Transcriptional Profile Corroborates that bml Mutant Plays likely Role in Premature Leaf Senescence of Rice (Oryza sativa L.)

Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization
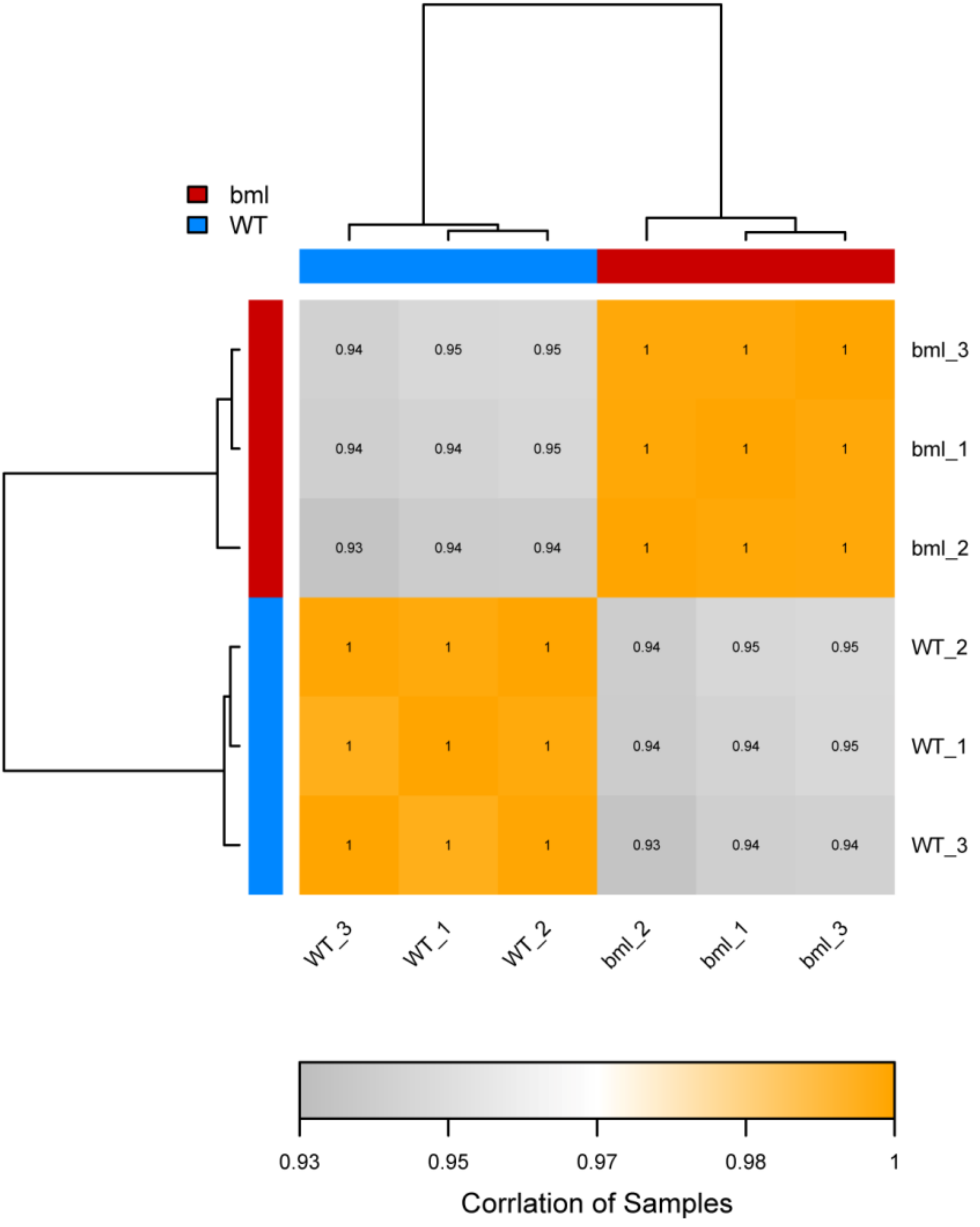
2.2. RNA-Seq Analysis
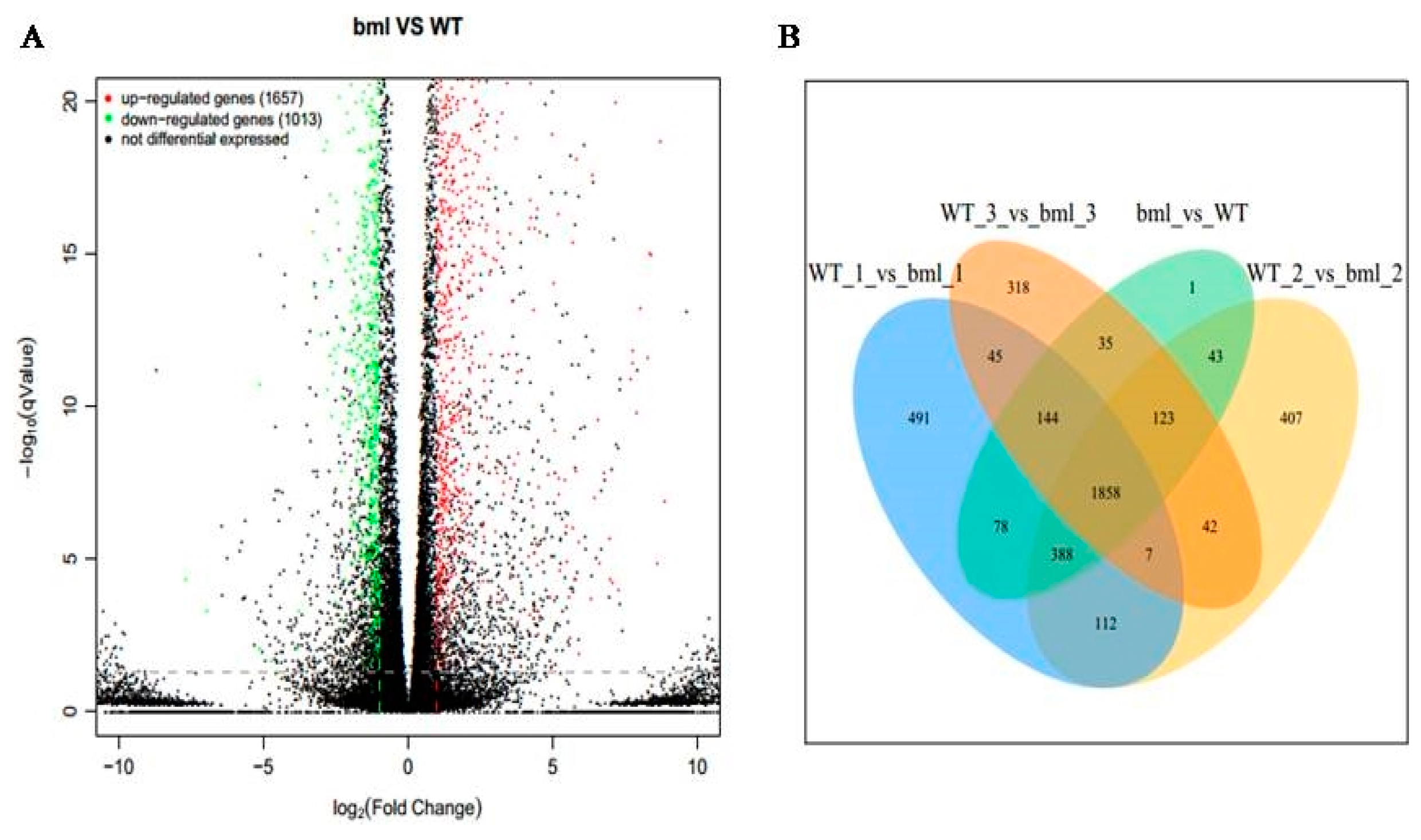
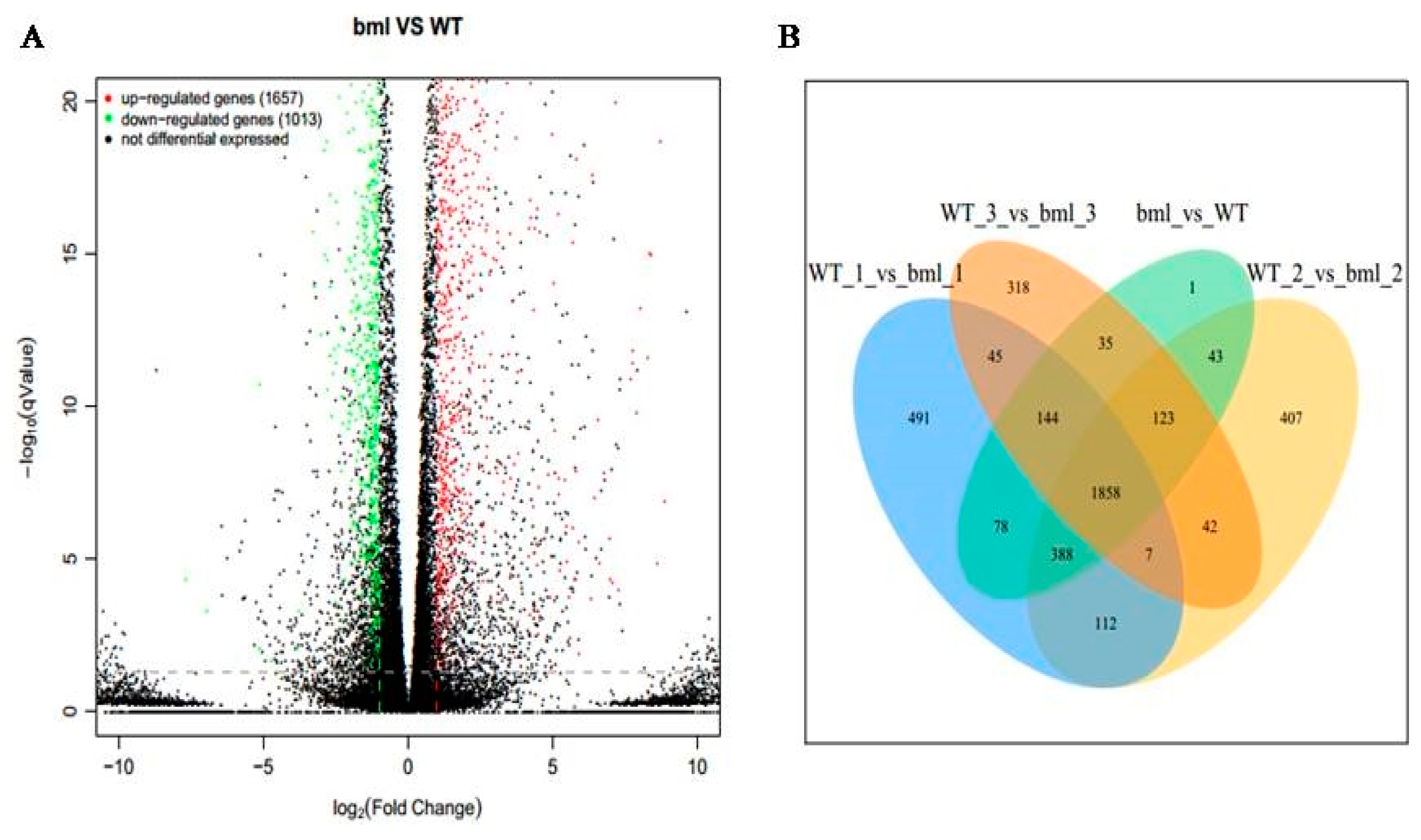
2.3. Identifications of DEGs
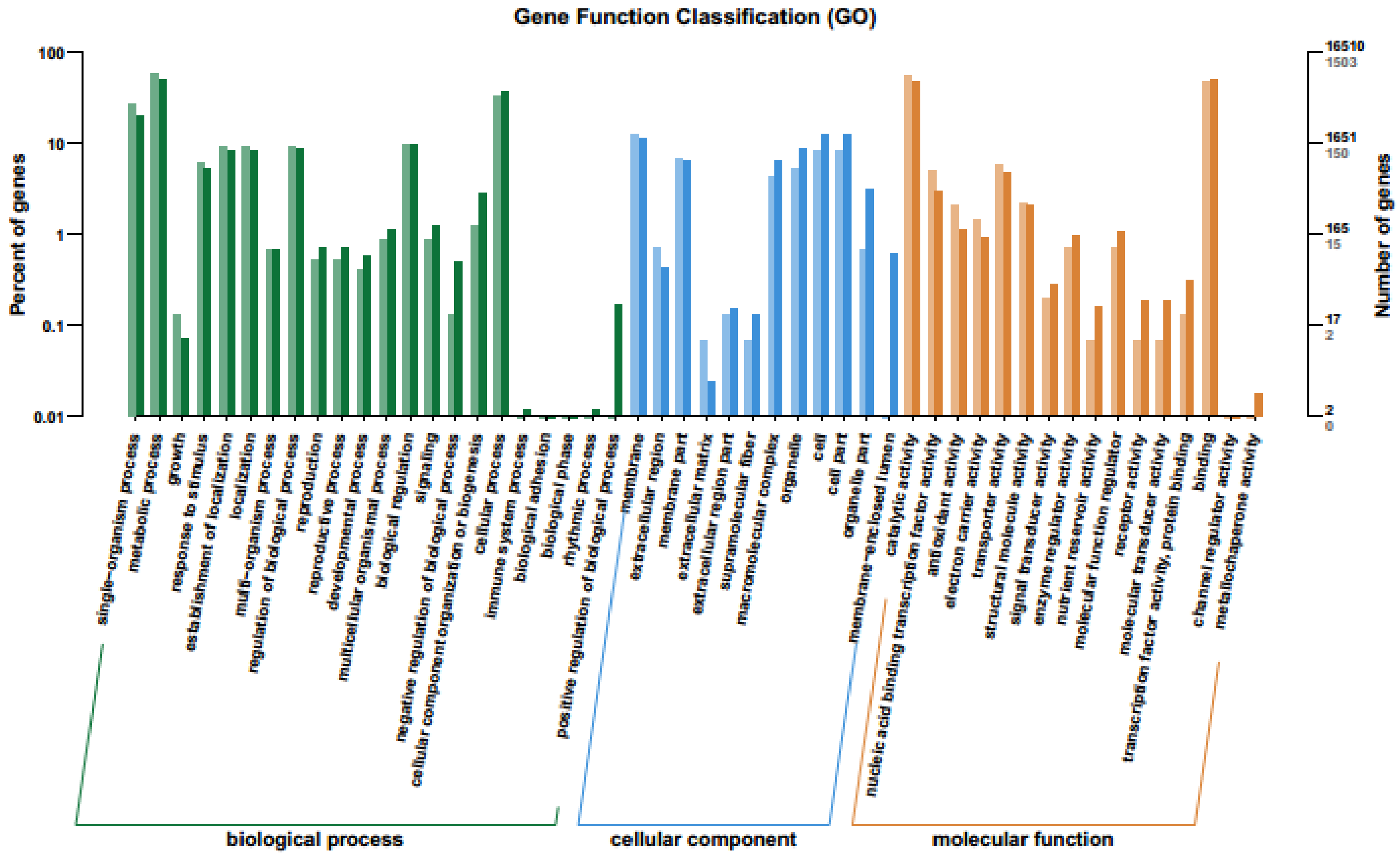
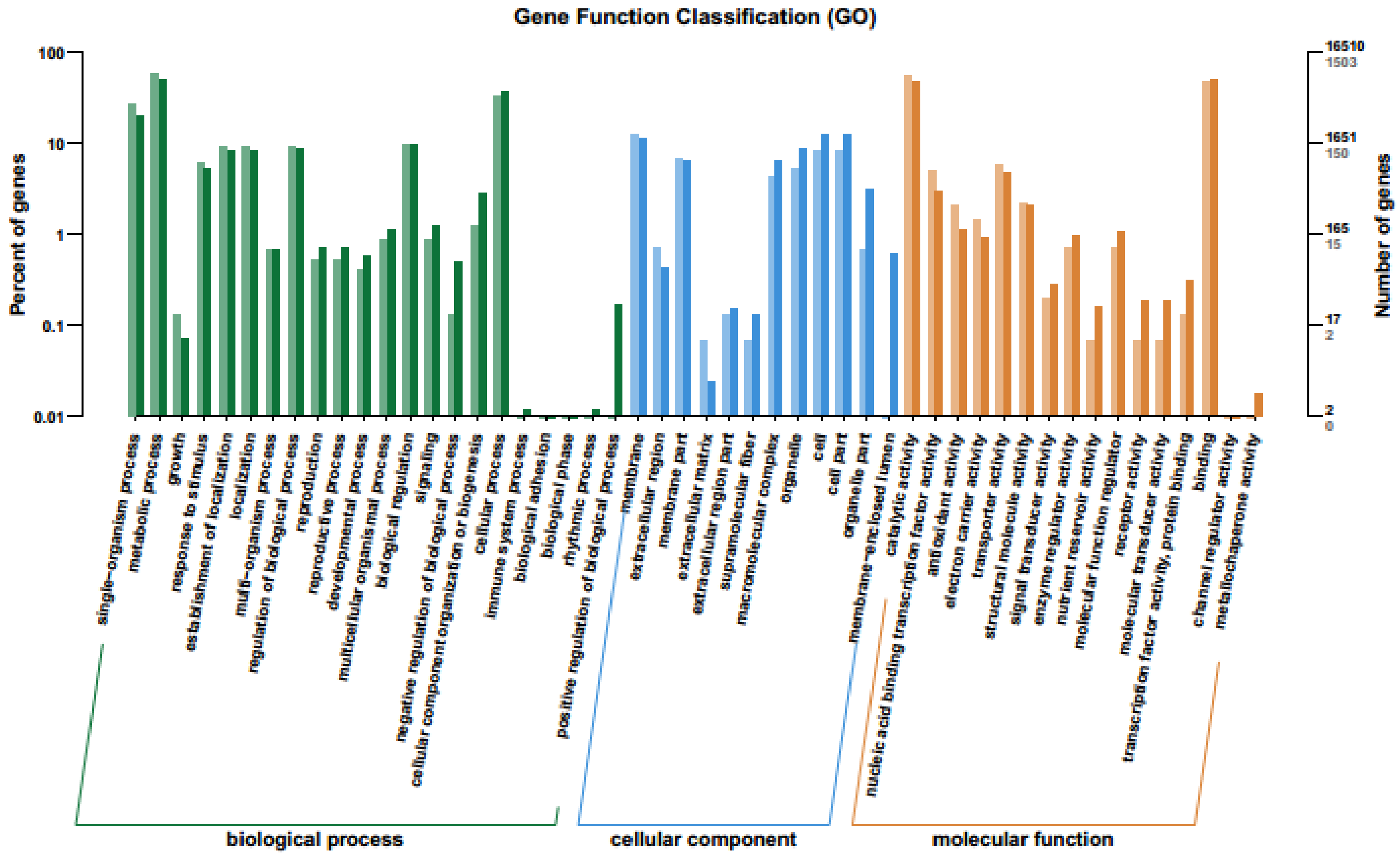
2.4. GO Enrichment Analysis
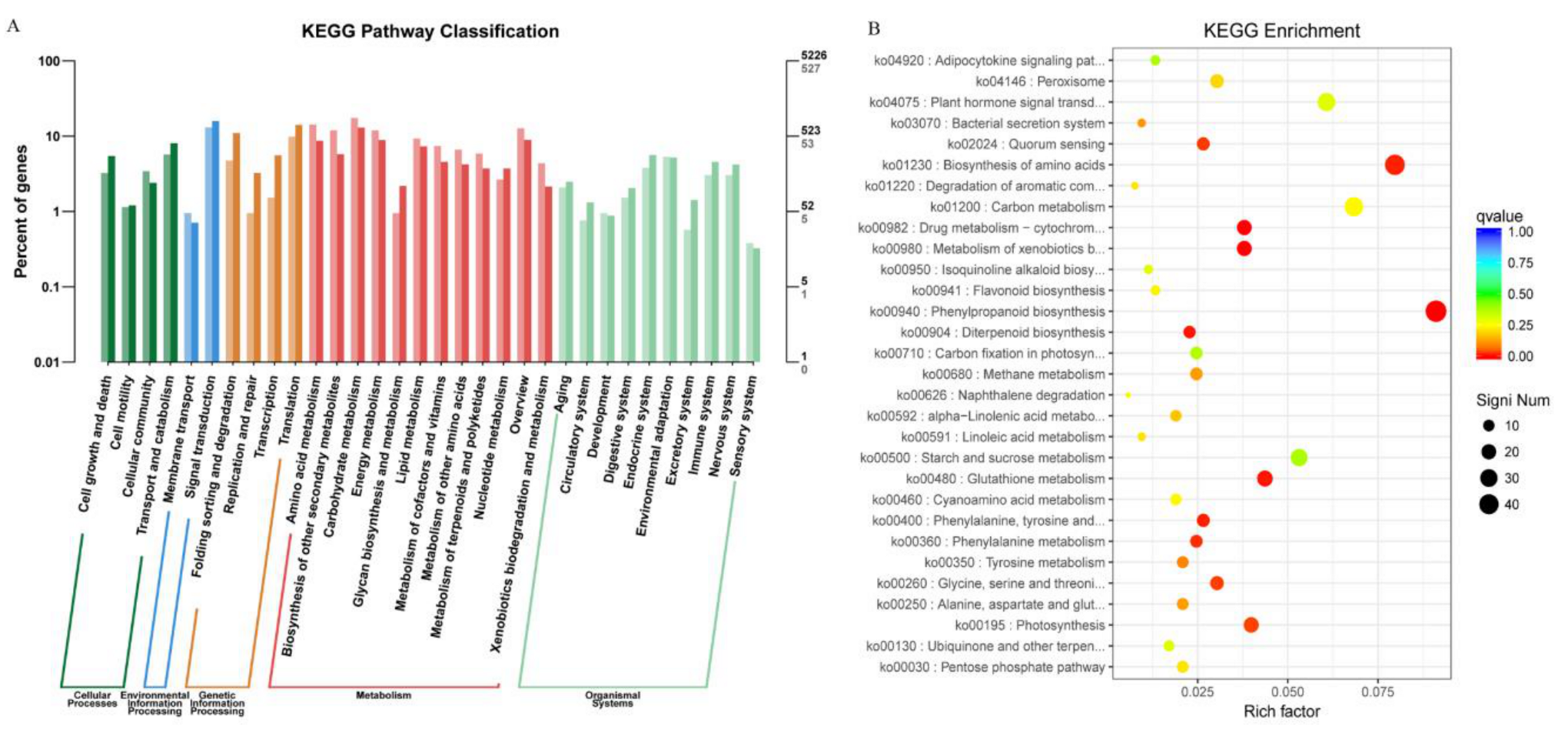
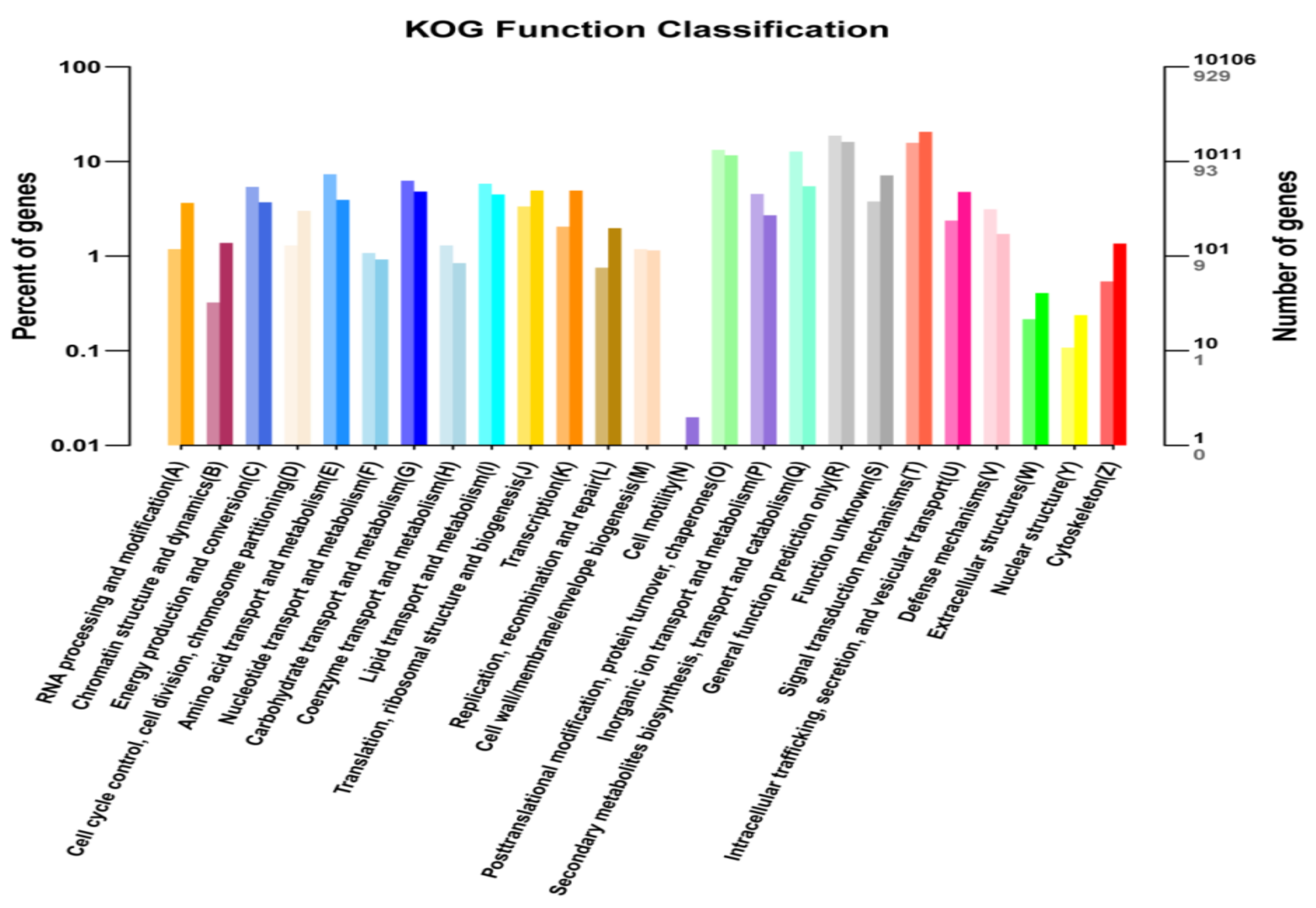
2.5. KEGG and KOG Pathway Analysis of DEGs
2.6. Confirmation of Gene Expression Profile through qRT-PCR
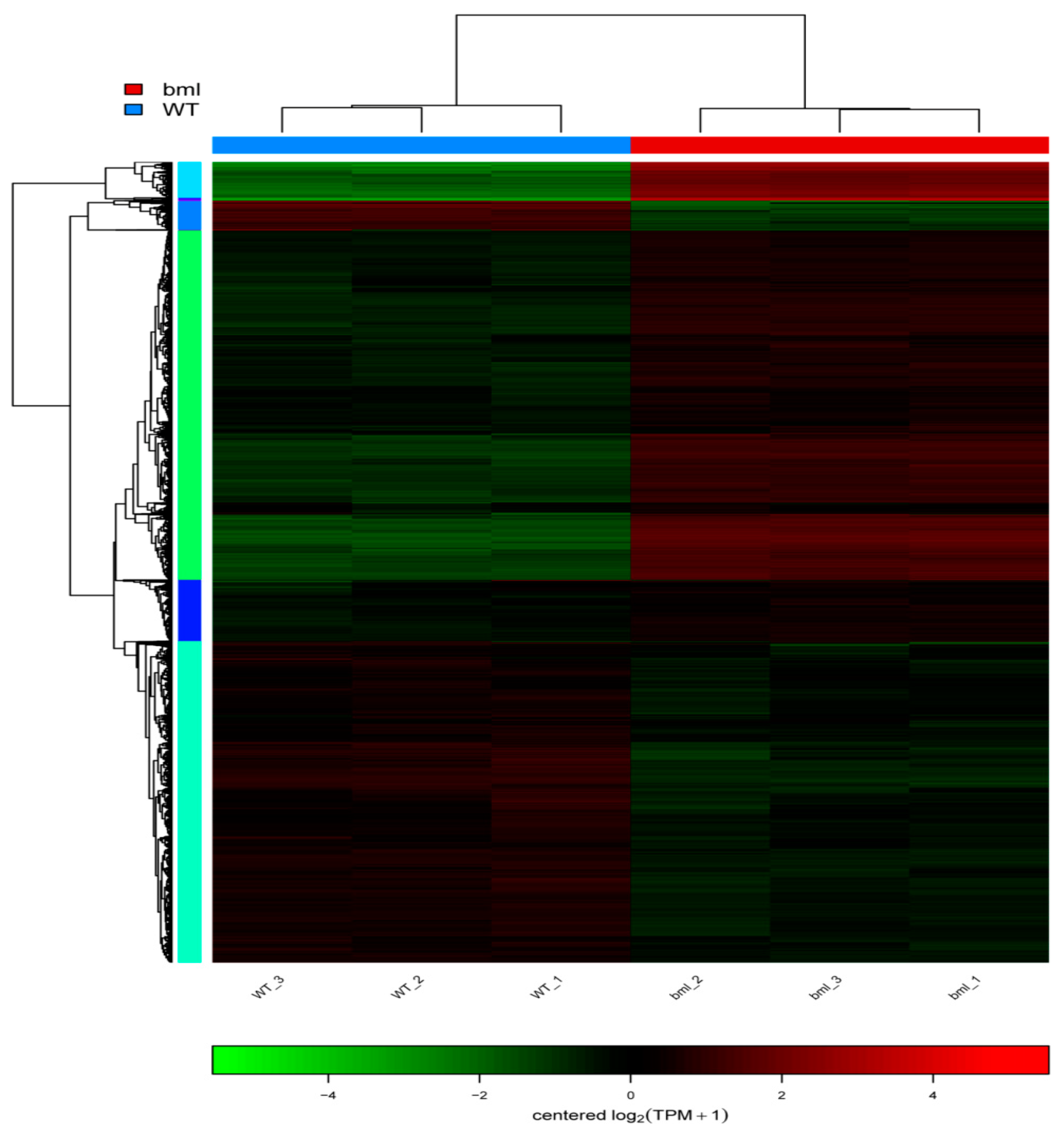
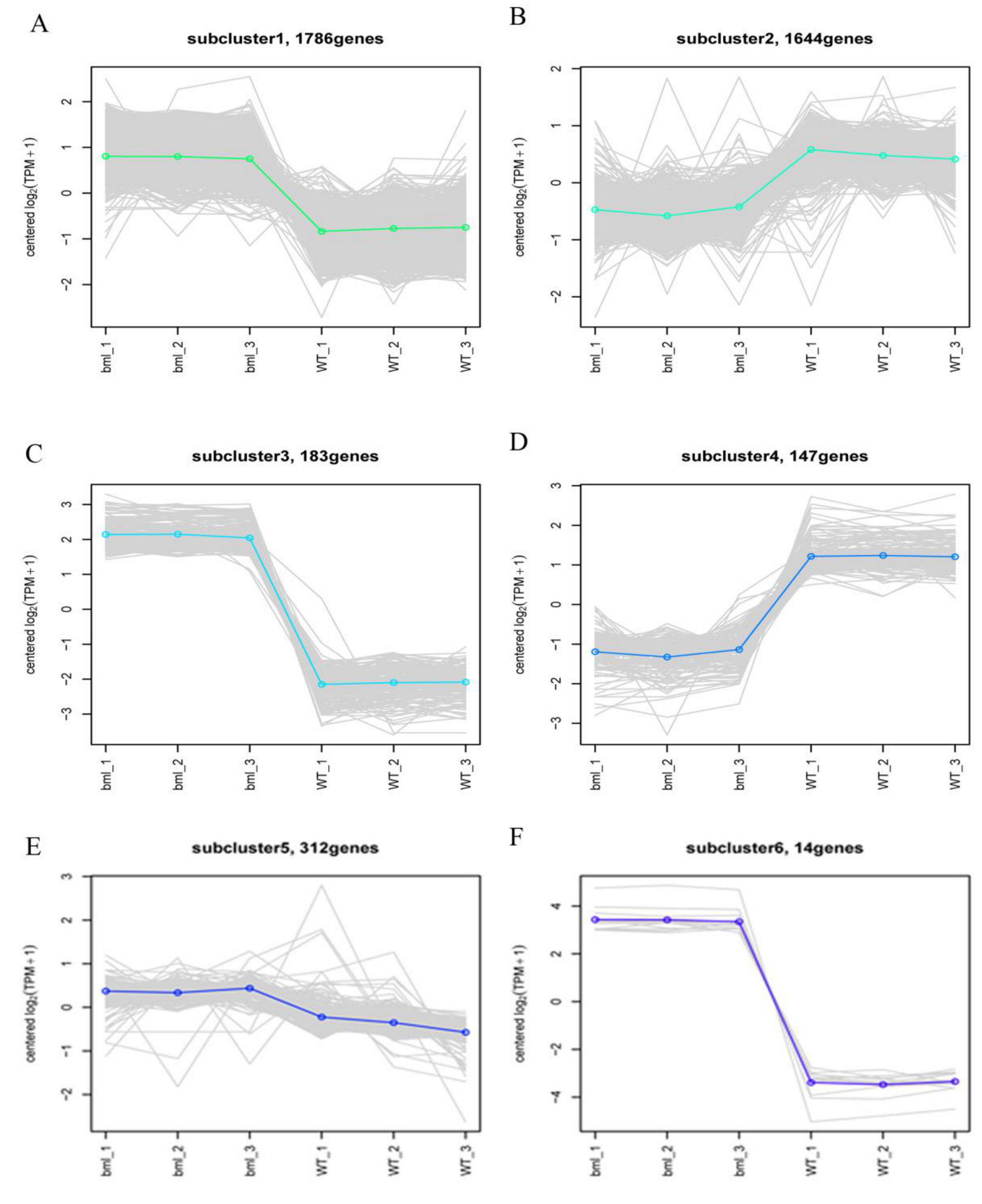
2.7. Cluster Analysis of DEGs
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Isolation, Library Preparation and Sequencing
4.3. Data Filtering, Reads Mapping and RNA-Seq Data Analysis
4.4. Transcriptomic Analysis
4.5. Quantitative Real-Time PCR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schippers, J.H.; Schmidt, R.; Wagstaff, C.; Jing, H.-C. Living to die and dying to live: The survival strategy behind leaf senescence. Plant Physiol. 2015, 169, 914–930. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.O.; Woo, H.R.; Nam, H.G. Molecular genetics of leaf senescence in Arabidopsis. Trends Plant Sci. 2003, 8, 272–278. [Google Scholar] [CrossRef]
- Buchanan-Wollaston, V.; Earl, S.; Harrison, E.; Mathas, E.; Navabpour, S.; Page, T.; Pink, D. The molecular analysis of leaf senescence–A genomics approach. Plant Biotechnol. J. 2003, 1, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, H.R.; Nam, H.G. Toward systems understanding of leaf senescence: An integrated multi-omics perspective on leaf senescence research. Mol. Plant 2016, 9, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Ye, G.; Zeng, D. Genetic dissection of leaf senescence in rice. Int. J. Mol. Sci. 2017, 18, 2686. [Google Scholar]
- Gregersen, P.L.; Culetic, A.; Boschian, L.; Krupinska, K. Plant senescence and crop productivity. Plant Mol. Biol. 2013, 82, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Distelfeld, A.; Avni, R.; Fischer, A.M. Senescence, nutrient remobilization, and yield in wheat and barley. J. Exp. Bot. 2014, 65, 3783–3798. [Google Scholar] [CrossRef] [PubMed]
- Bray, E.; BaileyYserres, J.; Weretilnyk, C. Responses to Abiotic Stress. In Biochemestry & Molecular Biology of Plants; Gruissem, W., Jones, R., Eds.; American Society of Plant Physiologists: Rockville, MD, USA, 2000; pp. 1158–1203. [Google Scholar]
- Lira, B.S.; Gramegna, G.; Trench, B.; Alves, F.R.R.; Silva, É.M.; Silva, G.F.F.; Thirumalaikumar, V.P.; Lupi, A.C.D.; Demarco, D.; Purgatto, E. Manipulation of a senescence-associated gene improves fleshy fruit yield. Plant Physiol. 2017, 175, 77–91. [Google Scholar] [CrossRef]
- Juvany, M.; Müller, M.; Munné-Bosch, S. Photo-oxidative stress in emerging and senescing leaves: A mirror image? J. Exp. Bot. 2013, 64, 3087–3098. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, C. Signal transduction in leaf senescence. Plant Mol. Biol. 2013, 82, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, G.; Liu, S.; An, Q.; Zheng, Q.; Li, B.; Li, Z. Comparative changes in the antioxidant system in the flag leaf of early and normally senescing near-isogenic lines of wheat (Triticum aestivum L.). Plant Cell Rep. 2014, 33, 1109–1120. [Google Scholar] [CrossRef]
- Buchanan-Wollaston, V. The molecular biology of leaf senescence. J. Exp. Bot. 1997, 48, 181–199. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, Y.; Zhou, G.; Ye, R.; Zhao, L.; Li, X.; Lin, Y. Identification of early senescence-associated genes in rice flag leaves. Plant Mol. Biol. 2008, 67, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, Z.; Jiang, Z.; Zhao, Y.; Peng, J.; Jin, J.; Guo, H.; Luo, J. LSD: A leaf senescence database. Nucleic Acids Res. 2010, 39, D1103–D1107. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, M.; Chen, Y.; Wu, P.; Wu, G.; Jiang, H. Knockdown of OsPAO and OsRCCR1 cause different plant death phenotypes in rice. J. Plant Physiol. 2011, 168, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Morita, R.; Katsuma, S.; Nishimura, M.; Tanaka, A.; Kusaba, M. Two short-chain dehydrogenase/reductases, NON-YELLOW COLORING 1 and NYC1-LIKE, are required for chlorophyll b and light-harvesting complex II degradation during senescence in rice. Plant J. 2009, 57, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, M.; Ito, H.; Morita, R.; Iida, S.; Sato, Y.; Fujimoto, M.; Kawasaki, S.; Tanaka, R.; Hirochika, H.; Nishimura, M. Rice NON-YELLOW COLORING1 is involved in light-harvesting complex II and grana degradation during leaf senescence. Plant Cell 2007, 19, 1362–1375. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Liang, Y.; Zhang, J.; Zheng, H.; Dong, G.; Qian, Q.; Zuo, J. Involvement of a putative bipartite transit peptide in targeting rice pheophorbide a oxygenase into chloroplasts for chlorophyll degradation during leaf senescence. J. Genet. Genom. 2016, 43, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, H.; Zhai, G.; Wang, L.; Shao, J.; Tao, Y. OspTAC2 encodes a pentatricopeptide repeat protein and regulates rice chloroplast development. J. Genet. Genom. 2016, 43, 601–608. [Google Scholar] [CrossRef]
- Uji, Y.; Akimitsu, K.; Gomi, K. Identification of OsMYC2-regulated senescence-associated genes in rice. Planta 2017, 245, 1241–1246. [Google Scholar] [CrossRef]
- Lee, S.H.; Sakuraba, Y.; Lee, T.; Kim, K.W.; An, G.; Lee, H.Y.; Paek, N.C. Mutation of Oryza sativa CORONATINE INSENSITIVE 1b (OsCOI1b) delays leaf senescence. J. Integr. Plant Biol. 2015, 57, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhang, H.; Wan, J.; Wu, Y.; Li, K.; Jin, C.; Chen, W.; Wang, S.; Wang, W.; Zhang, H. Control of leaf senescence by an MeOH-Jasmonates cascade that is epigenetically regulated by OsSRT1 in rice. Mol. Plant 2016, 9, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, W.; Gan, S.S. SAUR36, a small auxin up RNA gene, is involved in the promotion of leaf senescence in Arabidopsis. Plant Physiol. 2013, 161, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Kant, S.; Bi, Y.M.; Zhu, T.; Rothstein, S.J. SAUR39, a small auxin-up RNA gene, acts as a negative regulator of auxin synthesis and transport in rice. Plant Physiol. 2009, 151, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Quirino, B.F.; Normanly, J.; Amasino, R.M. Diverse range of gene activity during Arabidopsis thaliana leaf senescence includes pathogen-independent induction of defense-related genes. Plant Mol. Biol. 1999, 40, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Van der Graaff, E.; Schwacke, R.; Schneider, A.; Desimone, M.; Flügge, U.I.; Kunze, R. Transcription analysis of Arabidopsis membrane transporters and hormone pathways during developmental and induced leaf senescence. Plant Physiol. 2006, 141, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gong, P.; Li, K.; Huang, F.; Cheng, F.; Pan, G. A single cytosine deletion in the OsPLS1 gene encoding vacuolar-type H+-ATPase subunit A1 leads to premature leaf senescence and seed dormancy in rice. J. Exp. Bot. 2016, 67, 2761–2776. [Google Scholar] [CrossRef]
- Su, M.; Huang, G.; Zhang, Q.; Wang, X.; Li, C.; Tao, Y.; Zhang, S.; Lai, J.; Yang, C.; Wang, Y. The LEA protein, ABR, is regulated by ABI5 and involved in dark-induced leaf senescence in Arabidopsis thaliana. Plant Sci. 2016, 247, 93–103. [Google Scholar] [CrossRef]
- Woo, H.R.; Kim, H.J.; Nam, H.G.; Lim, P.O. Plant leaf senescence and death–regulation by multiple layers of control and implications for aging in general. J. Cell Sci. 2013, 126, 4823–4833. [Google Scholar] [CrossRef]
- Besseau, S.; Li, J.; Palva, E.T. WRKY54 and WRKY70 co-operate as negative regulators of leaf senescence in Arabidopsis thaliana. J. Exp. Bot. 2012, 63, 2667–2679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xia, C.; Zhang, L.; Dong, C.; Liu, X.; Kong, X. Transcriptome Analysis of a Premature Leaf Senescence Mutant of Common Wheat (Triticum aestivum L.). Int. J. Mol. Sci. 2018, 19, 782. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Kim, C.Y.; Lee, J.; Lee, S.K.; Jeon, J.S. OsWRKY42 represses OsMT1d and induces reactive oxygen species and leaf senescence in rice. Mol. Cells 2014, 37, 532. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.; Guevara, D.; Yaish, M.W.; Hannam, C.; Long, N.; Clarke, J.D.; Bi, Y.M.; Rothstein, S.J. GNC and CGA1 modulate chlorophyll biosynthesis and glutamate synthase (GLU1/Fd-GOGAT) expression in Arabidopsis. PLoS ONE 2011, 6, e26765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hou, Y.; Hao, Q.; Chen, H.; Chen, L.; Yuan, S.; Shan, Z.; Zhang, X.; Yang, Z.; Qiu, D. Genome-wide survey of the soybean GATA transcription factor gene family and expression analysis under low nitrogen stress. PLoS ONE 2015, 10, e0125174. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Casaretto, J.A.; Ying, S.; Mahmood, K.; Liu, F.; Bi, Y.M.; Rothstein, S.J. Overexpression of OsGATA12 regulates chlorophyll content, delays plant senescence and improves rice yield under high density planting. Plant Mol. Biol. 2017, 94, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Uauy, C.; Distelfeld, A.; Fahima, T.; Blechl, A.; Dubcovsky, J. A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 2006, 314, 1298–1301. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Piao, W.; Lim, J.H.; Han, S.H.; Kim, Y.S.; An, G.; Paek, N.C. Rice ONAC106 inhibits leaf senescence and increases salt tolerance and tiller angle. Plant Cell Physiol. 2015, 56, 2325–2339. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, L.; Huang, W.; Yuan, M.; Zhou, F.; Li, X.; Lin, Y. Overexpression of OsSWEET5 in rice causes growth retardation and precocious senescence. PLoS ONE 2014, 9, e94210. [Google Scholar] [CrossRef]
- Krattinger, S.; Lagudah, E.; Spielmeyer, W.; Singh, R.; Huerta-Espino, J.; Mc Fadden, H.; Bossolini, E.; Selter, L.; Keller, B. An ABC transporter confers durable resistance to multiple fungal pathogens in wheat. In Proceedings of the 3rd 19th International Triticeae Mapping Initiative, COST Tritigen, Clermont-Ferrand, France, 31 August–4 September 2009. [Google Scholar]
- Yang, S.S.; Tu, Z.J.; Cheung, F.; Xu, W.W.; Lamb, J.F.; Jung, H.J.G.; Vance, C.P.; Gronwald, J.W. Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems. BMC Genom. 2011, 12, 199. [Google Scholar] [CrossRef]
- Ekblom, R.; Galindo, J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity 2011, 107, 1. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Zody, M.C. Advancing RNA-seq analysis. Nat. Biotechnol. 2010, 28, 421. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Islam, F.; Li, L.; Long, M.; Yang, C.; Jin, X.; Ali, B.; Mao, B.; Zhou, W. Complementary RNA-sequencing based transcriptomics and iTRAQ proteomics reveal the mechanism of the alleviation of quinclorac stress by salicylic acid in Oryza sativa ssp. japonica. Int. J. Mol. Sci. 2017, 18, 1975. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef] [PubMed]
- Akhter, D.; Qin, R.; Nath, U.K.; Alamin, M.; Jin, X.; Shi, C. The Brown Midrib Leaf (bml) Mutation in Rice (Oryza sativa L.) Causes Premature Leaf Senescence and the Induction of Defense Responses. Genes 2018, 9, 203. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Tan, Y.; Cui, G.; Feng, Y.; Cui, Q.; Song, X. Transcriptome and gene expression analysis of DHA producer Aurantiochytrium under low temperature conditions. Sci. Rep. 2015, 5, 14446. [Google Scholar] [CrossRef] [PubMed]
- Schön, M.; Töller, A.; Diezel, C.; Roth, C.; Westphal, L.; Wiermer, M.; Somssich, I.E. Analyses of wrky18 wrky40 plants reveal critical roles of SA/EDS1 signaling and indole-glucosinolate biosynthesis for Golovinomyces orontii resistance and a loss-of resistance towards Pseudomonas syringae pv. tomato AvrRPS4. Mol. Plant-Microbe Interact. 2013, 26, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S. Molecular regulation of leaf senescence. Curr. Opin. Plant Biol. 2003, 6, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Kim, Y.; Park, S.; Back, K. Evaluation of light-harvesting complex proteins as senescence-related protein markers in detached rice leaves. Photosynthetica 2009, 47, 638–640. [Google Scholar] [CrossRef]
- Yamatani, H.; Sato, Y.; Masuda, Y.; Kato, Y.; Morita, R.; Fukunaga, K.; Nagamura, Y.; Nishimura, M.; Sakamoto, W.; Tanaka, A. NYC4, the rice ortholog of Arabidopsis THF1, is involved in the degradation of chlorophyll–protein complexes during leaf senescence. Plant J. 2013, 74, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Zoschke, R.; Watkins, K.P.; Miranda, R.G.; Barkan, A. The PPR-SMR protein PPR 53 enhances the stability and translation of specific chloroplast RNA s in maize. Plant J. 2016, 85, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Rozhon, W.; Poppenberger, B. The role of hormones in the aging of plants-a mini-review. Gerontology 2014, 60, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2009, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genom. Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Zeng, D.D.; Qin, R.; Li, M.; Alamin, M.; Jin, X.L.; Liu, Y.; Shi, C.H. The ferredoxin-dependent glutamate synthase (OsFd-GOGAT) participates in leaf senescence and the nitrogen remobilization in rice. Mol. Genet. Genom. 2017, 292, 385–395. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Average Raw Read Length (bp) | Clean Reads | Average Clean Read Length (bp) | Clean Base (bp) | Error (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|---|
| WT | 60,545,108 | 150 | 56,637,709 | 147.10 | 30,965 | 0.00 | 98.44 | 94.64 | 54.71 |
| Bml | 62,982,574 | 150 | 61, 147, 444 | 147.50 | 43,180 | 0.00 | 98.59 | 94.99 | 55.60 |
| Sample Name | Total Reads | Total Mapped (%) | Multiple Mapped (%) | Uniquely Mapped (%) | Non-Splice Reads (%) | Splice Reads (%) | Reads Mapped in Proper Pairs (%) |
|---|---|---|---|---|---|---|---|
| WT | 53579398 | 94.01 | 3.00 | 91.00 | 61.46 | 29.55 | 87.31 |
| bml | 57136394 | 94.27 | 2.56 | 93.45 | 63.36 | 28.33 | 88.06 |
| Gene ID | Gene Name | bml vs. WT | Gene Description | Pathway/Function Involved | |
|---|---|---|---|---|---|
| RNA-Seq log2 Fold Change | qRT PCR Fold Change | ||||
| LOC_Os01g12710 | NYC1 | −1.509 | −0.880 ± 0.02 | NAD(P)-binding Rossmann-fold superfamily protein | Porphyrin and chlorophyll metabolism |
| LOC_Os02g52650 | LHCA5 | −1.266 | −569 ± 0.207 | Photosystem I light harvesting complex gene 5 | Photosynthesis-antenna proteins |
| LOC_Os09g26810 | LHCA2.1 | −1.0265 | −0.879 ± 0.0890 | Photosystem I light harvesting complex gene 6 | Photosynthesis-antenna proteins |
| LOC_Os06g44010 | ATWRKY40 | 3.722 | 3.455 ± 0.418 | WRKY DNA-binding protein 40 | WRKY transcription factor |
| LOC_Os12g02470 | ATWRKY53 | 6.512 | 5.360 ± 0.1837 | WRKY family transcription factor | WRKY transcription factor |
| LOC_Os11g02540 | ATWRKY70 | 4.669 | 2.862 ± 0.2069 | WRKY DNA-binding protein 70 | WRKY transcription factor |
| LOC_Os07g04560 | anac042 | 1.566 | 2.641 ± 0.0344 | NAC domain containing protein 42 | NAC transcription factor |
| LOC_Os10g42130 | anac071 | 2.630 | 2.798 ± 0.0182 | NAC domain containing protein 71 | NAC transcription factor |
| LOC_Os09g36200 | ATNYE | 1.437 | 1.375 ± 0.0246 | Non-yellowing 1 | NAC transcription factor |
| LOC_Os01g21250 | AtLEA5 | 4.182 | 3.947 ± 0.0286 | Senescence-linked gene 21 | Biological process |
| LOC_Os03g0529 | SAG12 | 1.043 | 3.139 ± 0.0128 | Senescence-associated gene 12 | Proteolysis (Biological process) |
| LOC_Os06g16370 | ATCOL2 | −2.334 | −0.194 ± 0.0378 | CONSTANS-like 2 | GATA transcription factor |
| LOC_Os03g08330 | JAZ10 | 1.336 | 0.893 ± 0.0239 | Jasmonate-zim-domain protein 1 | Plant hormone signal transduction |
| LOC_Os01g09450 | IAA26 | −3.341 | −1.985 ± 0.0462 | Phytochrome-associated protein 1 | Plant hormone signal transduction |
| LOC_Os02g57250 | IAA13 | −1.316 | −0.639 ± 0.0302 | Auxin-induced protein 13 | Plant hormone signal transduction |
| LOC_Os01g28450 | ATPR1 | 6.095 | 5.015 ± 0.0108 | Pathogenesis-related gene 1 | Plant hormone signal transduction |
| LOC_Os07g03730 | PR1a | 10.420 | 4.106 ± 0.105 | Pathogenesis-related gene 1 | Plant hormone signal transduction |
| LOC_Os01g09800 | ATNPR1 | 1.140 | 1.893 ± 0.1017 | Pathogenesis-related gene 1 | Plant hormone signal transduction |
| LOC_Os04g32480 | JAZ5 | 3.746 | 2.084 ± 0.143 | Jasmonate-zim-domain protein 10 | Plant hormone signal transduction |
| LOC_Os09g29660 | ABCG11 | 1.868343 | 1.901 ± 0.041 | ABC super family | ABC transporter |
| LOC_Os07g48260 | ATWRKY54 | 3.980698 | 4.035 ± 0.051 | WRKY DNA-binding protein 54 | WRKY transcription factor |
| LOC_Os01g60700 | ATSIK | 1.036138 | 1.241 ± 0.312 | Serine/threonine protein kinase | Protein kinase superfamily protein |
| LOC_Os04g33630 | ATFD3 | −1.74014 | −0.934 ± 0.063 | Ferredoxin 3 | Photosynthesis related |
| LOC_Os09g36250 | AtMYB42 | 1.8693 | 1.012 ± 0.082 | MYB domain protein 42 | MYB domain transcripton factor |
| LOC_Os04g43070 | AMT1;1 | 1.9946169 | 1.512 ± 0.101 | Ammonium transporter 1;1 | Ammonium transporter |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, D.; Qin, R.; Nath, U.K.; Eshag, J.; Jin, X.; Shi, C. Transcriptional Profile Corroborates that bml Mutant Plays likely Role in Premature Leaf Senescence of Rice (Oryza sativa L.). Int. J. Mol. Sci. 2019, 20, 1708. https://doi.org/10.3390/ijms20071708
Akhter D, Qin R, Nath UK, Eshag J, Jin X, Shi C. Transcriptional Profile Corroborates that bml Mutant Plays likely Role in Premature Leaf Senescence of Rice (Oryza sativa L.). International Journal of Molecular Sciences. 2019; 20(7):1708. https://doi.org/10.3390/ijms20071708
Chicago/Turabian StyleAkhter, Delara, Ran Qin, Ujjal Kumar Nath, Jamal Eshag, Xiaoli Jin, and Chunhai Shi. 2019. "Transcriptional Profile Corroborates that bml Mutant Plays likely Role in Premature Leaf Senescence of Rice (Oryza sativa L.)" International Journal of Molecular Sciences 20, no. 7: 1708. https://doi.org/10.3390/ijms20071708
APA StyleAkhter, D., Qin, R., Nath, U. K., Eshag, J., Jin, X., & Shi, C. (2019). Transcriptional Profile Corroborates that bml Mutant Plays likely Role in Premature Leaf Senescence of Rice (Oryza sativa L.). International Journal of Molecular Sciences, 20(7), 1708. https://doi.org/10.3390/ijms20071708