Cerebrospinal Fluid (CSF) Exchange with Artificial CSF Enriched with Mesenchymal Stem Cell Secretions Ameliorates Experimental Autoimmune Encephalomyelitis

Abstract
1. Introduction
2. Results
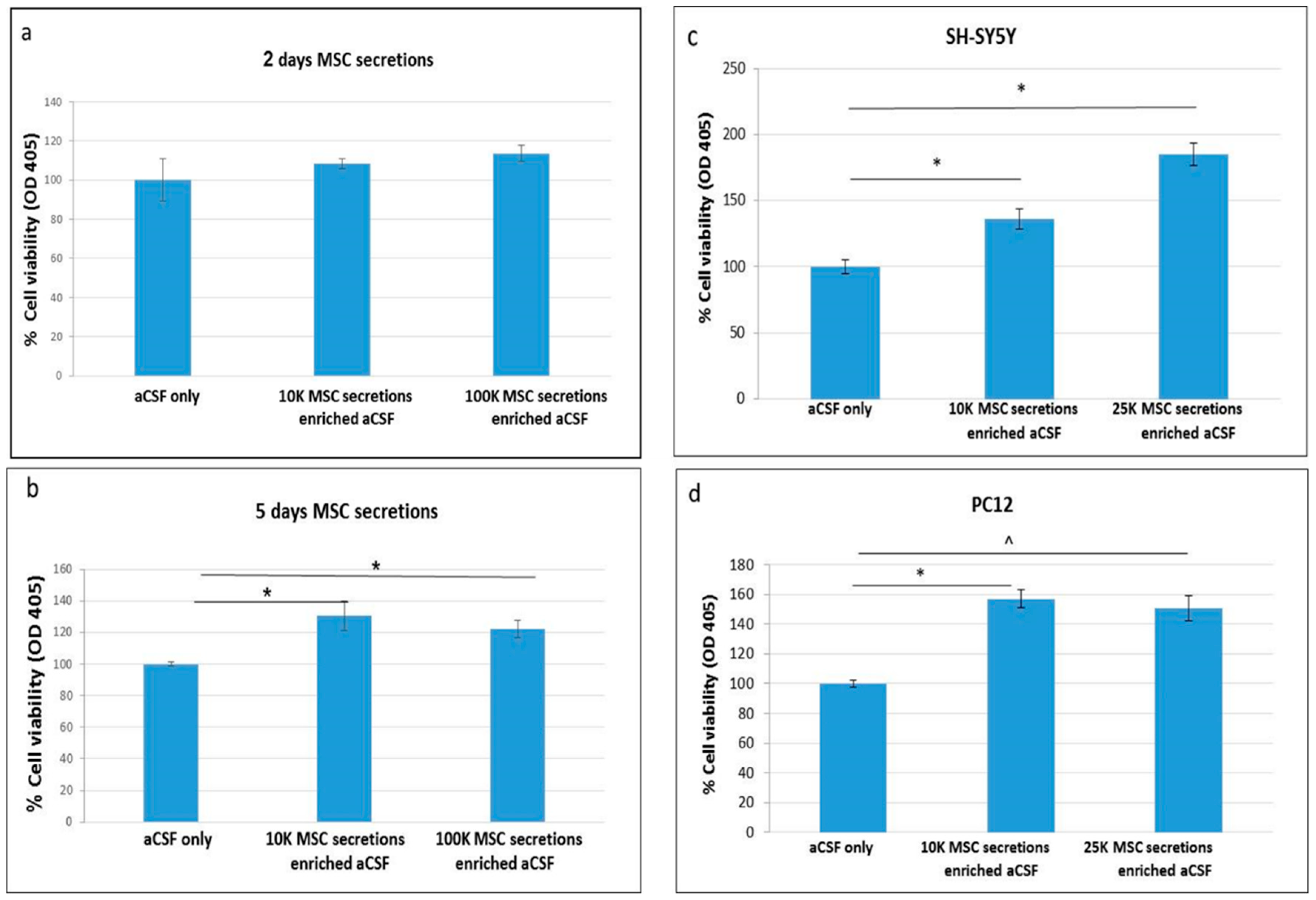
2.1. Enriched-aCSF Increases Cell Viability of Neuronal Cell Lines
2.2. Enriched-aCSF Increases Cell Viability of Neuronal Cell Lines Exposed to Neurotoxins
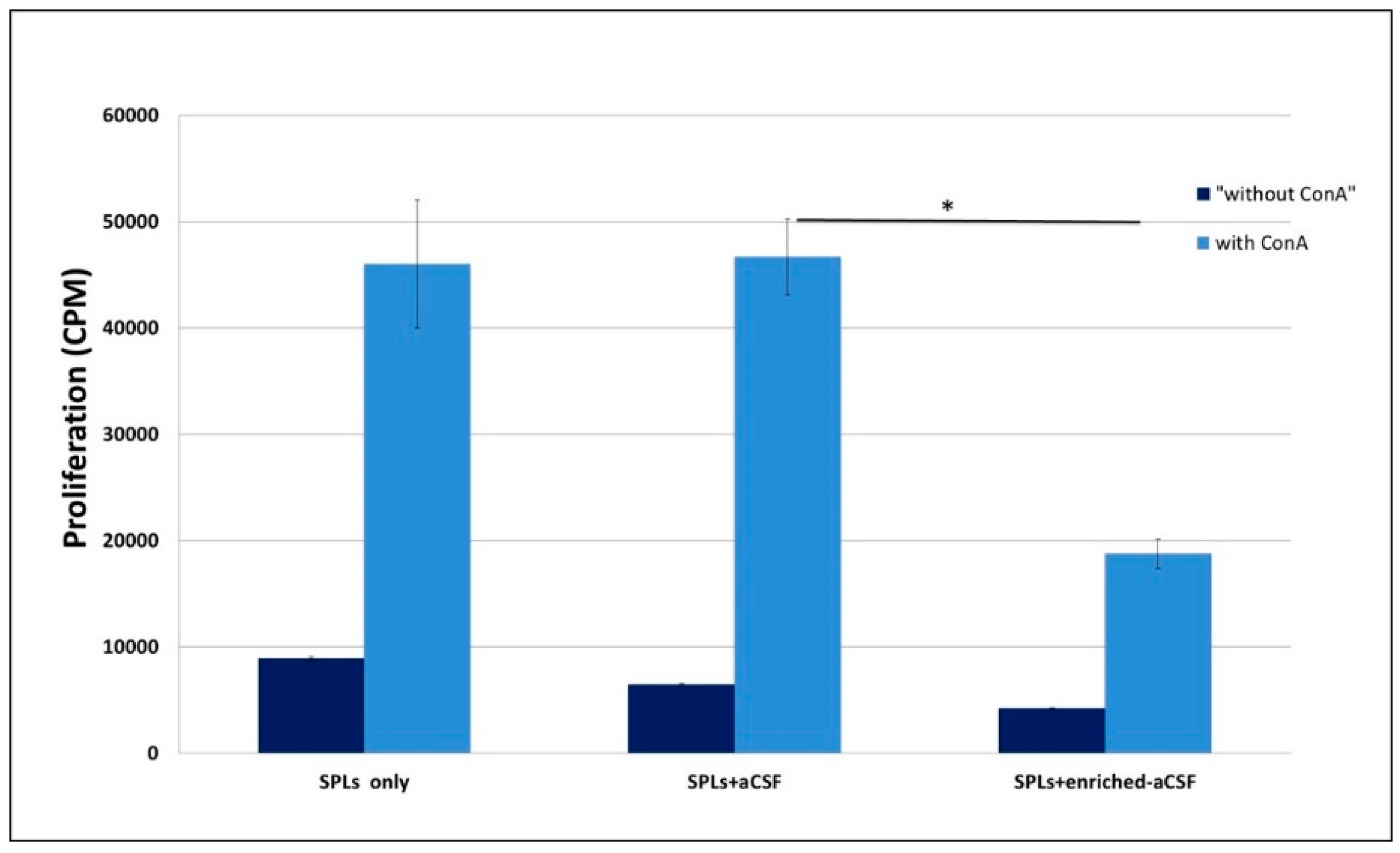
2.3. Enriched-aCSF Suppressed ConA Induced Splenocytes Proliferation
2.4. Enriched-aCSF Contains Neurotrophic Factors, Anti-Inflammatory Cytokines, and Anti-Oxidants
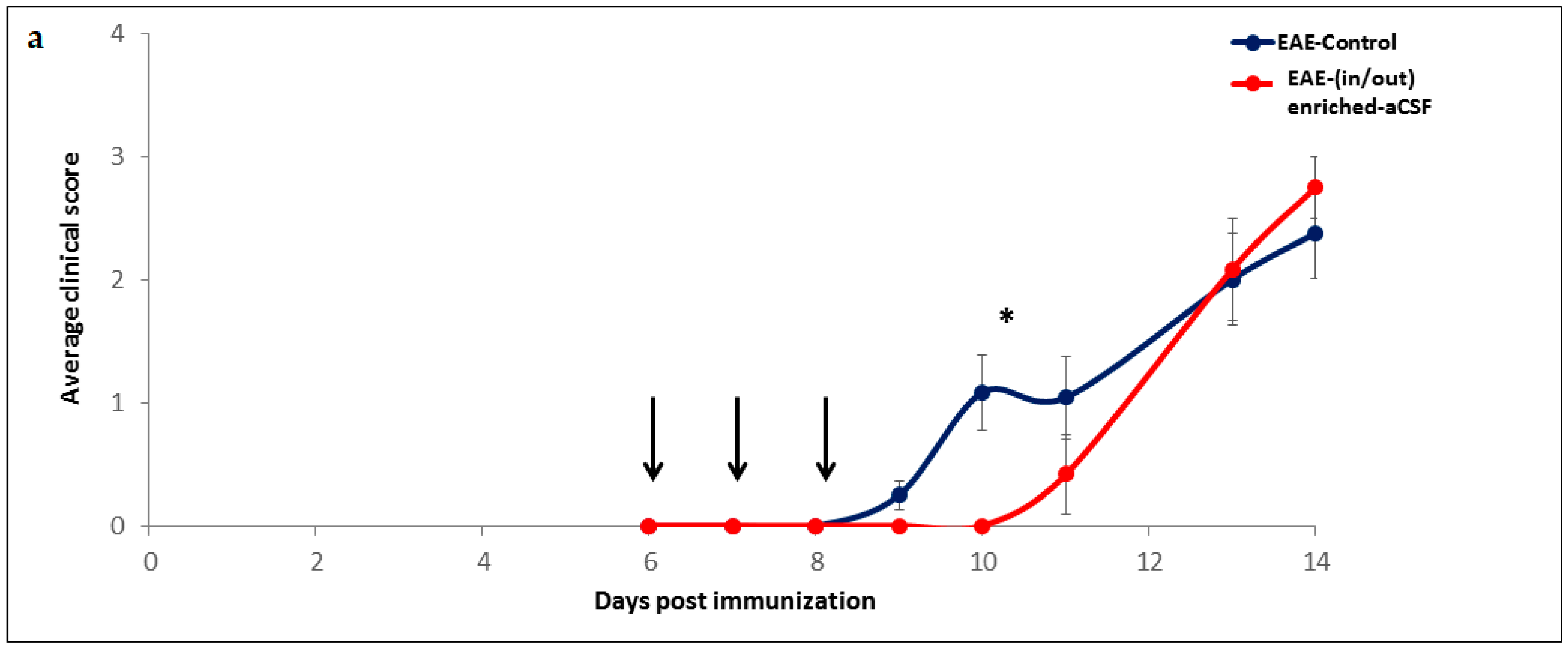
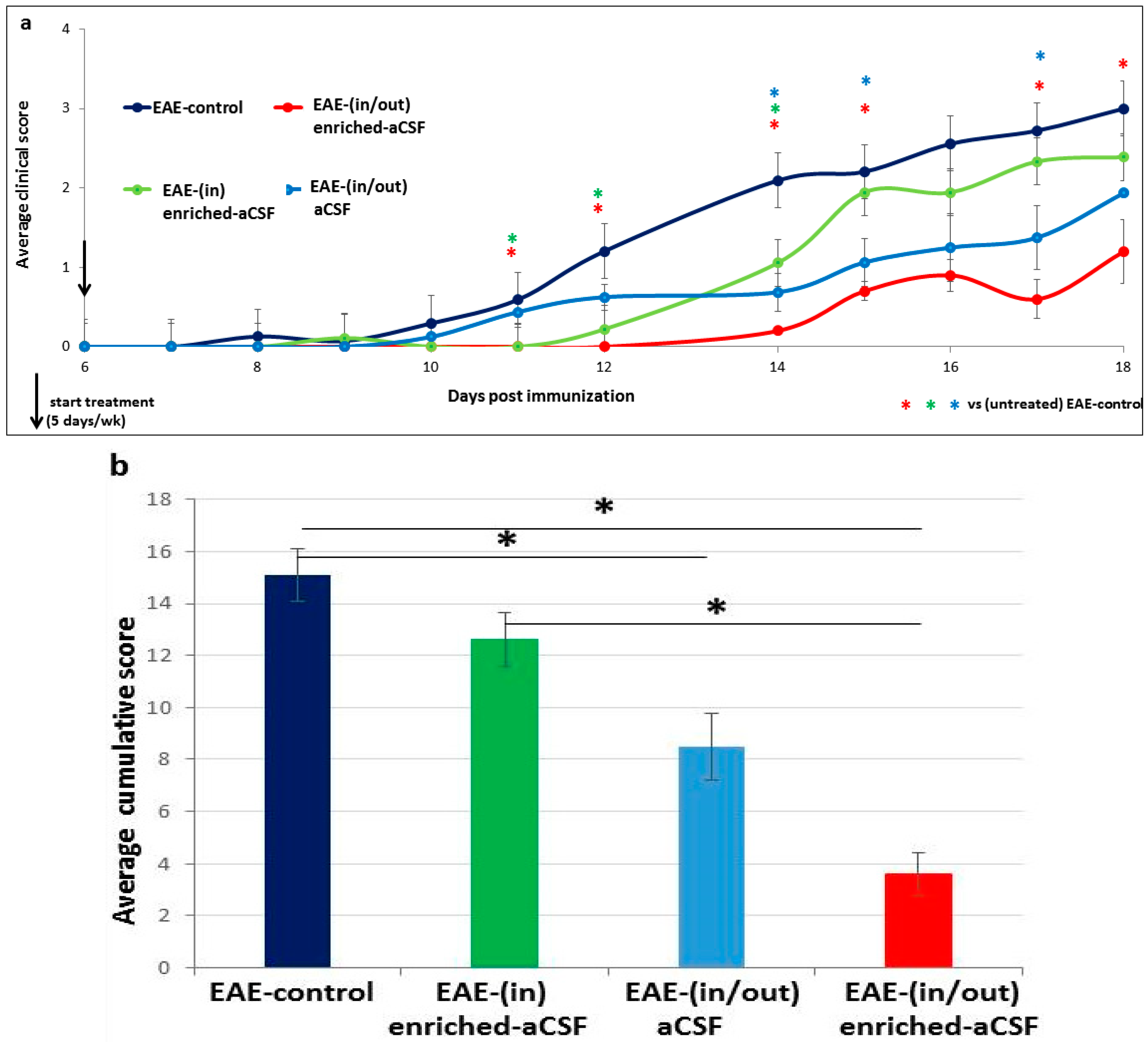
2.5. Short-Term MSC Secretions Enriched-aCSF (in/out) Exchange Therapy Induced a Short Term Amelioration of EAE Clinical Symptoms
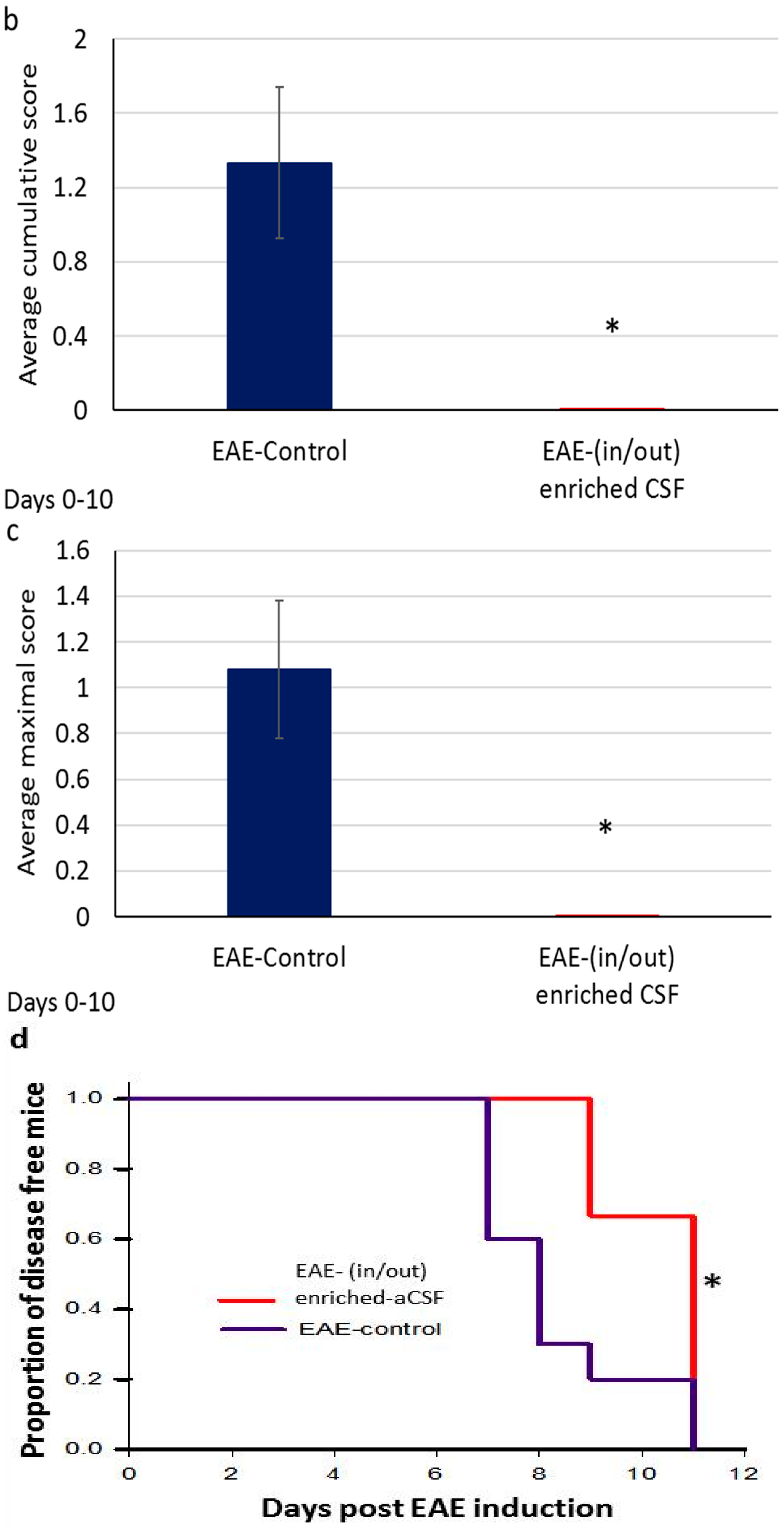
2.6. Prolonged Amelioration of the EAE Clinical Symptoms during a Prolonged CSF Exchange Therapy
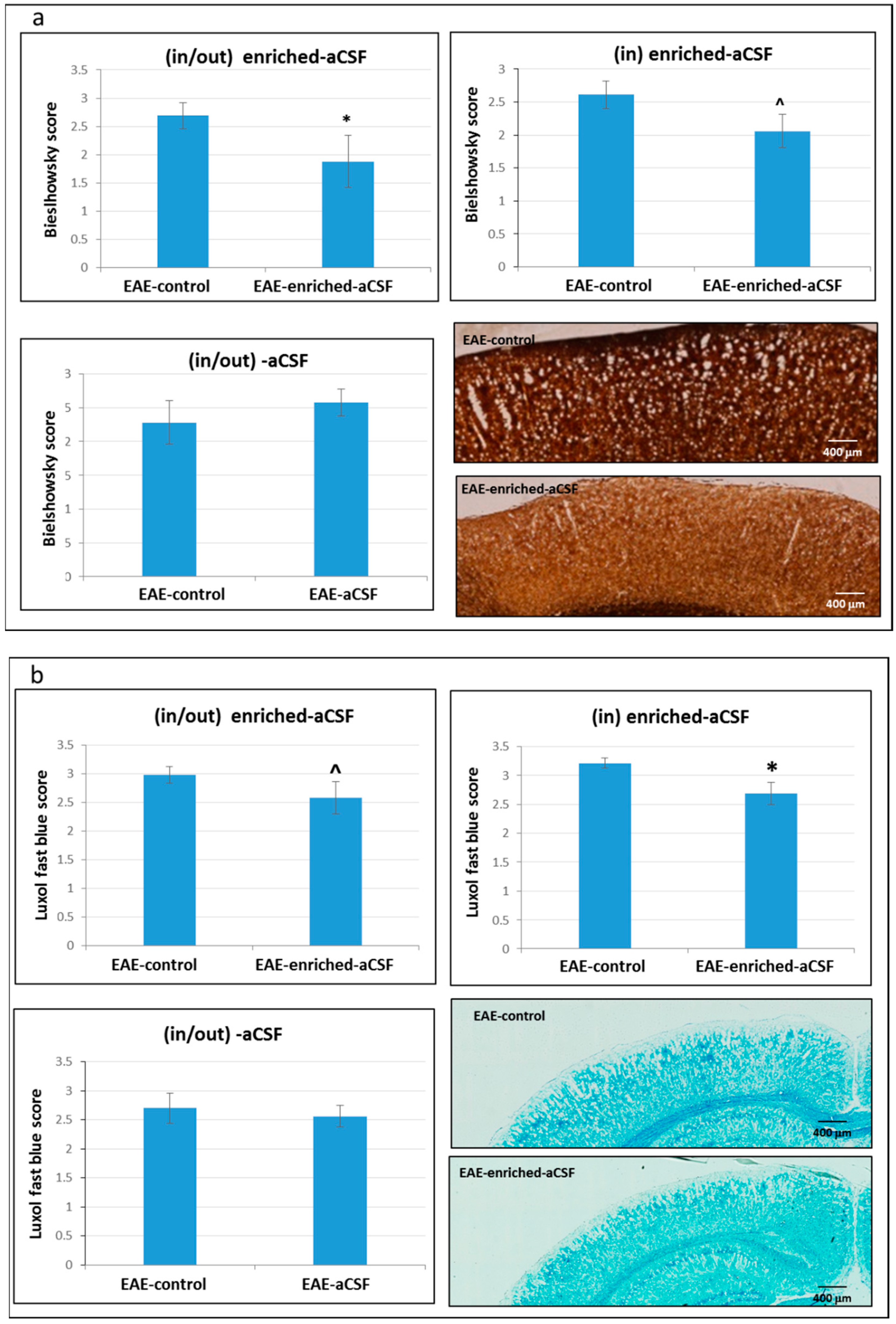
2.7. CSF Exchange Therapy Reduced Axonal Damage and Demyelination
3. Discussion
4. Materials and Methods
4.1. Cell Culture Studies
4.1.1. Preparation of MSCs were Performed According to Our Reported Protocol
Bone Marrow (BM) Aspiration
Separation of Mononuclear Cells (MNCs) from the Whole BM
Propagation of MSCs
Characterization of Isolated Human MSCs
4.1.2. Preparation of aCSF Enriched with Secretions of MSCs
4.1.3. Testing the Viability of Neuronal Cell Lines Treated with the Enriched-aCSF
4.1.4. Measurement of Secreted Neurotrophic Factors, Anti-inflammatory Cytokines, and Anti-oxidant Capacity in the Enriched-aCSF
4.1.5. In Vitro Proliferation of Spleen Lymphocytes (Splenocytes) Treated with the Enriched-aCSF
4.2. Animal Studies
4.2.1. Insertion of the CSF Exchange Device
4.2.2. Induction of EAE
4.2.3. CSF Exchange Therapy
4.2.4. Histological Examination
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sheikh, S.; Haque, E.; Mir, S.S. Neurodegenerative Dieases: Multifctorial, Conformational Diseases and Their Therapeutic Interventions. J. Neurodegener. Dis. 2013, 2013, 563481. [Google Scholar] [PubMed]
- Irani, D.N. CSF in Clinical Practice. Saunders 2009; Section 1: Normal Anatomy and Phisiology of the CSF Compartment; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Rosenmann, H. CSF biomarkers for amyloid and tau pathology in Alzheimer’s disease. J. Mol. Neurosci. 2012, 47, 1–14. [Google Scholar] [CrossRef]
- Tarasiuk, J.; Kułakowska, A.; Drozdowski, W.; Lewczuk, P.; Kornhuber, J. CSF markers in amyotrophic lateral sclerosis. J. Transm. 2012, 119, 747–757. [Google Scholar] [CrossRef]
- Martin, N.A.; Nawrocki, A.; Molnar, V.; Elkjaer, M.L.; Thygesen, E.K.; Palkovits, M.; Acs, P.; Sejbaek, T.; Nielsen, H.H.; Hegedus, Z.; et al. Orthologous proteins of experimental de- and remyelination are differentially regulated in the CSF proteome of multiple sclerosis subtypes. PLoS ONE 2018, 13, e0202530. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Nalini, A.; Gourie-Devi, M.; Raju, T.R. Reactive Astrogliosis in Neonatal Rat Spinal Cord after Exposure to Cerebrospinal Fluid from Patients with Amyotrophic Lateral Sclerosis. Exp. Neurol. 1998, 149, 295–298. [Google Scholar] [CrossRef][Green Version]
- Ramamohan, P.Y.; Gourie-Devi, M.; Nalini, A.; Shobha, K.; Ramamohan, Y.; Joshi, P.; Raju, T.R. Cerebrospinal fluid from amyotrophic lateral sclerosis patients causes fragmentation of the Golgi apparatus in the neonatal rat spinal cord. Amyotroph. Scler. 2007, 8, 79–82. [Google Scholar] [CrossRef]
- Gwathmey, K.; Burns, T. Neurologic indications for therapeutic plasma exchange: 2013 update. J. Clin. Apher. 2014, 29, 211–219. [Google Scholar] [CrossRef]
- Zappaterra, M.W.; Lehtinen, M.K. The cerebrospinal fluid: Regulator of neurogenesis, behavior, and beyond. Cell. Mol. Life Sci. 2012, 69, 2863–2878. [Google Scholar] [CrossRef]
- Wollinsky, K.H.; Weindler, M.; Hülser, P.-J.; Geiger, P.; Matzek, N.; Mehrkens, H.-H.; Kornhuber, H.H. Liquorpheresis (CSF-filtration): An effective treatment in acute and chronic severe autoimmune polyradiculoneuritis (Guillain-Barré syndrome). Eur. Arch. Psychiatry Clin. Neurosci. 1991, 241, 73–76. [Google Scholar] [CrossRef]
- Finsterer, J.; Mamoli, B. Liquorpheresis (CSF filtration) in familial amyotrophic lateral sclerosis. Spinal Cord 1999, 37, 592–593. [Google Scholar] [CrossRef][Green Version]
- Lescot, T.; Boroli, F.; Reina, V.; Chauvet, D.; Boch, A.-L.; Puybasset, L. Effect of continuous cerebrospinal fluid drainage on therapeutic intensity in severe traumatic brain injury. Neurochirurgie 2012, 58, 235–240. [Google Scholar] [CrossRef]
- Houle, P.J.; Vender, J.R.; Fountas, K.; McDonnell, D.E.; Fick, J.R.; Robinson, J.-S. Pump-regulated Lumbar Subarachnoid Drainage. Neurosurgery 2000, 46, 929–932. [Google Scholar]
- Kristof, R.A.; Clusmann, H.; Koehler, W.; Fink, K.B.; Schramm, J. Treatment of accidental high dose intraventricular mezlocillin application by cerebrospinal fluid exchange. J. Neurol. Neurosurg. 1998, 64, 379–381. [Google Scholar] [CrossRef]
- Di Nicola, M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Meisel, R.; Zibert, A.; Laryea, M.; Göbel, U.; Däubener, W.; Dilloo, D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase–mediated tryptophan degradation. Blood 2004, 103, 4619–4621. [Google Scholar] [CrossRef]
- Sotiropoulou, P.A.; Perez, S.A.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. Interactions Between Human Mesenchymal Stem Cells and Natural Killer Cells. Stem Cells 2006, 24, 74–85. [Google Scholar] [CrossRef]
- Krampera, M.; Cosmi, L.; Angeli, R.; Pasini, A.; Liotta, F.; Andreini, A.; Santarlasci, V.; Mazzinghi, B.; Pizzolo, G.; Vinante, F.; et al. Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells 2006, 24, 386–398. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Immunoregulatory function of mesenchymal stem cells. Eur. J. Immunol. 2006, 36, 2566–2573. [Google Scholar] [CrossRef]
- Kassis, I.; Vaknin-Dembinsky, A.; Karussis, D. Bone Marrow Mesenchymal Stem Cells: Agents of Immunomodulation and Neuroprotection. Curr. Stem Cell Res. Ther. 2011, 6, 63–68. [Google Scholar] [CrossRef]
- Asami, T.; Ishii, M.; Fujii, H.; Namkoong, H.; Tasaka, S.; Matsushita, K.; Ishii, K.; Yagi, K.; Fujiwara, H.; Funatsu, Y.; et al. Modulation of Murine Macrophage TLR7/8-Mediated Cytokine Expression by Mesenchymal Stem Cell-Conditioned Medium. Mediat. Inflamm. 2013, 2013, 1–13. [Google Scholar] [CrossRef]
- Kassis, I.; Grigoriadis, N.; Gowda-Kurkalli, B.; Mizrachi-Kol, R.; Ben-Hur, T.; Slavin, S.; Abramsky, O.; Karussis, D. Neuroprotection and Immunomodulation with Mesenchymal Stem Cells in Chronic Experimental Autoimmune Encephalomyelitis. Arch. Neurol. 2008, 65, 753–761. [Google Scholar] [CrossRef]
- Kassis, I.; Petrou, P.; Halimi, M.; Karussis, D. Mesenchymal stem cells (MSC) derived from mice with experimental autoimmune encephalomyelitis (EAE) suppress EAE and have similar biological properties with MSC from healthy donors. Immunol. Lett. 2013, 154, 70–76. [Google Scholar] [CrossRef]
- Zappia, E.; Casazza, S.; Benvenuto, F.; Bonanni, I.; Gerdoni, E.; Giunti, D.; Ceravolo, A.; Cazzanti, F.; Mancardi, G.; Uccelli, A.; et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 2005, 106, 1755–1761. [Google Scholar] [CrossRef]
- Rafei, M.; Campeau, P.M.; Aguilar-Mahecha, A.; Buchanan, M.; Williams, P.; Birman, E.; Yuan, S.; Young, Y.K.; Boivin, M.-N.; Forner, K.; et al. Mesenchymal Stromal Cells Ameliorate Experimental Autoimmune Encephalomyelitis by Inhibiting CD4 Th17 T Cells in a CC Chemokine Ligand 2-Dependent Manner. J. Immunol. 2009, 182, 5994–6002. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.; Chen, X.G.; Wang, L.; Gautam, S.C.; Xu, Y.X.; Katakowski, M.; Zhang, L.J.; Lu, M.; Janakiraman, N.; et al. Human marrow stromal cell therapy for stroke in rat: Neurotrophins and functional recovery. Neurology 2002, 59, 514–523. [Google Scholar] [CrossRef]
- Li, N.; Li, X.R.; Yuan, J.Q. Effects of bone-marrow mesenchymal stem cells transplanted into vitreous cavity of rat injured by ischemia/reperfusion. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 503–514. [Google Scholar]
- Mahmood, A.; Lu, D.; Chopp, M. Marrow Stromal Cell Transplantation after Traumatic Brain Injury Promotes Cellular Proliferation within the Brain. Neurosurgery 2004, 55, 1185–1193. [Google Scholar] [CrossRef]
- Park, H.J.; Lee, P.H.; Bang, O.Y.; Lee, G.; Ahn, Y.H. Mesenchymal stem cells therapy exerts neuroprotection in a progressive animal model of Parkinson’s disease. J. Neurochem. 2008, 107, 141–151. [Google Scholar] [CrossRef]
- Barzilay, R.; Ganz, J.; Sadan, O.; Ben-Zur, T.; Bren, Z.; Hinden, N.; Taler, M.; Lev, N.; Gil-Ad, I.; Weizman, A.; et al. Mesenchymal stem cells protect from sub-chronic phencyclidine insult in vivo and counteract changes in astrocyte gene expression in vitro. Eur. Neuropsychopharmacol. 2013, 23, 1115–1123. [Google Scholar] [CrossRef]
- Segal-Gavish, H.; Karvat, G.; Barak, N.; Barzilay, R.; Ganz, J.; Edry, L.; Aharony, I.; Offen, D.; Kimchi, T. Mesenchymal Stem Cell Transplantation Promotes Neurogenesis and Ameliorates Autism Related Behaviors in BTBR Mice. Autism Res. 2016, 9, 17–32. [Google Scholar] [CrossRef]
- Kan, I.; Barhum, Y.; Melamed, E.; Offen, D. Mesenchymal stem cells stimulate endogenous neurogenesis in the subventricular zone of adult mice. Stem Cell Rev. 2011, 7, 404–412. [Google Scholar] [CrossRef]
- Kassis, I.; Vaknin-Dembinsky, A.; Bulte, J.; Karussis, D. Effects of Supermagnetic Iron Oxide Labeling on the Major Functional Properties of Human Mesenchymal Stem Cells from Multiple Sclerosis Patients. Int. J. Stem Cells 2010, 3, 144–153. [Google Scholar] [CrossRef][Green Version]
- Petrou, P.; Gothelf, Y.; Argov, Z.; Gotkine, M.; Levy, Y.S.; Kassis, I.; Vaknin-Dembinsky, A.; Ben-Hur, T.; Offen, D.; Abramsky, O.; et al. Safety and Clinical Effects of Mesenchymal Stem Cells Secreting Neurotrophic Factor Transplantation in Patients with Amyotrophic Lateral Sclerosis: Results of Phase 1/2 and 2a Clinical Trials. JAMA Neurol. 2016, 3, 337–344. [Google Scholar] [CrossRef]
- Syková, E.; Rychmach, P.; Drahorádová, I.; Konrádová, Š.; Růžičková, K.; Voříšek, I.; Forostyak, S.; Homola, A.; Bojar, M. Transplantation of Mesenchymal Stromal Cells in Patients with Amyotrophic Lateral Sclerosis: Results of Phase I/IIa Clinical Trial. Cell Transplant. 2017, 26, 647–658. [Google Scholar] [CrossRef]
- Potian, J.A.; Aviv, H.; Ponzio, N.M.; Harrison, J.S.; Rameshwar, P. Veto-Like Activity of Mesenchymal Stem Cells: Functional Discrimination Between Cellular Responses to Alloantigens and Recall Antigens. J. Immunol. 2003, 171, 3426–3434. [Google Scholar] [CrossRef]
- Coulson-Thomas, V.J.; Gesteira, T.F.; Hascall, V.; Kao, W. Umbilical cord mesenchymal stem cells suppress host rejection: The role of the glycocalyx. J. Biol. Chem. 2014, 289, 23465–23481. [Google Scholar] [CrossRef]
- Pardridge, W.M. Transnasal and intraventricular delivery. In Peptide Drug Delivery to the Brain (Table 4.2); Raven Press: New York, NY, USA, 1991; p. 112. [Google Scholar]
- Nessler, J.; Bénardais, K.; Gudi, V.; Hoffmann, A.; Tejedor, L.S.; Janßen, S.; Prajeeth, C.K.; Baumgärtner, W.; Kavelaars, A.; Heijnen, C.J.; et al. Effects of Murine and Human Bone Marrow-Derived Mesenchymal Stem Cells on Cuprizone Induced Demyelination. PLoS ONE 2013, 8, e69795. [Google Scholar] [CrossRef]
- Ravasi, M.; Scuteri, A.; Pasini, S.; Bossi, M.; Menendez, V.R.; Maggioni, D.; Tredici, G. Undifferentiated MSCs are able to myelinate DRG neuron processes through p75. Exp. Cell Res. 2013, 319, 2989–2999. [Google Scholar] [CrossRef]
- Makar, T.K.; Trisler, D.; Sura, K.T.; Sultana, S.; Patel, N.; Bever, C.T. Brain derived neurotrophic factor treatment reduces inflammation and apoptosis in experimental allergic encephalomyelitis. J. Neurol. Sci. 2008, 270, 70–76. [Google Scholar] [CrossRef]
- Makar, T.K.; Bever, C.T.; Singh, I.S.; Royal, W.; Sahu, S.N.; Sura, T.P.; Sultana, S.; Sura, K.T.; Patel, N.; Dhib-Jalbut, S.; et al. Brain-derived neurotrophic factor gene delivery in an animal model of multiple sclerosis using bone marrow stem cells as a vehicle. J. Neuroimmunol. 2009, 210, 40–51. [Google Scholar] [CrossRef]
- Yang, J.; Yan, Y.; Xia, Y.; Kang, T.; Li, X.; Ciric, B.; Xu, H.; Rostami, A.; Zhang, G.X. Neurotrophin 3 transduction augments remyelinating and immunomodulatory capacity of neural stem cells. Mol. Ther. 2014, 22, 440–450. [Google Scholar] [CrossRef]
- Ariyannur, P.S.; Ribeiro, R.; Tanaka, M.; Moffett, J.R.; Kirmani, B.F.; Namboodiri, A.M.; Zhang, Y.; Wen, J. Efficacy of N-Acetylserotonin and Melatonin in the EAE Model of Multiple Sclerosis. J. Neuroimmune Pharmacol. 2016, 11, 763–773. [Google Scholar]
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci. Rep. 2011, 1, 201. [Google Scholar] [CrossRef]
- Hou, H.; Cao, R.; Miao, J.; Sun, Y.; Liu, X.; Song, X.; Guo, L. Fingolimod ameliorates the development of experimental autoimmune encephalomyelitis by inhibiting Akt–mTOR axis in mice. Int. Immunopharmacol. 2016, 30, 171–178. [Google Scholar] [CrossRef]
- Ginsburg, I.; Rozenstein-Tsalkovich, L.; Koren, E.; Rosenmann, H. The Herbal Preparation Padma® 28 Protects Against Neurotoxicity in PC12 Cells. Phytother. Res. 2011, 25, 740–743. [Google Scholar] [CrossRef]
- Rosenmann, H.; Grigoriadis, N.; Karussis, D.; Boimel, M.; Touloumi, O.; Ovadia, H.; Abramsky, O. Tauopathy-like Abnormalities and Neurologic Deficits in Mice Immunized with Neuronal Tau Protein. Arch. Neurol. 2006, 63, 1459–1467. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors Detected in the Enriched-aCSF (eCSF) | Concentration | |
|---|---|---|
| BDNF | 22.06 ± 4.83 pg/mL | |
| CNTF | 16.87 ± 12.50 pg/mL | |
| TGF-β | 14.13 ± 6.26 pg/mL | |
| Anti-oxidant capacity | Enzymatic + non-enzymatic | 0.46 ± 0.108 nmol/μL |
| Non-enzymatic only | 0.07 ± 0.012 nmol/μL | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valitsky, M.; Benhamron, S.; Nitzan, K.; Karussis, D.; Ella, E.; Abramsky, O.; Kassis, I.; Rosenmann, H. Cerebrospinal Fluid (CSF) Exchange with Artificial CSF Enriched with Mesenchymal Stem Cell Secretions Ameliorates Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci. 2019, 20, 1793. https://doi.org/10.3390/ijms20071793
Valitsky M, Benhamron S, Nitzan K, Karussis D, Ella E, Abramsky O, Kassis I, Rosenmann H. Cerebrospinal Fluid (CSF) Exchange with Artificial CSF Enriched with Mesenchymal Stem Cell Secretions Ameliorates Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences. 2019; 20(7):1793. https://doi.org/10.3390/ijms20071793
Chicago/Turabian StyleValitsky, Michael, Sandrine Benhamron, Keren Nitzan, Dimitrios Karussis, Ezra Ella, Oded Abramsky, Ibrahim Kassis, and Hanna Rosenmann. 2019. "Cerebrospinal Fluid (CSF) Exchange with Artificial CSF Enriched with Mesenchymal Stem Cell Secretions Ameliorates Experimental Autoimmune Encephalomyelitis" International Journal of Molecular Sciences 20, no. 7: 1793. https://doi.org/10.3390/ijms20071793
APA StyleValitsky, M., Benhamron, S., Nitzan, K., Karussis, D., Ella, E., Abramsky, O., Kassis, I., & Rosenmann, H. (2019). Cerebrospinal Fluid (CSF) Exchange with Artificial CSF Enriched with Mesenchymal Stem Cell Secretions Ameliorates Experimental Autoimmune Encephalomyelitis. International Journal of Molecular Sciences, 20(7), 1793. https://doi.org/10.3390/ijms20071793