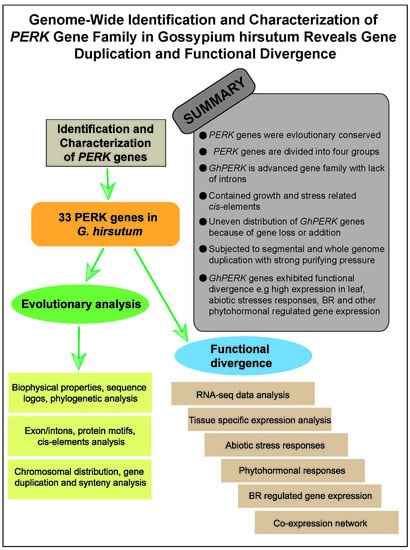
Genome-Wide Identification and Characterization of the PERK Gene Family in Gossypium hirsutum Reveals Gene Duplication and Functional Divergence

 , , and
, , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Identification of PERKs
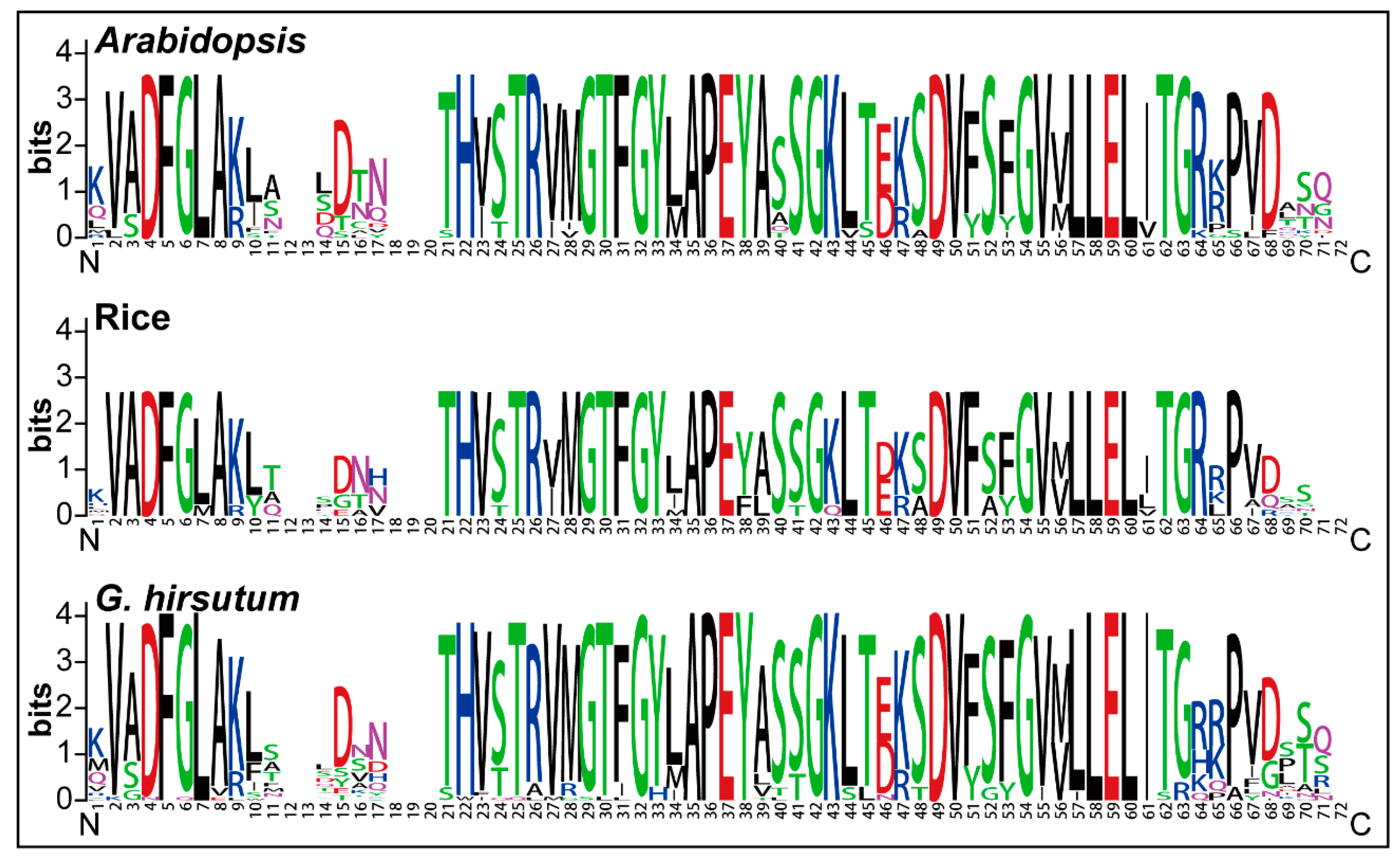
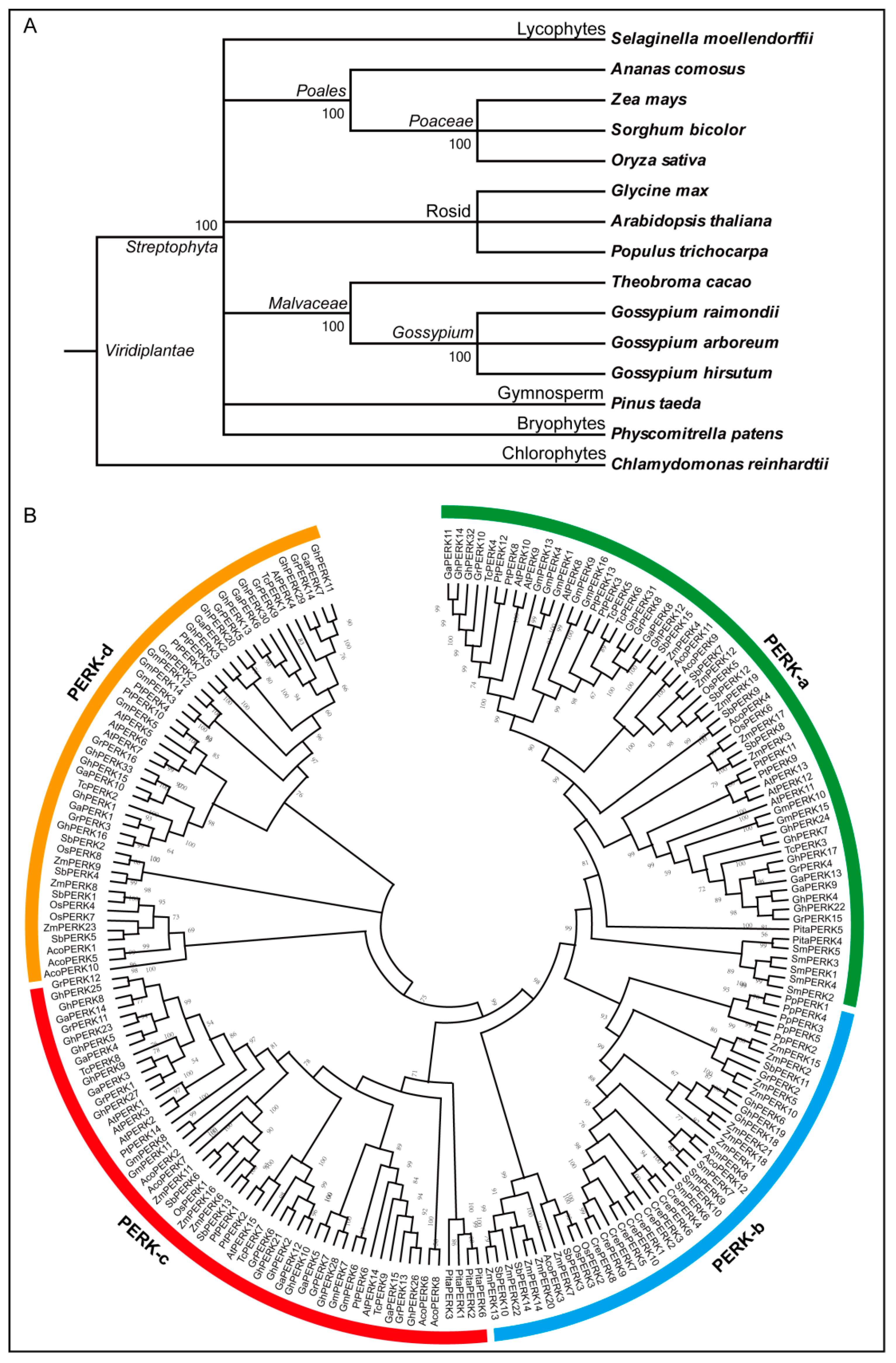
2.2. Sequence Logos and Phylogenetic Analysis
2.3. Exon/Intron, Protein Motif and cis-element Analysis
2.4. Chromosomal Distribution, Gene Duplication and Synteny Analysis
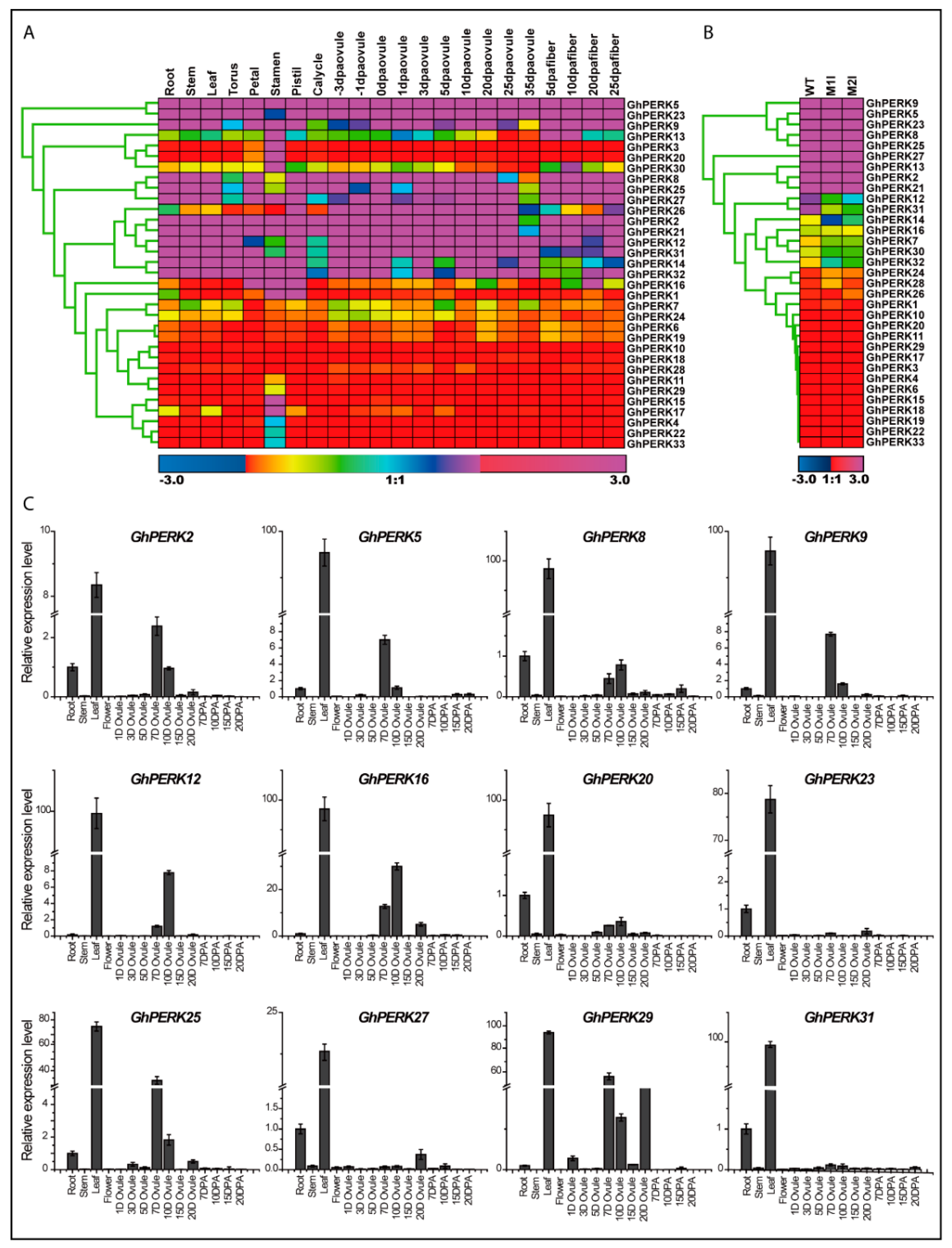
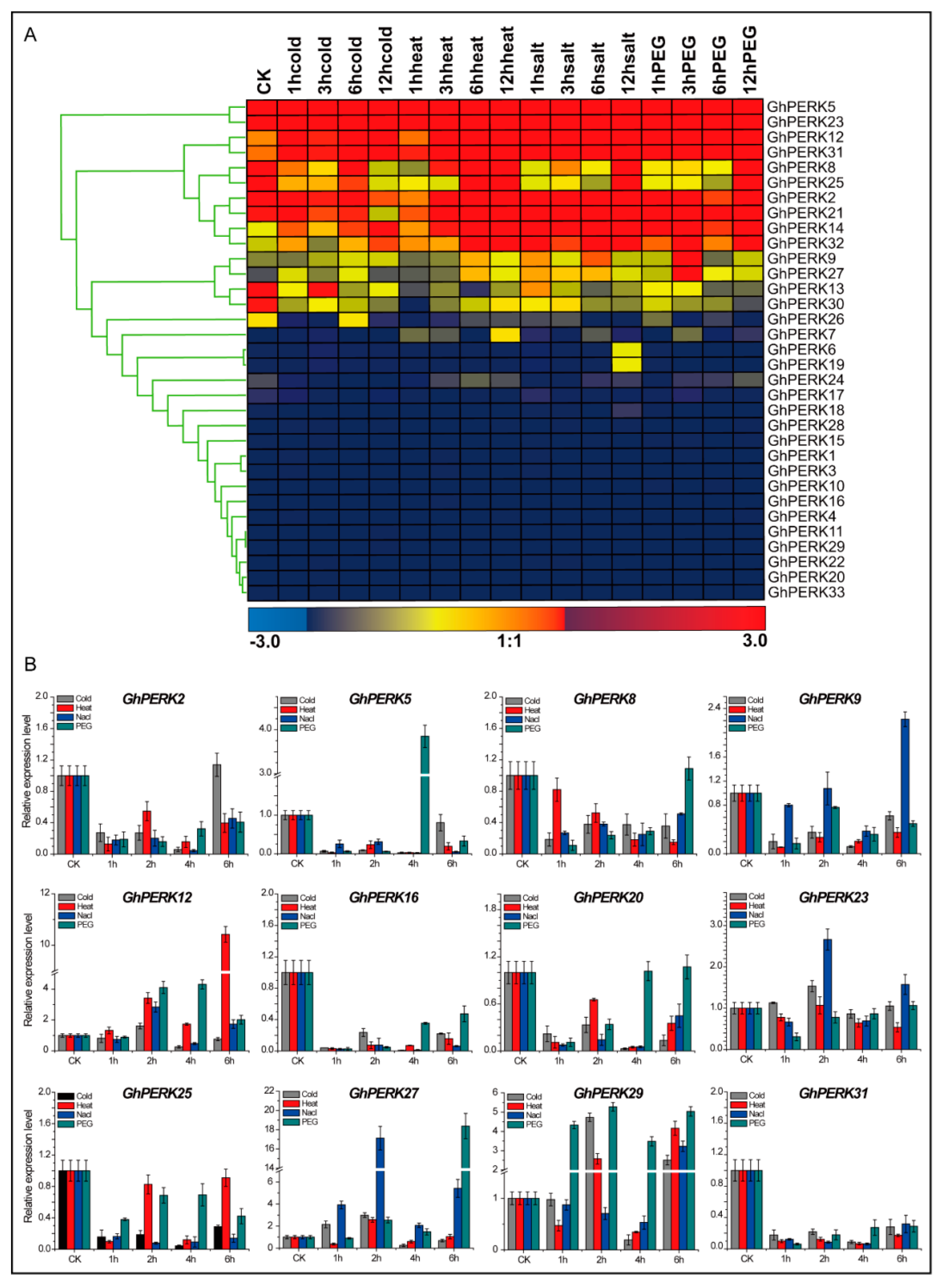
2.5. Tissue-Specific Gene Expression Patterns and Abiotic Stress Responses
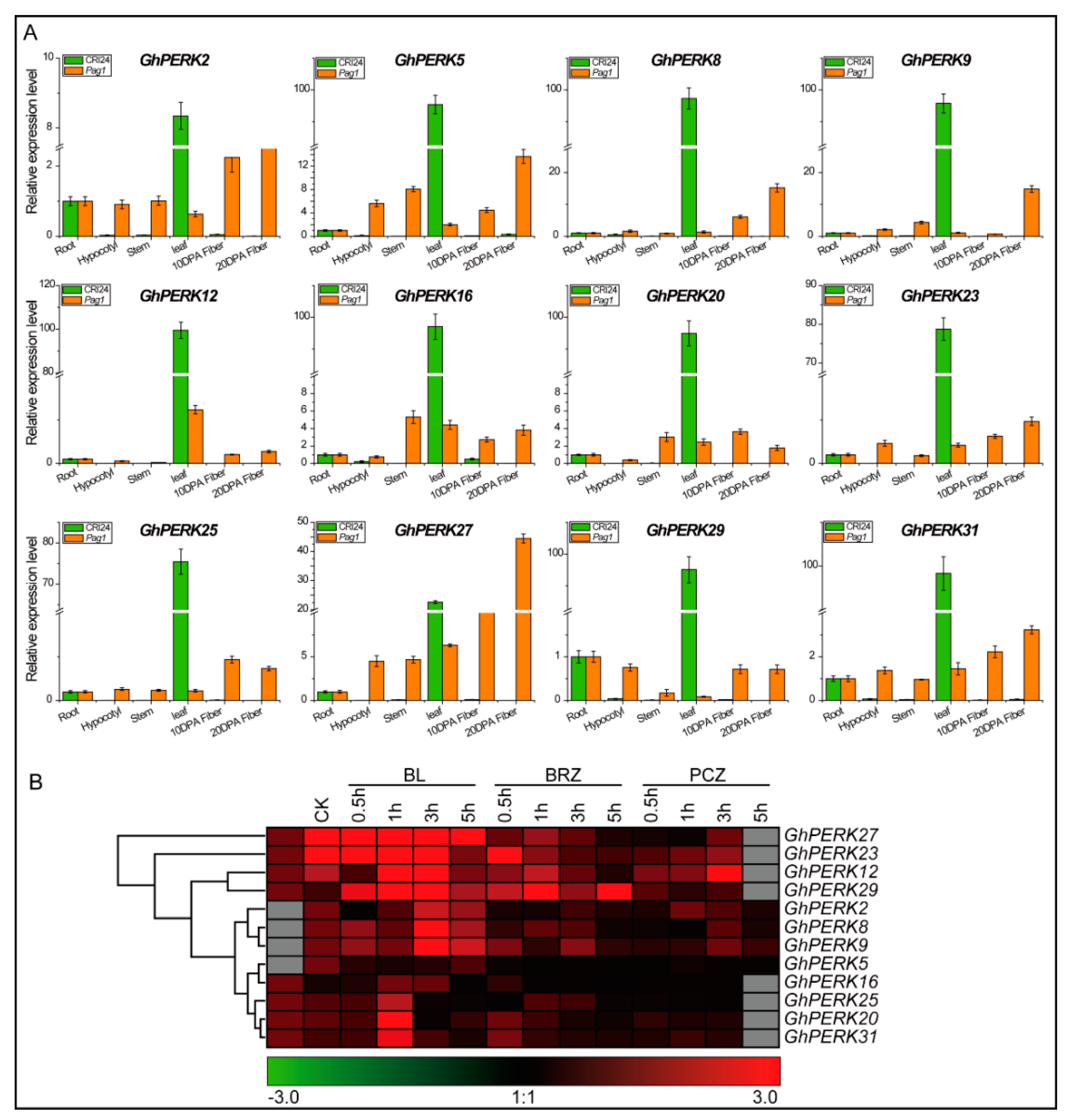
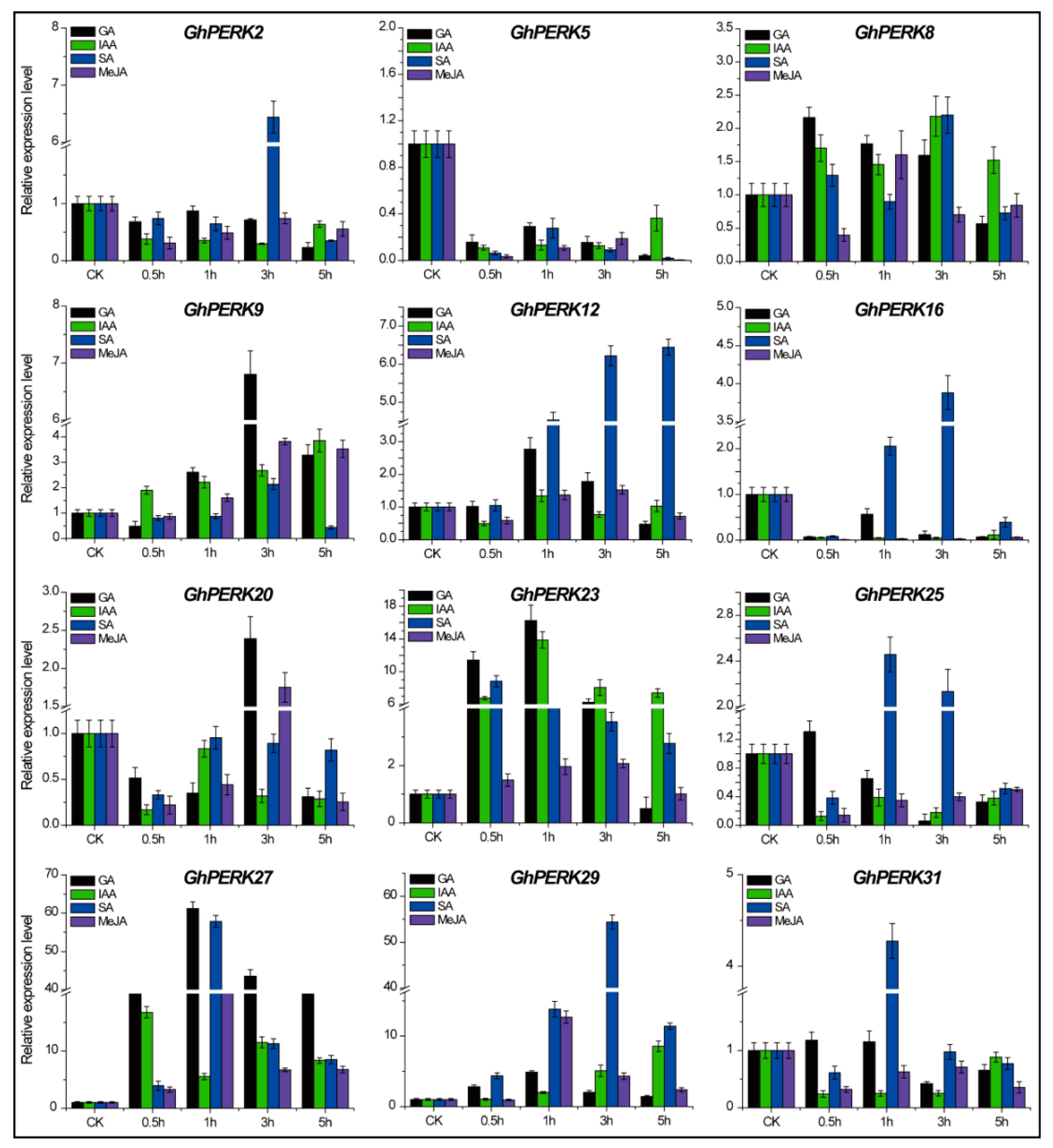
2.6. GhPERK Gene Expression in Response to Phytohormone Treatments
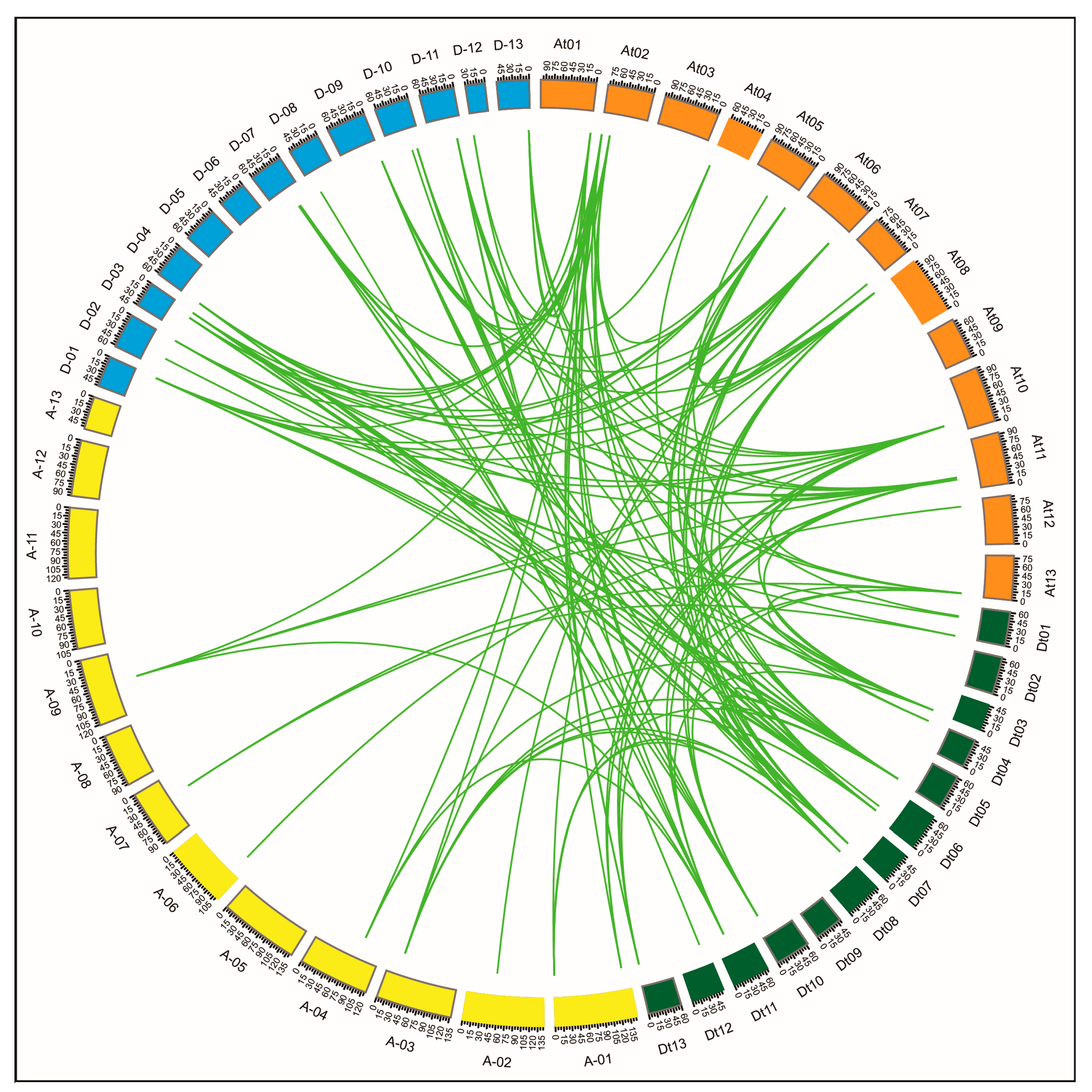
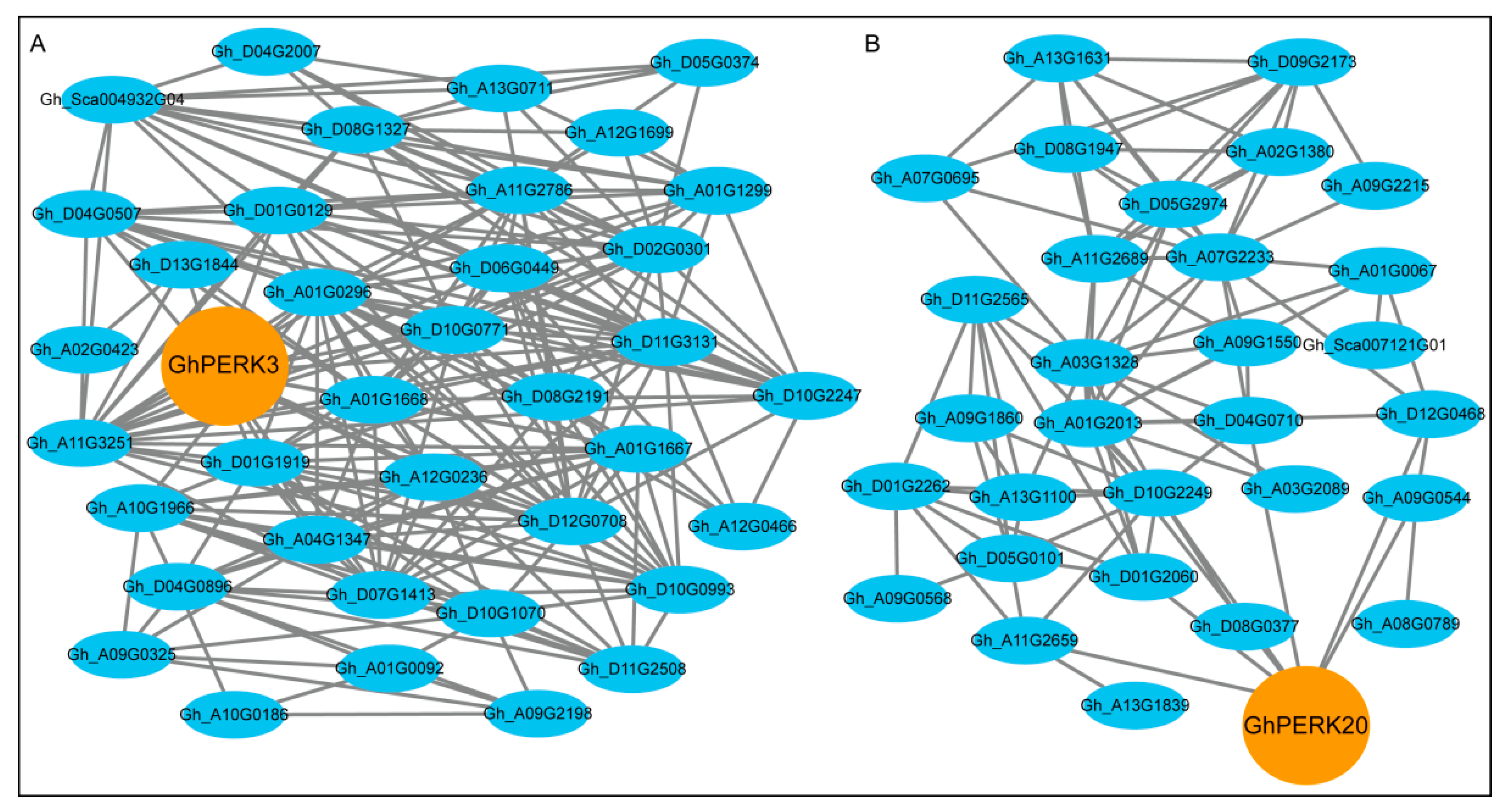
2.7. Regulatory Sub-Networks Involving GhPERK and Other G. hirsutum Genes
3. Discussion
3.1. GhPERK Genes Show Evolutionary Conservation
3.2. GhPERK Gene Family Duplication and Expansion
3.3. GhPERK Genes are Regulated by Abiotic and Hormonal Stresses
3.4. GhPERK co-expressed Genes Showed Functional Divergance
4. Materials and Methods
4.1. Identification of PERK Protein Family Members
4.2. Sequence Logos and Phylogenetic Analysis
4.3. Exon/Intron, Protein Motifs and Promoter cis-elements Analysis
4.4. Genomic Distribution, Gene Duplication and Synteny Analysis
4.5. Expression Analysis, Stress Treatments, qRT-PCR and Co-expression Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NaCl | Sodium chloride |
| PEG | Polyethylene glycol |
| GA | Gibberellic acid |
| IAA | Indole-3-acetic acid |
| BL | Brassinolide |
| BRZ | Brassinazole |
| PCZ | Propiconazole |
| MeJA | Methyl jasmonate |
| SA | Salicylic acid |
| DPA | Days post-anthesis |
| µM | Micro-molar |
| Mya | Million years ago |
| G. arboreum | Gossypium arboreum |
| G. hirsutum | Gossypium hirsutum |
| G. raimondii | Gossypium raimondii |
| Z. mays | Zea mays |
| O. sativa | Oryza sativa |
| G. max | Glycine max |
| A. thaliana | Arabidopsis thaliana |
| P. trichocarpa | Populus trichocarpa |
| T. cacao | Theobroma cacao |
| S. bicolor | Sorghum bicolor |
References
- Morris, E.R.; Walker, J.C. Receptor-like protein kinases: The keys to response. Curr. Opin. Plant Biol. 2003, 6, 339–342. [Google Scholar] [CrossRef]
- Shiu, S.H.; Bleecker, A.B. Receptor-like kinases from arabidopsis form a monophyletic gene family related to animal receptor kinases. Proc. Natl. Acad. Sci. USA 2001, 98, 10763–10768. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Karlowski, W.M.; Pan, R.; Tzeng, Y.H.; Mayer, K.F.; Li, W.H. Comparative analysis of the receptor-like kinase family in arabidopsis and rice. Plant Cell 2004, 16, 1220–1234. [Google Scholar] [CrossRef] [PubMed]
- Dievart, A.; Clark, S.E. Lrr-containing receptors regulating plant development and defense. Development 2004, 131, 251–261. [Google Scholar] [CrossRef]
- Shiu, S.H.; Bleecker, A.B. Plant receptor-like kinase gene family: Diversity, function and signaling. Sci. STKE 2001, 2001, re22. [Google Scholar] [CrossRef] [PubMed]
- Shiu, S.H.; Bleecker, A.B. Expansion of the receptor-like kinase/pelle gene family and receptor-like proteins in arabidopsis. Plant Physiol. 2003, 132, 530–543. [Google Scholar]
- Li, J.; Chory, J. A putative leucine-rich repeat receptor kinase involved in brassinosteroid signal transduction. Cell 1997, 90, 929–938. [Google Scholar] [CrossRef]
- Li, J.; Wen, J.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. Bak1, an arabidopsis lrr receptor-like protein kinase, interacts with bri1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef]
- Nam, K.H.; Li, J. Bri1/bak1, a receptor kinase pair mediating brassinosteroid signaling. Cell 2002, 110, 203–212. [Google Scholar] [CrossRef]
- Champion, A.; Kreis, M.; Mockaitis, K.; Picaud, A.; Henry, Y. Arabidopsis kinome: After the casting. Funct. Integr. Genom. 2004, 4, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Shpak, E.D.; Berthiaume, C.T.; Hill, E.J.; Torii, K.U. Synergistic interaction of three erecta-family receptor-like kinases controls arabidopsis organ growth and flower development by promoting cell proliferation. Development 2004, 131, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Dievart, A.; Dalal, M.; Tax, F.E.; Lacey, A.D.; Huttly, A.; Li, J.; Clark, S.E. CLAVATA1 dominant-negative alleles reveal functional overlap between multiple receptor kinases that regulate meristem and organ development. Plant Cell 2003, 15, 1198–1211. [Google Scholar] [CrossRef]
- Shpak, E.D.; Lakeman, M.B.; Torii, K.U. Dominant-negative receptor uncovers redundancy in the arabidopsis erecta leucine-rich repeat receptor-like kinase signaling pathway that regulates organ shape. Plant Cell 2003, 15, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.F.; Goring, D.R. The proline-rich, extensin-like receptor kinase-1 (perk1) gene is rapidly induced by wounding. Plant Mol. Biol. 2002, 50, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Nakhamchik, A.; Zhao, Z.; Provart, N.J.; Shiu, S.H.; Keatley, S.K.; Cameron, R.K.; Goring, D.R. A comprehensive expression analysis of the arabidopsis proline-rich extensin-like receptor kinase gene family using bioinformatic and experimental approaches. Plant Cell Physiol. 2004, 45, 1875–1881. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, G.; Zhou, Y.; Zhang, Z.; Wang, W.; Du, Y.; Wu, Z.; Song, C.P. Plasma membrane-associated proline-rich extensin-like receptor kinase 4, a novel regulator of ca signalling, is required for abscisic acid responses in arabidopsis thaliana. Plant J. 2009, 60, 314–327. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, Y.; Song, C.P. Arabidopsis proline-rich extensin-like receptor kinase 4 modulates the early event toward abscisic acid response in root tip growth. Plant Signal. Behav. 2009, 4, 1075–1077. [Google Scholar] [CrossRef]
- Kim, H.J.; Triplett, B.A. Cotton fiber germin-like protein. I. Molecular cloning and gene expression. Planta 2004, 218, 516–524. [Google Scholar]
- Sun, Y.; Veerabomma, S.; Abdel-Mageed, H.A.; Fokar, M.; Asami, T.; Yoshida, S.; Allen, R.D. Brassinosteroid regulates fiber development on cultured cotton ovules. Plant Cell Physiol. 2005, 46, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.R.; Zhang, C.J.; Yang, X.J.; Liu, K.; Wu, Z.X.; Zhang, X.Y.; Zheng, W.; Xun, Q.Q.; Liu, C.L.; Lu, L.L.; et al. Pag1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol. 2014, 203, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Du, X.M.; Huang, G.; He, S.P.; Yang, Z.E.; Sun, G.F.; Ma, X.F.; Li, N.; Zhang, X.Y.; Sun, J.L.; Liu, M.; et al. Resequencing of 243 diploid cotton accessions based on an updated a genome identifies the genetic basis of key agronomic traits. Nat. Genet. 2018, 50, 796–802. [Google Scholar] [CrossRef]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef]
- Wang, M.; Wang, P.; Lin, M.; Ye, Z.; Li, G.; Tu, L.; Shen, C.; Li, J.; Yang, Q.; Zhang, X. Evolutionary dynamics of 3d genome architecture following polyploidization in cotton. Nat. Plants 2018, 4, 90–97. [Google Scholar] [CrossRef]
- Zhang, T.Z.; Hu, Y.; Jiang, W.K.; Fang, L.; Guan, X.Y.; Chen, J.D.; Zhang, J.B.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Li, F.G.; Fan, G.Y.; Wang, K.B.; Sun, F.M.; Yuan, Y.L.; Song, G.L.; Li, Q.; Ma, Z.Y.; Lu, C.R.; Zou, C.S.; et al. Genome sequence of the cultivated cotton gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.C.; Llewellyn, D.; Showmaker, K.C.; Shu, S.Q.; Udall, J.; et al. Repeated polyploidization of gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Korenaga, T. Plant cis-acting regulatory DNA elements (place) database: 1999. Nucleic Acids Res. 1999, 27, 297–300. [Google Scholar] [CrossRef]
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003, 78, 139–186. [Google Scholar]
- Ruprecht, C.; Proost, S.; Hernandez-Coronado, M.; Ortiz-Ramirez, C.; Lang, D.; Rensing, S.A.; Becker, J.D.; Vandepoele, K.; Mutwil, M. Phylogenomic analysis of gene co-expression networks reveals the evolution of functional modules. Plant J. 2017, 90, 447–465. [Google Scholar] [CrossRef]
- Bao, Y.; Hu, G.J.; Flagel, L.E.; Salmon, A.; Bezanilla, M.; Paterson, A.H.; Wang, Z.N.; Wendel, J.F. Parallel up-regulation of the profilin gene family following independent domestication of diploid and allopolyploid cotton (Gossypium). Proc. Natl. Acad. Sci. USA 2011, 108, 21152–21157. [Google Scholar] [CrossRef]
- Yang, Z.E.; Gong, Q.; Wang, L.L.; Jin, Y.Y.; Xi, J.P.; Li, Z.; Qin, W.Q.; Yang, Z.R.; Lu, L.L.; Chen, Q.J.; et al. Genome-wide study of yabby genes in upland cotton and their expression patterns under different stresses. Front. Genet. 2018, 9, 33. [Google Scholar] [CrossRef]
- Maestrini, P.; Cavallini, A.; Rizzo, M.; Giordani, T.; Bernardi, R.; Durante, M.; Natali, L. Isolation and expression analysis of low temperature-induced genes in white poplar (Populus alba). J. Plant Physiol. 2009, 166, 1544–1556. [Google Scholar] [CrossRef]
- Roy, S.W.; Gilbert, W. The evolution of spliceosomal introns: Patterns, puzzles and progress. Nat. Rev. Genet. 2006, 7, 211–221. [Google Scholar]
- Serrano, M.; Parra, S.; Alcaraz, L.D.; Guzman, P. The ATL gene family from Arabidopsis thaliana and Oryza sativa comprises a large number of putative ubiquitin ligases of the ring-h2 type. J. Mol. Evol. 2006, 62, 434–445. [Google Scholar] [CrossRef]
- Roy, S.W.; Gilbert, W. Complex early genes. Proc. Natl. Acad. Sci. USA 2005, 102, 1986–1991. [Google Scholar] [CrossRef]
- Chothia, C.; Gough, J.; Vogel, C.; Teichmann, S.A. Evolution of the protein repertoire. Science 2003, 300, 1701–1703. [Google Scholar] [CrossRef]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938–950. [Google Scholar] [CrossRef]
- Yang, S.H.; Zhang, X.H.; Yue, J.X.; Tian, D.C.; Chen, J.Q. Recent duplications dominate nbs-encoding gene expansion in two woody species. Mol. Genet. Genom. 2008, 280, 187–198. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Higo, K.; Ugawa, Y.; Iwamoto, M.; Higo, H. PLACE: A database of plant cis-acting regulatory DNA elements. Nucleic Acids Res. 1998, 26, 358–359. [Google Scholar] [CrossRef]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef]
- Prince, V.E.; Pickett, F.B. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 2002, 3, 827–837. [Google Scholar] [CrossRef]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D. Rna-seq analysis of short fiber mutants ligon-lintless-1 (li 1) and –2 (li 2) revealed important role of aquaporins in cotton (Gossypium hirsutum L.) fiber elongation. BMC Plant Biol. 2015, 15, 65. [Google Scholar] [CrossRef]
- Aoki, K.; Ogata, Y.; Shibata, D. Approaches for extracting practical information from gene co-expression networks in plant biology. Plant Cell Physiol. 2007, 48, 381–390. [Google Scholar] [CrossRef]
- Yang, Z.E.; Gong, Q.; Qin, W.Q.; Yang, Z.R.; Cheng, Y.; Lu, L.L.; Ge, X.Y.; Zhang, C.J.; Wu, Z.X.; Li, F.G. Genome-wide analysis of wox genes in upland cotton and their expression pattern under different stresses. Bmc Plant Biol. 2017, 17, 113. [Google Scholar] [CrossRef]
- Fankhauser, C.; Chory, J. Light control of plant development. Annu. Rev. Cell Dev. Biol. 1997, 13, 203–229. [Google Scholar] [CrossRef]
- Wen, F.; Zhu, H.; Li, P.; Jiang, M.; Mao, W.Q.; Ong, C.; Chu, Z.Q. Genome-wide evolutionary characterization and expression analyses of wrky family genes in brachypodium distachyon. DNA Res. 2014, 21, 327–339. [Google Scholar] [CrossRef]
- Song, C.P.; Agarwal, M.; Ohta, M.; Guo, Y.; Halfter, U.; Wang, P.C.; Zhu, J.K. Role of an arabidopsis ap2/erebp-type transcriptional repressor in abscisic acid and drought stress responses. Plant Cell 2005, 17, 2384–2396. [Google Scholar] [CrossRef]
- Narusaka, Y.; Nakashima, K.; Shinwari, Z.K.; Sakuma, Y.; Furihata, T.; Abe, H.; Narusaka, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Interaction between two cis-acting elements, abre and dre, in aba-dependent expression of Arabidopsis rd29A gene in response to dehydration and high-salinity stresses. Plant J. 2003, 34, 137–148. [Google Scholar] [CrossRef]
- Diaz-Martin, J.; Almoguera, C.N.; Prieto-Dapena, P.; Espinosa, J.M.; Jordano, J. Functional interaction between two transcription factors involved in the developmental regulation of a small heat stress protein gene promoter. Plant Physiol. 2005, 139, 1483–1494. [Google Scholar] [CrossRef]
- Singh, K.B.; Foley, R.C.; Onate-Sanchez, L. Transcription factors in plant defense and stress responses. Curr. Opin. Plant Biol. 2002, 5, 430–436. [Google Scholar] [CrossRef]
- Qanmber, G.; Yu, D.; Li, J.; Wang, L.; Ma, S.; Lu, L.; Yang, Z.; Li, F. Genome-wide identification and expression analysis of gossypium ring-h2 finger e3 ligase genes revealed their roles in fiber development and phytohormone and abiotic stress responses. J. Cotton Res. 2018, 1, 1. [Google Scholar] [CrossRef]
- Pandey, A.; Misra, P.; Alok, A.; Kaur, N.; Sharma, S.; Lakhwani, D.; Asif, M.H.; Tiwari, S.; Trivedi, P.K. Genome-wide identification and expression analysis of homeodomain leucine zipper subfamily iv (hdz iv) gene family from musa accuminata. Front. Plant Sci. 2016, 7, 20. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Yang, Z.E.; Chen, E.Y.; Zhang, C.J.; Zhang, X.Y.; Li, F.G. Genome-wide analysis of gras transcription factor gene family in Gossypium hirsutum L. BMC Genom. 2018, 19, 348. [Google Scholar] [CrossRef]
- Lecharny, A.; Boudet, N.; Gy, I.; Aubourg, S.; Kreis, M. Introns in, introns out in plant gene families: A genomic approach of the dynamics of gene structure. J. Struct. Funct. Genom. 2003, 3, 111–116. [Google Scholar] [CrossRef]
- Roy, S.W.; Penny, D. A very high fraction of unique intron positions in the intron-rich diatom thalassiosira pseudonana indicates widespread intron gain. Mol. Biol. Evol. 2007, 24, 1447–1457. [Google Scholar] [CrossRef]
- Iwamoto, M.; Maekawa, M.; Saito, A.; Higo, H.; Higo, K. Evolutionary relationship of plant catalase genes inferred from exon-intron structures: Isozyme divergence after the separation of monocots and dicots. Theor. Appl. Genet. 1998, 97, 9–19. [Google Scholar] [CrossRef]
- Frugoli, J.A.; McPeek, M.A.; Thomas, T.L.; McClung, C.R. Intron loss and gain during evolution of the catalase gene family in angiosperms. Genetics 1998, 149, 355–365. [Google Scholar]
- Tiwari, S.C.; Wilkins, T.A. Cotton (Gossypium hirsutum) seed trichomes expand via diffuse growing mechanism. Can. J. Bot.-Rev. Can. De Bot. 1995, 73, 746–757. [Google Scholar] [CrossRef]
- He, H.S.; Dong, Q.; Shao, Y.H.; Jiang, H.Y.; Zhu, S.W.; Cheng, B.J.; Xiang, Y. Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 2012, 31, 1199–1217. [Google Scholar] [CrossRef]
- Schauser, L.; Wieloch, W.; Stougaard, J. Evolution of nin-like proteins in arabidopsis, rice and lotus japonicus. J. Mol. Evol. 2005, 60, 229–237. [Google Scholar] [CrossRef]
- Barakat, A.; Bagniewska-Zadworna, A.; Choi, A.; Plakkat, U.; DiLoreto, D.S.; Yellanki, P.; Carlson, J.E. The cinnamyl alcohol dehydrogenase gene family in populus: Phylogeny, organization and expression. BMC Plant Biol. 2009, 9, 26. [Google Scholar] [CrossRef]
- Wang, Z.C.; Zhang, H.Z.; Yang, J.L.; Chen, Y.L.; Xu, X.M.; Mao, X.L.; Li, C.H. Phylogenetic, expression and bioinformatic analysis of the ABC1 gene family in Populus trichocarpa. Sci. World J. 2013, 2013, 785070. [Google Scholar]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef]
- Ramsey, J.; Schemske, D.W. Pathways, mechanisms and rates of polyploid formation in flowering plants. Annu. Rev. Ecol. Syst. 1998, 29, 467–501. [Google Scholar] [CrossRef]
- Gaeta, R.T.; Pires, J.C.; Iniguez-Luy, F.; Leon, E.; Osborn, T.C. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 2007, 19, 3403–3417. [Google Scholar] [CrossRef]
- Woodhouse, M.R.; Schnable, J.C.; Pedersen, B.S.; Lyons, E.; Lisch, D.; Subramaniam, S.; Freeling, M. Following tetraploidy in maize, a short deletion mechanism removed genes preferentially from one of the two homologs. PLoS Biol. 2010, 8, e1000409. [Google Scholar] [CrossRef]
- Baumberger, N.; Doesseger, B.; Guyot, R.; Diet, A.; Parsons, R.L.; Clark, M.A.; Simmons, M.P.; Bedinger, P.; Goff, S.A.; Ringli, C.; et al. Whole-genome comparison of leucine-rich repeat extensins in arabidopsis and rice. A conserved family of cell wall proteins form a vegetative and a reproductive clade. Plant Physiol. 2003, 131, 1313–1326. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Y.H.; Wu, C.G.; Yang, G.D.; Li, Y.Y.; Zheng, C.C. Genome-wide analysis of ccch zinc finger family in arabidopsis and rice. BMC Genom. 2008, 9, 44. [Google Scholar] [CrossRef]
- Dossa, K.; Diouf, D.; Cisse, N. Genome-wide investigation of hsf genes in sesame reveals their segmental duplication expansion and their active role in drought stress response. Front. Plant Sci. 2016, 7, 1522. [Google Scholar] [CrossRef]
- Yin, G.J.; Xu, H.L.; Xiao, S.Y.; Qin, Y.J.; Li, Y.X.; Yan, Y.M.; Hu, Y.K. The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 2013, 13, 148. [Google Scholar] [CrossRef]
- Ren, Z.; Yu, D.; Yang, Z.; Li, C.; Qanmber, G.; Li, Y.; Li, J.; Liu, Z.; Lu, L.; Wang, L.; et al. Genome-wide identification of the MIKC-type MADS-box gene family in Gossypium hirsutum L. Unravels their roles in flowering. Front Plant Sci 2017, 8, 384. [Google Scholar] [CrossRef]
- Charon, C.; Bruggeman, Q.; Thareau, V.; Henry, Y. Gene duplication within the green lineage: The case of tel genes. J. Exp. Bot. 2012, 63, 5061–5077. [Google Scholar] [CrossRef]
- Wang, X.W.; Wang, H.Z.; Wang, J.; Sun, R.F.; Wu, J.; Liu, S.Y.; Bai, Y.Q.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Letunic, I.; Doerks, T.; Bork, P. Smart: Recent updates, new developments and status in 2015. Nucleic Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. Interproscan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. Meme: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Li, J.; Yu, D.; Qanmber, G.; Lu, L.; Wang, L.; Zheng, L.; Liu, Z.; Wu, H.; Liu, X.; Chen, Q.; et al. GhKLCR1, a kinesin light chain-related gene, induces drought-stress sensitivity in Arabidopsis. Sci. China Life Sci. 2018, 62, 63–75. [Google Scholar] [CrossRef]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. Plantcare, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Jia, J.T.; Zhao, P.C.; Cheng, L.Q.; Yuan, G.X.; Yang, W.G.; Liu, S.; Chen, S.Y.; Qi, D.M.; Liu, G.S.; Li, X.X. Mads-box family genes in sheepgrass and their involvement in abiotic stress responses. BMC Plant Biol. 2018, 18, 42. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Suyama, M.; Torrents, D.; Bork, P. Pal2nal: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef]
- Yang, Z.H. Paml 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- You, Q.; Xu, W.; Zhang, K.; Zhang, L.; Yi, X.; Yao, D.; Wang, C.; Zhang, X.; Zhao, X.; Provart, N.J.; et al. ccNET: Database of co-expression networks with functional modules for diploid and polyploid Gossypium. Nucleic Acids Res. 2017, 45, D1090–D1099. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qanmber, G.; Liu, J.; Yu, D.; Liu, Z.; Lu, L.; Mo, H.; Ma, S.; Wang, Z.; Yang, Z. Genome-Wide Identification and Characterization of the PERK Gene Family in Gossypium hirsutum Reveals Gene Duplication and Functional Divergence. Int. J. Mol. Sci. 2019, 20, 1750. https://doi.org/10.3390/ijms20071750
Qanmber G, Liu J, Yu D, Liu Z, Lu L, Mo H, Ma S, Wang Z, Yang Z. Genome-Wide Identification and Characterization of the PERK Gene Family in Gossypium hirsutum Reveals Gene Duplication and Functional Divergence. International Journal of Molecular Sciences. 2019; 20(7):1750. https://doi.org/10.3390/ijms20071750
Chicago/Turabian StyleQanmber, Ghulam, Ji Liu, Daoqian Yu, Zhao Liu, Lili Lu, Huijuan Mo, Shuya Ma, Zhi Wang, and Zuoren Yang. 2019. "Genome-Wide Identification and Characterization of the PERK Gene Family in Gossypium hirsutum Reveals Gene Duplication and Functional Divergence" International Journal of Molecular Sciences 20, no. 7: 1750. https://doi.org/10.3390/ijms20071750
APA StyleQanmber, G., Liu, J., Yu, D., Liu, Z., Lu, L., Mo, H., Ma, S., Wang, Z., & Yang, Z. (2019). Genome-Wide Identification and Characterization of the PERK Gene Family in Gossypium hirsutum Reveals Gene Duplication and Functional Divergence. International Journal of Molecular Sciences, 20(7), 1750. https://doi.org/10.3390/ijms20071750