.-Y.; Sen, Y.-P.; Bay, B.-H.; Yip, G.W. Targeting heparan sulfate proteoglycans in breast cancer treatment. Recent Pat. Anticancer Drug Discov. 2008, 3, 151–158. [Google Scholar] [CrossRef]
- Bogdanovich, S.; Krag, T.O.B.; Barton, E.R.; Morris, L.D.; Whittemore, L.-A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Sin, T.K.; Yu, A.P.; Yung, B.Y.; Yip, S.P.; Chan, L.W.; Wong, C.S.; Ying, M.; Rudd, J.A.; Siu, P.M. Modulating effect of SIRT1 activation induced by resveratrol on Foxo1-associated apoptotic signalling in senescent heart. J. Physiol. 2014, 592, 2535–2548. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cachexia and sarcopenia: Mechanisms and potential targets for intervention. Curr. Opin. Pharmacol. 2015, 22, 100–106. [Google Scholar] [CrossRef]
- Jiang, G.; Huang, C.; Li, J.; Huang, H.; Jin, H.; Zhu, J.; Wu, X.-R.; Huang, C. Role of STAT3 and FOXO1 in the Divergent Therapeutic Responses of Non-metastatic and Metastatic Bladder Cancer Cells to miR-145. Mol. Cancer Ther. 2017, 16, 924–935. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, C.; Zhang, A.; Cai, H.; Price, S.R.; Wang, X.H. MicroRNA-23a and MicroRNA-27a Mimic Exercise by Ameliorating CKD-Induced Muscle Atrophy. J. Am. Soc. Nephrol. 2017, 28, 2631–2640. [Google Scholar] [CrossRef] [PubMed]
- Ash, G.I.; Kostek, M.A.; Lee, H.; Angelopoulos, T.J.; Clarkson, P.M.; Gordon, P.M.; Moyna, N.M.; Visich, P.S.; Zoeller, R.F.; Price, T.B.; et al. Glucocorticoid Receptor (NR3C1) Variants Associate with the Muscle Strength and Size Response to Resistance Training. PLoS ONE 2016, 11, e0148112. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Zhao, Y.; Li, T.; Zhang, Y.; Zhu, D. miR-30e is negatively regulated by myostatin in skeletal muscle and is functionally related to fiber-type composition. Acta Biochim. Biophys. Sin. (Shanghai) 2017, 49, 392–399. [Google Scholar] [CrossRef]
- Zhu, H.; Han, C.; Wu, T. MiR-17-92 cluster promotes hepatocarcinogenesis. Carcinogenesis 2015, 36, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Trabulo, S.M.; Sanchez-Ripoll, Y.; Miranda-Lorenzo, I.; Lonardo, E.; Dorado, J.; Reis Vieira, C.; Ramirez, J.C.; Hidalgo, M.; Aicher, A.; et al. The miR-17-92 cluster counteracts quiescence and chemoresistance in a distinct subpopulation of pancreatic cancer stem cells. Gut 2015, 64, 1936–1948. [Google Scholar] [CrossRef]
- Chatterjee, A.; Chattopadhyay, D.; Chakrabarti, G. miR-17-5p downregulation contributes to paclitaxel resistance of lung cancer cells through altering beclin1 expression. PLoS ONE 2014, 9, e95716. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, W.; Liao, H.; Dai, L.; Jiang, Z.; Pan, Y.; Huang, H.; Mo, Y.; Li, S.; Yang, G.; et al. The regulatory and predictive functions of miR-17 and miR-92 families on cisplatin resistance of non-small cell lung cancer. BMC Cancer 2015, 15, 731. [Google Scholar] [CrossRef]
- Dong, C.; Yang, X.-Z.; Zhang, C.-Y.; Liu, Y.-Y.; Zhou, R.-B.; Cheng, Q.-D.; Yan, E.-K.; Yin, D.-C. Myocyte enhancer factor 2C and its directly-interacting proteins: A review. Prog. Biophys. Mol. Biol. 2017, 126, 22–30. [Google Scholar] [CrossRef]
- Chinchilla, A.; Lozano, E.; Daimi, H.; Esteban, F.J.; Crist, C.; Aranega, A.E.; Franco, D. MicroRNA profiling during mouse ventricular maturation: A role for miR-27 modulating Mef2c expression. Cardiovasc. Res. 2011, 89, 98–108. [Google Scholar] [CrossRef]
- Shen, L.; Chen, L.; Zhang, S.; Du, J.; Bai, L.; Zhang, Y.; Jiang, Y.; Li, X.; Wang, J.; Zhu, L. MicroRNA-27b Regulates Mitochondria Biogenesis in Myocytes. PLoS ONE 2016, 11, e0148532. [Google Scholar] [CrossRef]
- McFarlane, C.; Vajjala, A.; Arigela, H.; Lokireddy, S.; Ge, X.; Bonala, S.; Manickam, R.; Kambadur, R.; Sharma, M. Negative auto-regulation of myostatin expression is mediated by Smad3 and microRNA-27. PLoS ONE 2014, 9, e87687. [Google Scholar] [CrossRef]
- Rodriguez, J.; Vernus, B.; Chelh, I.; Cassar-Malek, I.; Gabillard, J.C.; Hadj Sassi, A.; Seiliez, I.; Picard, B.; Bonnieu, A. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell. Mol. Life Sci. 2014, 71, 4361–4371. [Google Scholar] [CrossRef]
- Argilés, J.M.; Orpí, M.; Busquets, S.; López-Soriano, F.J. Myostatin: More than just a regulator of muscle mass. Drug Discov. Today 2012, 17, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Han, H.Q.; Mitch, W.E. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr. Opin. Support. Palliat. Care 2011, 5, 334–341. [Google Scholar] [CrossRef]
- Smith, R.C.; Lin, B.K. Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr. Opin. Support. Palliat. Care 2013, 7, 352–360. [Google Scholar] [CrossRef]
- Gallot, Y.S.; Durieux, A.-C.; Castells, J.; Desgeorges, M.M.; Vernus, B.; Plantureux, L.; Rémond, D.; Jahnke, V.E.; Lefai, E.; Dardevet, D.; et al. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res. 2014, 74, 7344–7356. [Google Scholar] [CrossRef]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Counteracting inflammation: A promising therapy in cachexia. Crit. Rev. Oncog. 2012, 17, 253–262. [Google Scholar] [CrossRef]
- Brzoska, E.; Kowalewska, M.; Markowska-Zagrajek, A.; Kowalski, K.; Archacka, K.; Zimowska, M.; Grabowska, I.; Czerwińska, A.M.; Czarnecka-Góra, M.; Stremińska, W.; et al. Sdf-1 (CXCL12) improves skeletal muscle regeneration via the mobilisation of Cxcr4 and CD34 expressing cells. Biol. Cell 2012, 104, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Dileepan, M.; Sarver, A.E.; Rao, S.P.; Panettieri, R.A.; Subramanian, S.; Kannan, M.S. MicroRNA Mediated Chemokine Responses in Human Airway Smooth Muscle Cells. PLoS ONE 2016, 11, e0150842. [Google Scholar] [CrossRef]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef]
- Diao, Y.; Wang, X.; Wu, Z. SOCS1, SOCS3, and PIAS1 promote myogenic differentiation by inhibiting the leukemia inhibitory factor-induced JAK1/STAT1/STAT3 pathway. Mol. Cell. Biol. 2009, 29, 5084–5093. [Google Scholar] [CrossRef]
- Lieskovska, J.; Guo, D.; Derman, E. Growth impairment in IL-6-overexpressing transgenic mice is associated with induction of SOCS3 mRNA. Growth Horm. IGF Res. 2003, 13, 26–35. [Google Scholar] [CrossRef]
- Mehic, D.; Bakiri, L.; Ghannadan, M.; Wagner, E.F.; Tschachler, E. Fos and jun proteins are specifically expressed during differentiation of human keratinocytes. J. Investig. Dermatol. 2005, 124, 212–220. [Google Scholar] [CrossRef]
- Zhang, P.; Cheng, J.; Zou, S.; D’Souza, A.D.; Koff, J.L.; Lu, J.; Lee, P.J.; Krause, D.S.; Egan, M.E.; Bruscia, E.M. Pharmacological modulation of the AKT/microRNA-199a-5p/CAV1 pathway ameliorates cystic fibrosis lung hyper-inflammation. Nat. Commun. 2015, 6, 6221. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Kulyté, A.; Lorente-Cebrián, S.; Gao, H.; Mejhert, N.; Agustsson, T.; Arner, P.; Rydén, M.; Dahlman, I. MicroRNA profiling links miR-378 to enhanced adipocyte lipolysis in human cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E267–E274. [Google Scholar] [CrossRef]
- Moraes, L.N.; Fernandez, G.J.; Vechetti-Júnior, I.J.; Freire, P.P.; Souza, R.W.A.; Villacis, R.A.R.; Rogatto, S.R.; Reis, P.P.; Dal-Pai-Silva, M.; Carvalho, R.F. Integration of miRNA and mRNA expression profiles reveals microRNA-regulated networks during muscle wasting in cardiac cachexia. Sci. Rep. 2017, 7, 6998. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Chou, C.-H.; Chang, N.-W.; Shrestha, S.; Hsu, S.-D.; Lin, Y.-L.; Lee, W.-H.; Yang, C.-D.; Hong, H.-C.; Wei, T.-Y.; Tu, S.-J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk–database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Dong, Q.; Muruganujan, A.; Gaudet, P.; Lewis, S.; Thomas, P.D. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D204–D210. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Lazareva-Ulitsky, B.; Loo, R.; Kejariwal, A.; Vandergriff, J.; Rabkin, S.; Guo, N.; Muruganujan, A.; Doremieux, O.; Campbell, M.J.; et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005, 33, D284–D288. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Snel, B.; Lehmann, G.; Bork, P.; Huynen, M.A. STRING: A web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000, 28, 3442–3444. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wagner, A.H.; Coffman, A.C.; Ainscough, B.J.; Spies, N.C.; Skidmore, Z.L.; Campbell, K.M.; Krysiak, K.; Pan, D.; McMichael, J.F.; Eldred, J.M.; et al. DGIdb 2.0: Mining clinically relevant drug-gene interactions. Nucleic Acids Res. 2016, 44, D1036–D1044. [Google Scholar] [CrossRef]
- Griffith, M.; Griffith, O.L.; Coffman, A.C.; Weible, J.V.; McMichael, J.F.; Spies, N.C.; Koval, J.; Das, I.; Callaway, M.B.; Eldred, J.M.; et al. DGIdb: Mining the druggable genome. Nat. Methods 2013, 10, 1209–1210. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Gene Expression Platforms | Validation Platforms | Muscle | Cancer Cachexia Study (Rodent Model or Cancer Type) | Number of Samples (Cachectic/Control) | Ref. |
|---|---|---|---|---|---|---|
| Mus Musculus | ||||||
| Tseng et al. 2015 | Illumina HiSeq 2500 | RT | Gastrocnemius | Colon-26 adenocarcinoma tumor-bearing mice | 3/3 | [36] |
| Roberts et al. 2013 | Illumina Genome Analyzer II | RT, WB | Quadriceps | Pancreatic adenocarcinoma-bearing mice | 2/2 | [37] |
| Bonetto et al. 2011 | Illumina MouseWG-6 v2.0 expression beadchip | RT, WB | Quadriceps | Colon-26 adenocarcinoma tumor-bearing mice | 4/4 | [38] |
| Gilabert et al. 2014 | Affymetrix Mouse Gene 1.0 ST Array | RT | Biceps femoris | Pancreatic adenocarcinoma-bearing mice | 3/3 | [39] |
| Shum et al. 2015 | Affymetrix Mouse Gene 1.0 ST Array | RT | Gastrocnemius | Colon-26 carcinoma tumor-bearing mice | 3/3 | [40] |
| Fontes-Oliveira et al. 2014 | Affymetrix RAE230Plus | RT | Extensor Digitorum Longus | Rats injected with AH-130 Yoshida ascites hepatoma cells | 7/6 | [41] |
| Homo Sapiens | ||||||
| Martinelli et al. 2016 | Agilent-014850 Whole Human Genome Microarray | RT, WB | Rectus abdominis | Pancreatic, colorectal, Hepatic, and renal cancers | 115 | [43] |
| Stephens et al. 2010 | Affymetrix GeneChip Human Genome U133 Plus 2.0 Array | RT | Rectus abdominis | Gastrointestinal cancer | 18/3 | [28] |
| Gallagher et al. 2012 | Affymetrix Human Genome U133 Plus 2.0 Array | RT | Quadriceps | Upper gastrointestinal cancer | 12/6 | [29] |
| Official Symbol | Species | Function | Location |
|---|---|---|---|
| Up-Regulated | |||
| TIE1 | H | Regulation of angiogenesis | Cell Membrane |
| EIF3I | H | Cell proliferation, including cell cycling, differentiation and apoptosis | Cytoplasm |
| HGS | H | Intracellular signal transduction mediated by cytokines and growth factors | Cytoplasm |
| NUDC | H | Neurogenesis and neuronal migration | Cytoplasm |
| PCK1 | H | Metabolic pathway that produces glucose | Cytoplasm |
| TSC2 | H | Negatively regulating mTORC1 signaling and playing a role in microtubule-mediated protein transport | Cytoplasm |
| CAMK2B | H | Regulation of sarcoplasmic reticulum Ca2+ transport in skeletal muscle | Cytoplasm; Sarcoplasmic reticulum membrane |
| POLRMT | H | Transcription of mitochondrial DNA into RNA | Mitochondrion |
| COMP | H | Suppressor of apoptosis; interact with extracellular matrix proteins such as the collagens and fibronectin | Secreted - ECM |
| MMP3 | H | Degrade fibronectin, laminin, gelatins, collagens, and cartilage proteoglycans | Secreted - ECM |
| ADIPOQ | H | Control of fat metabolism, insulin sensitivity, cell growth, angiogenesis and tissue remodeling | Secreted - ER |
| ANGPTL7 | H | Anti-angiogenic protein and play a role in extracellular matrix formation | Secreted - ER |
| Kcnip4 | M | Modulates channel expression at the cell membrane | Cell Membrane |
| Pnpla2 | M | Response of the organism to starvation, enhancing hydrolysis of triglycerides and providing free fatty acids to other tissues | Cell Membrane |
| Socs3 | M | Negative regulation of cytokines that signal through the JAK/STAT pathway | Cytoplasm |
| Foxo1 | M | Autophagic cell death induction in response to starvation or oxidative stress | Cytoplasm and Nucleus |
| Stat3 | M | Signal transducer and transcription activator that mediates cellular responses to interleukins and growth factors | Cytoplasm and Nucleus |
| Ufd1 | M | Promote ubiquitination and degradation | Cytoplasm and Nucleus |
| C1s1 | M | Serine protease | Extracellular exosome; Extracellular space |
| Ucp3 | M | Thermogenesis and energy balance | Mitochondrion |
| Cebpd | M | Immune and inflammatory responses. Transcriptional activator that enhances IL6 transcription | Nucleus |
| Junb | M | Regulate gene activity following the primary growth factor response | Nucleus |
| Fgg | M | Guide cell migration during re-epithelialization | Secreted |
| HP | M | Antibacterial activity and plays a role in modulating many aspects of the acute phase response | Secreted |
| Mstn | M | Acts specifically as a negative regulator of skeletal muscle growth | Secreted |
| Saa1 | M | Major acute phase protein | Secreted |
| Serpina3n | M | Response to cytokine | Secreted |
| FBXO32 | H, M | Proteasomal degradation of target proteins during skeletal muscle atrophy | Cytoplasm and Nucleus |
| TRIM63 | H, M | Regulates the proteasomal degradation of muscle proteins and inhibits de novo skeletal muscle protein synthesis | Cytoplasm and Nucleus |
| Down-Regulated | |||
| APCDD1 | H | Negative regulator of the Wnt signaling pathway | Cell Membrane |
| ADCY7 | H | Membrane-bound, calcium-inhibitable adenylyl cyclase | Cell Membrane |
| GABARAPL1 | H | Autophagosome maturation | Cytoplasm |
| HINT3 | H | Hydrolyzes phosphoramidate and acyl-adenylate substrates | Cytoplasm and Nucleus |
| NR3C1 | H | Affects inflammatory responses, cellular proliferation, and differentiation in target tissues | Cytoplasm and Nucleus |
| RCAN1 | H | Central nervous system development | Cytoplasm and Nucleus |
| HSP90AB1 | H | Maturation, structural maintenance, and proper regulation of specific target proteins involved, for instance in cell cycle control and signal transduction | Cytoplasm, nucleus, cell membrane and secreted |
| HSD11B1 | H | Reversibly catalyzes the conversion of cortisol to the inactive metabolite cortisone | Endoplasmic reticulum membrane |
| BNIP3 | H | Mitochondrial protein catabolic process; cell death pathway | Mitochondrion |
| SLC25A37 | H | Mitochondrial iron transporter that specifically mediates iron uptake | Mitochondrion |
| PAK1 | H | Cell adhesion, migration, proliferation, apoptosis, mitosis, and vesicle-mediated transport processes | Cytoplasm and Cell Membrane |
| PROX1 | H | Cell fate determination, gene transcriptional regulation, and progenitor cell regulation | Nucleus |
| Cav1 | M | T-cell proliferation and NF-kappa-B activation | Cell Membrane |
| Fap | M | Extracellular matrix degradation, tissue remodeling, fibrosis, wound healing, inflammation and tumor growth | Cell Membrane |
| Actc1 | M | Cell motility | Cytoplasm |
| Myh8 | M | Muscle contraction | Cytoplasm |
| Tuba1a | M | Constituent of microtubules | Cytoplasm |
| Tuba4a | M | Constituent of microtubules | Cytoplasm |
| Mef2c | M | Controls cardiac morphogenesis and myogenesis, and is also involved in vascular development | Nucleus |
| Tfcp2 | M | Binds a variety of cellular promoters, including fibrinogen and alpha-globin promoters | Nucleus |
| Lama2 | M | Attachment, migration, and organization of cells by interacting with extracellular matrix components | Secreted (ECM) |
| Fst | M | Specific inhibitor of the biosynthesis and secretion of pituitary follicle stimulating hormone (FSH) | Secreted (ER) |
| CXCL12 | H, M | Immune surveillance, inflammation response, tissue homeostasis, and tumor growth and metastasis | Secreted |
| Study | microRNA/mRNA Interactions | |
|---|---|---|
| microRNA | mRNA * | |
| Soares et al. [35] | ↓ miR-27b | ↓ Cxcl12 |
| ↑ miR-140 | ↓ Cxcl12 | |
| ↓ miR-27a | ↑ Foxo1 | |
| ↓ miR-27a | ↓ Mef2c | |
| ↓ miR-27b | ↓ Mef2c | |
| Narasimhan et al. [34] | ↑ miR-199a | ↑ Junb |
| ↑ miR-199a | ↓ Cav1 | |
| Gene Symbol | Gene Name | Selected Target Agent | Activity | Clinical Relevance | Ref. |
|---|---|---|---|---|---|
| ADIPOQ | Adiponectin | Spironolactone, Fenofibrate, Nevirapine | Aldosterone blocker; anti-diabetic and anti-atherosclerotic; antiretroviral therapy | Type 2 diabetes mellitus; cardiovascular disease; HIV infection | [44,45,46] |
| CAMK2B | Calcium/calmodulin-dependent protein kinase II, beta | Mibefradil, Nifedipine, Nisoldipine | Block T-type calcium channel | Smooth muscle; cardiac muscle | [47,48,49] |
| COMP | Cartilage oligomeric matrix protein | Tadalafil | Tumor cell growth inhibitory | Breast cancer | [50] |
| CXCL12 | Chemokine ligand 12 | Tinzaparin Sodium | Regulate the proteoglycan core proteins | Breast cancer | [51] |
| MSTN | Myostatin | Stamulumab | Growth/differentiation factor 8 inhibitor | Myopathies | [52,53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freire, P.P.; Fernandez, G.J.; Cury, S.S.; de Moraes, D.; Oliveira, J.S.; de Oliveira, G.; Dal-Pai-Silva, M.; dos Reis, P.P.; Carvalho, R.F. The Pathway to Cancer Cachexia: MicroRNA-Regulated Networks in Muscle Wasting Based on Integrative Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 1962. https://doi.org/10.3390/ijms20081962
Freire PP, Fernandez GJ, Cury SS, de Moraes D, Oliveira JS, de Oliveira G, Dal-Pai-Silva M, dos Reis PP, Carvalho RF. The Pathway to Cancer Cachexia: MicroRNA-Regulated Networks in Muscle Wasting Based on Integrative Meta-Analysis. International Journal of Molecular Sciences. 2019; 20(8):1962. https://doi.org/10.3390/ijms20081962
Chicago/Turabian StyleFreire, Paula Paccielli, Geysson Javier Fernandez, Sarah Santiloni Cury, Diogo de Moraes, Jakeline Santos Oliveira, Grasieli de Oliveira, Maeli Dal-Pai-Silva, Patrícia Pintor dos Reis, and Robson Francisco Carvalho. 2019. "The Pathway to Cancer Cachexia: MicroRNA-Regulated Networks in Muscle Wasting Based on Integrative Meta-Analysis" International Journal of Molecular Sciences 20, no. 8: 1962. https://doi.org/10.3390/ijms20081962
APA StyleFreire, P. P., Fernandez, G. J., Cury, S. S., de Moraes, D., Oliveira, J. S., de Oliveira, G., Dal-Pai-Silva, M., dos Reis, P. P., & Carvalho, R. F. (2019). The Pathway to Cancer Cachexia: MicroRNA-Regulated Networks in Muscle Wasting Based on Integrative Meta-Analysis. International Journal of Molecular Sciences, 20(8), 1962. https://doi.org/10.3390/ijms20081962