Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes

 ,
,  and
and
Abstract
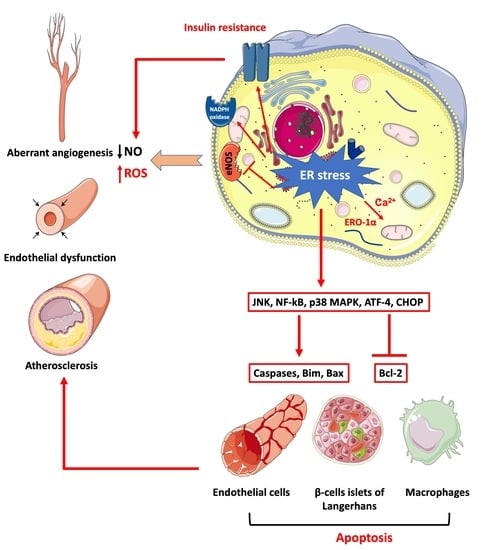
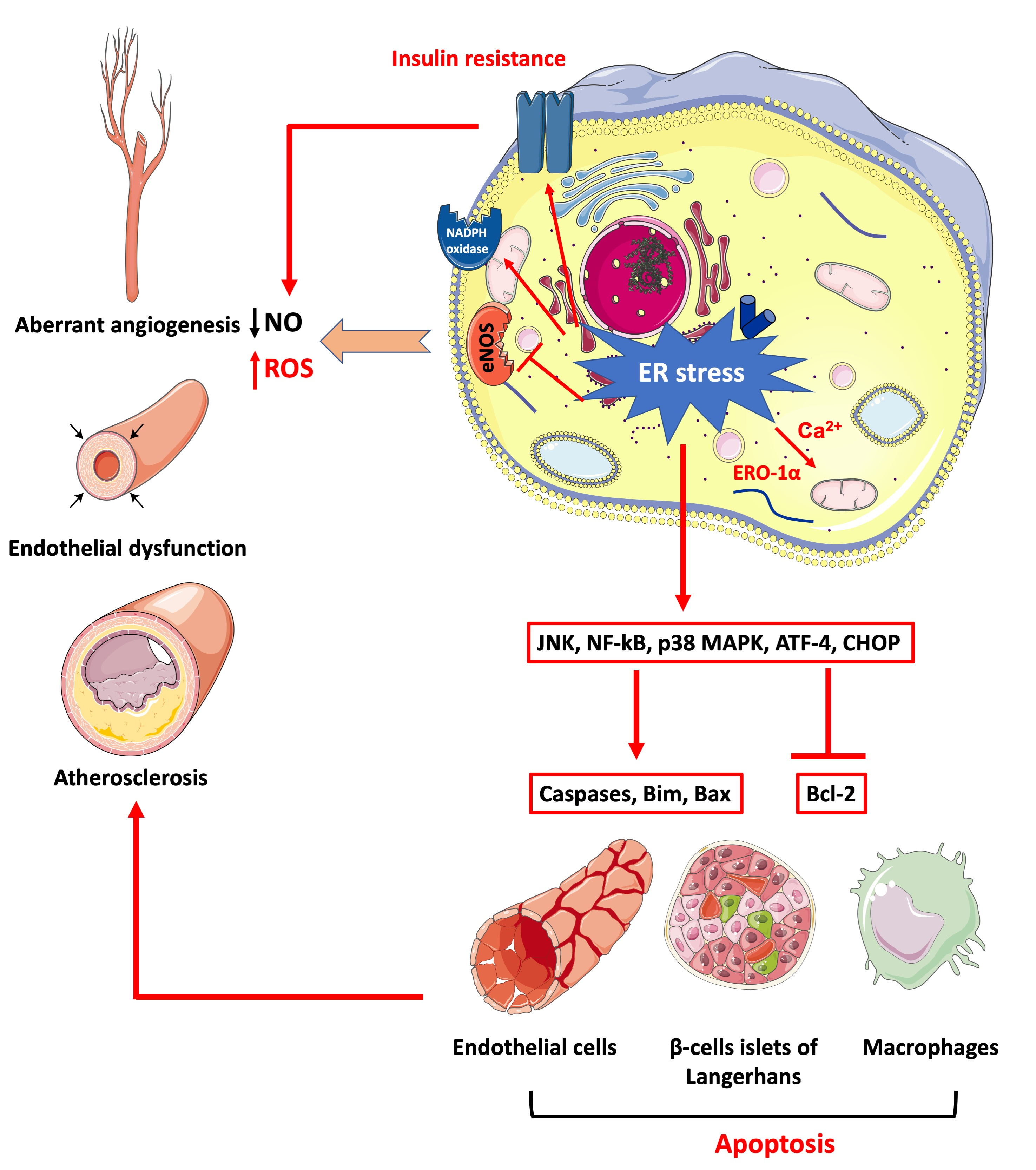{kind=link}
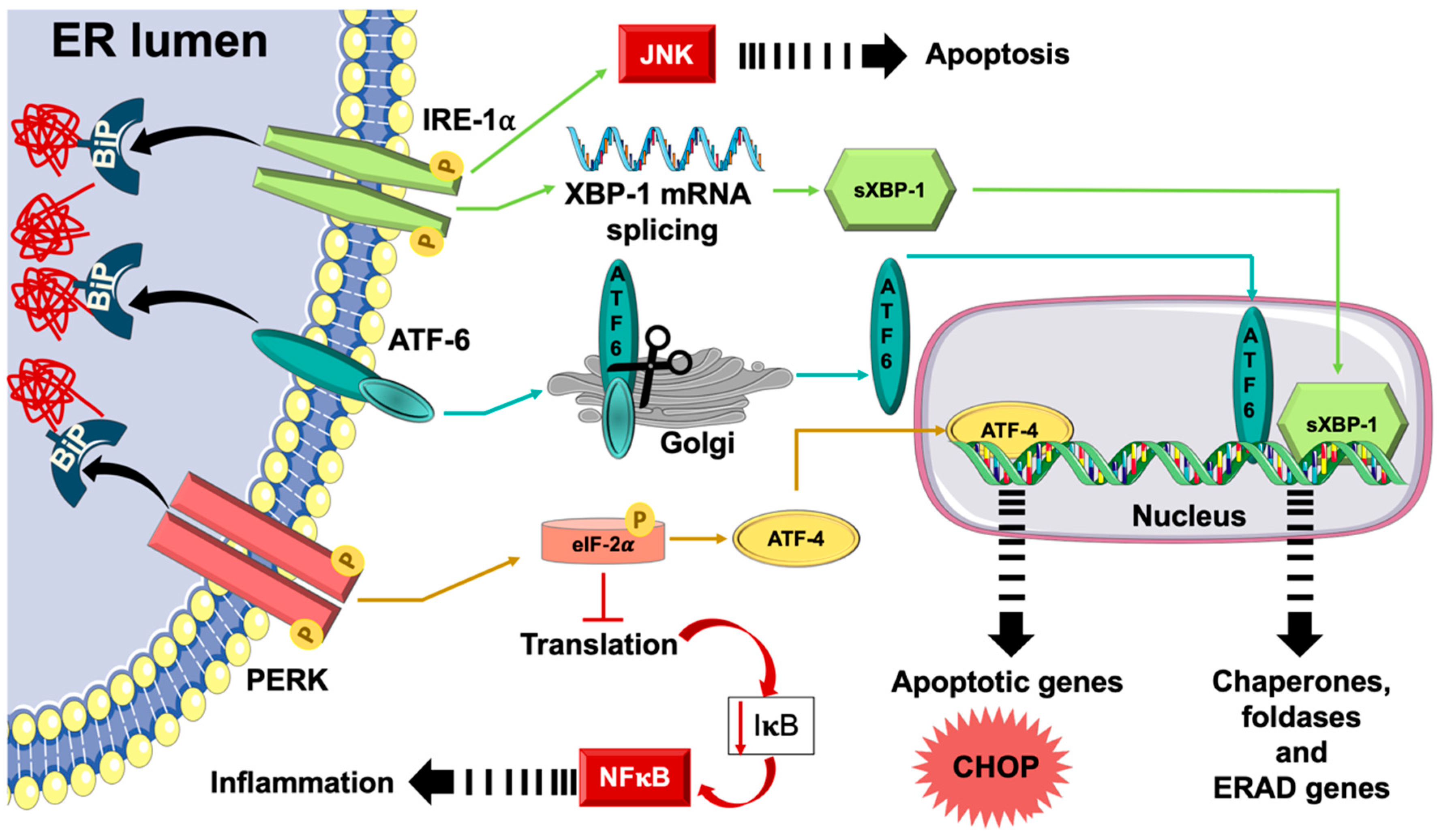{kind=link}
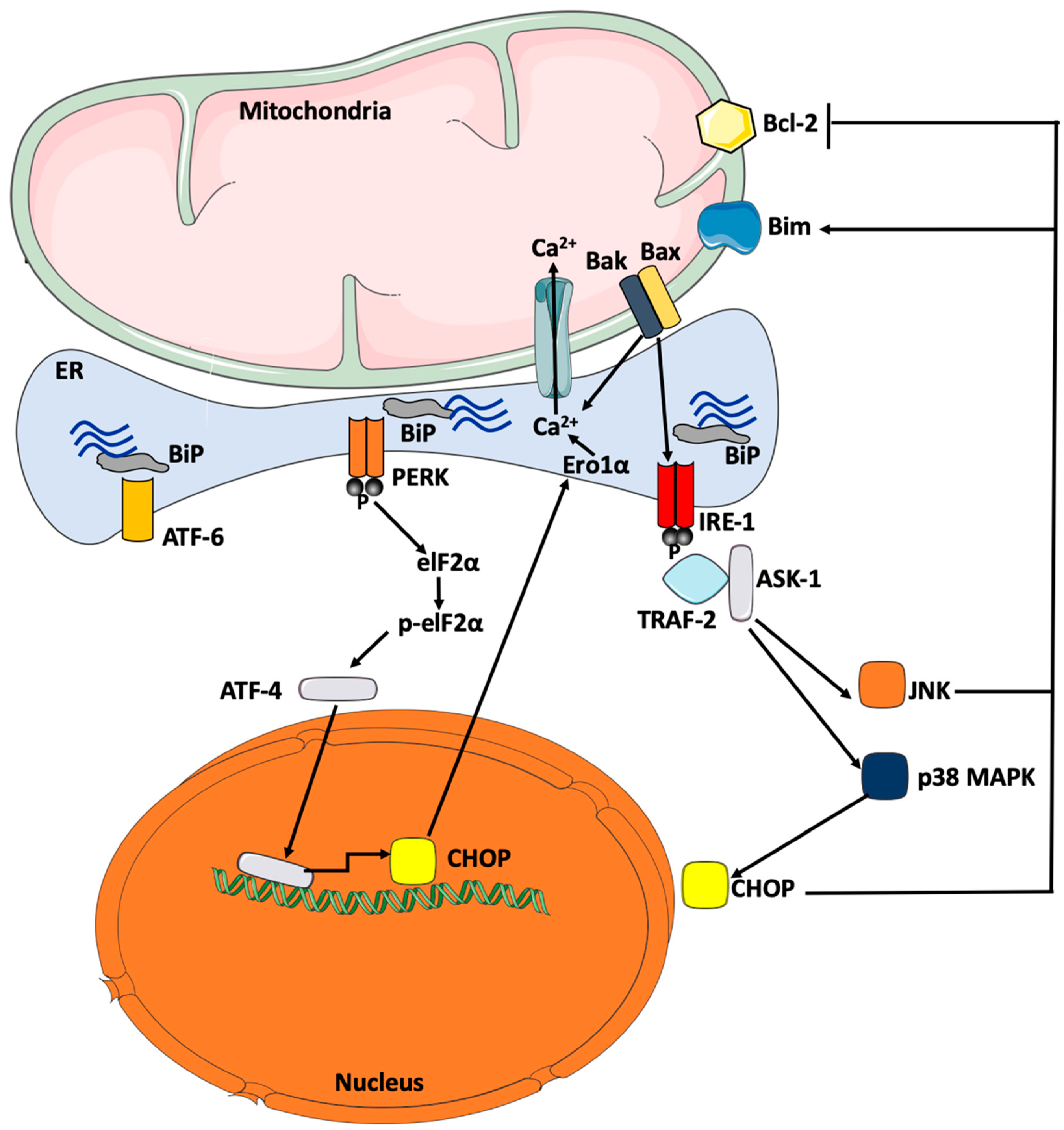{kind=link}
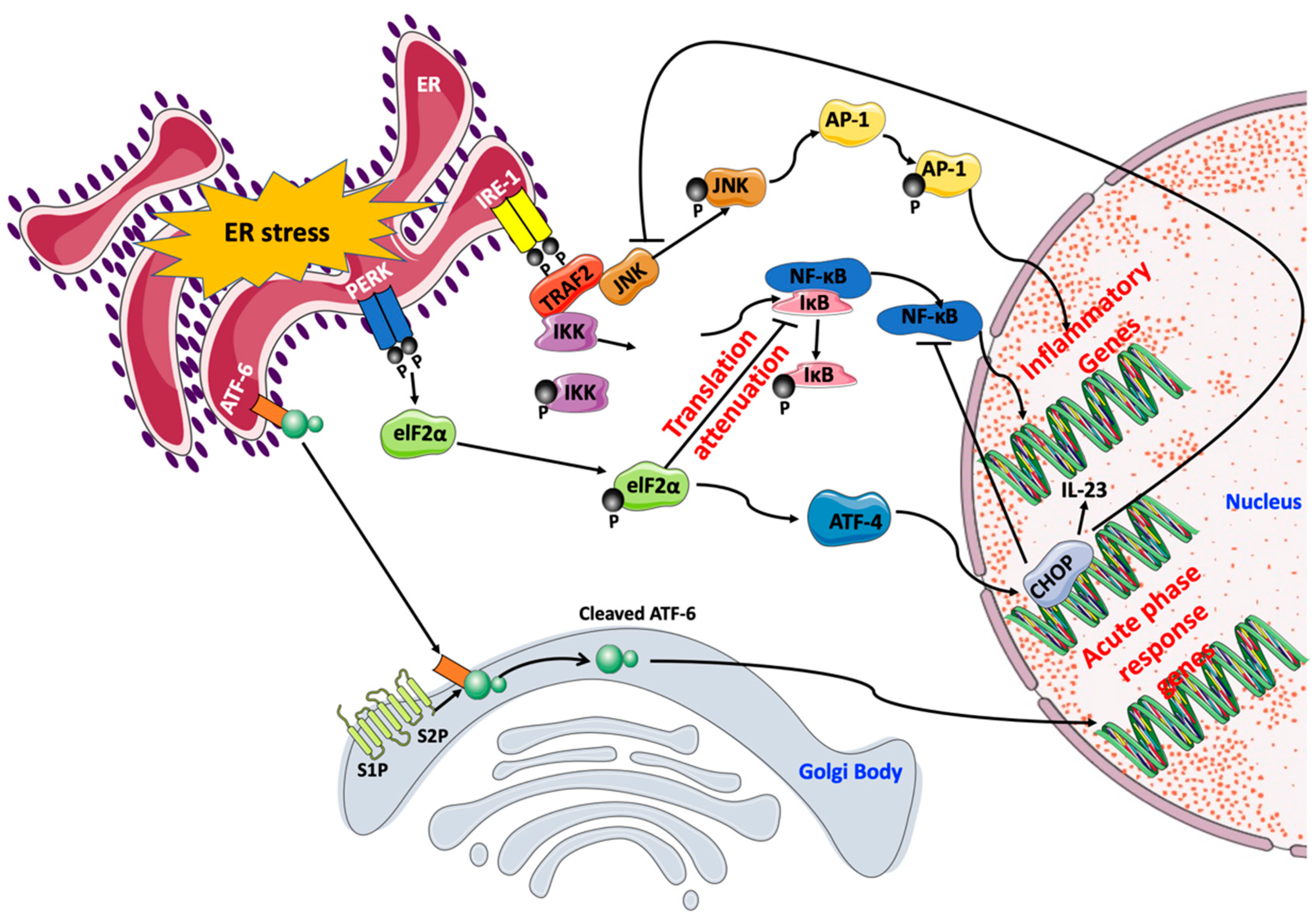{kind=link}
1. Introduction
2. Physiological Roles of Endothelium and Endothelial Dysfunction
2.1. Physiological Roles of Endothelium
2.2. Endothelial Dysfunction
3. Unfolded Protein Response (UPR) and Endoplasmic Reticulum (ER) Stress
3.1. The Physiological Roles of ER
3.2. UPR and ER Stress Response
4. Contribution of ER Stress Response to Cardiovascular Disease
4.1. The Role of ER Stress in Endothelial Dysfunction
4.2. The Role of ER Stress in the Development of Atherosclerosis
5. Major Mechanisms Underpinning ER Stress-Mediated Endothelial Dysfunction
5.1. ER Stress-Mediated Insulin Resistance
5.2. ER Stress-Mediated Oxidative Stress
5.3. ER Stress-Mediated Cellular Inflammation And Apoptosis
6. Clinical Utility of Targeting ER Stress
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mandviwala, T.; Khalid, U.; Deswal, A. Obesity and Cardiovascular Disease: A Risk Factor or a Risk Marker? Curr. Atheroscler. Rep. 2016, 18, 21. [Google Scholar] [CrossRef]
- Navarro Díaz, M. Consequences of morbid obesity on the kidney. Where are we going? Clin. Kidney J. 2016, 9, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Vascular complications in diabetes mellitus: The role of endothelial dysfunction. Clin. Sci. 2005, 109, 143–159. [Google Scholar] [CrossRef]
- Csige, I.; Ujvarosy, D.; Szabo, Z.; Lorincz, I.; Paragh, G.; Harangi, M.; Somodi, S. The Impact of Obesity on the Cardiovascular System. J. Diabetes Res. 2018, 2018, 3407306. [Google Scholar] [CrossRef] [PubMed]
- McGill, H.C., Jr.; McMahan, C.A.; Herderick, E.E.; Zieske, A.W.; Malcom, G.T.; Tracy, R.E.; Strong, J.P.; Pathobiological Determinants of Atherosclerosis in Youth Research Group. Obesity accelerates the progression of coronary atherosclerosis in young men. Circulation 2002, 105, 2712–2718. [Google Scholar] [CrossRef]
- Chawla, A.; Chawla, R.; Jaggi, S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J. Endocrinol. Metab. 2016, 20, 546–551. [Google Scholar] [CrossRef]
- WHO. DIABETES; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017.
- Liu, J.; Liu, Z. Muscle Insulin Resistance and the Inflamed Microvasculature: Fire from Within. Int. J. Mol. Sci. 2019, 20, 562. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, E.; DeFronzo, R.A. Insulin resistance and endothelial dysfunction: The road map to cardiovascular diseases. Diabetes/Metab. Res. Rev. 2006, 22, 423–436. [Google Scholar] [CrossRef]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef]
- Galán, M.; Kassan, M.; Choi, S.-K.; Partyka, M.; Trebak, M.; Henrion, D.; Matrougui, K. A Novel Role for Epidermal Growth Factor Receptor Tyrosine Kinase and Its Downstream Endoplasmic Reticulum Stress in Cardiac Damage and Microvascular Dysfunction in Type 1 Diabetes Mellitus. Hypertension 2012, 60, 71–80. [Google Scholar] [CrossRef]
- Kassan, M.; Galán, M.; Partyka, M.; Saifudeen, Z.; Henrion, D.; Trebak, M.; Matrougui, K. Endoplasmic Reticulum Stress Is Involved in Cardiac Damage and Vascular Endothelial Dysfunction in Hypertensive Mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M. The unfolded protein response. Mol. Biotechnol. 2006, 34, 279–290. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Ron, D. Endoplasmic Reticulum Stress Signaling in Disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial Cells in Physiology and in the Pathophysiology of Vascular Disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [PubMed]
- Zhang, H.-N.; Xu, Q.-Q.; Thakur, A.; Alfred, M.O.; Chakraborty, M.; Ghosh, A.; Yu, X.-B. Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sci. 2018, 213, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Khazaei, M.; Moien-Afshari, F.; Laher, I. Vascular endothelial function in health and diseases. Pathophysiology 2008, 15, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405. [Google Scholar] [CrossRef]
- Tronc, F.; Wassef, M.; Esposito, B.; Henrion, D.; Glagov, S.; Tedgui, A. Role of NO in flow-induced remodeling of the rabbit common carotid artery. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Bohm, F.; Pernow, J. The importance of endothelin-1 for vascular dysfunction in cardiovascular disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Granger, J.P.; Spradley, F.T.; Bakrania, B.A. The Endothelin System: A Critical Player in the Pathophysiology of Preeclampsia. Curr. Hypertens. Rep. 2018, 20, 32. [Google Scholar] [CrossRef]
- Varin, R.; Mulder, P.; Tamion, F.; Richard, V.; Henry, J.P.; Lallemand, F.; Lerebours, G.; Thuillez, C. Improvement of endothelial function by chronic angiotensin-converting enzyme inhibition in heart failure: Role of nitric oxide, prostanoids, oxidant stress, and bradykinin. Circulation 2000, 102, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M.; Huang, Y.; Vanhoutte, P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br. J. Pharmacol. 2011, 164, 894–912. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Wong, W.T.; Tian, X.Y.; Lau, C.W.; Huang, Y. Prostaglandins in action indispensable roles of cyclooxygenase-1 and -2 in endothelium-dependent contractions. Adv. Pharmacol. 2010, 60, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Balzan, S. Roles of LOX-1 in microvascular dysfunction. Microvasc. Res. 2016, 105, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335. [Google Scholar] [CrossRef]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk Between Oxidative Stress and Endoplasmic Reticulum (ER) Stress in Endothelial Dysfunction and Aberrant Angiogenesis Associated With Diabetes: A Focus on the Protective Roles of Heme Oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Battaglia-Hsu, S.-F.; Arnold, C. Endoplasmic Reticulum Stress in Metabolic Disorders. Cells 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. CMLS 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Pandey, V.K.; Mathur, A.; Kakkar, P. Emerging role of Unfolded Protein Response (UPR) mediated proteotoxic apoptosis in diabetes. Life Sci. 2019, 216, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Komuro, I.; Kitakaze, M. Endoplasmic Reticulum Stress As a Therapeutic Target in Cardiovascular Disease. Circ. Res. 2010, 107, 1071–1082. [Google Scholar] [CrossRef]
- Hu, M.; Phan, F.; Bourron, O.; Ferre, P.; Foufelle, F. Steatosis and NASH in type 2 diabetes. Biochimie 2017, 143, 37–41. [Google Scholar] [CrossRef]
- Bailey, D.; O’Hare, P. Transmembrane bZIP transcription factors in ER stress signaling and the unfolded protein response. Antioxid. Redox Signal. 2007, 9, 2305–2321. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.; Barreca, C.; O’Hare, P. Trafficking of the bZIP transmembrane transcription factor CREB-H into alternate pathways of ERAD and stress-regulated intramembrane proteolysis. Traffic 2007, 8, 1796–1814. [Google Scholar] [CrossRef] [PubMed]
- Hassler, J.; Cao, S.S.; Kaufman, R.J. IRE1, a double-edged sword in pre-miRNA slicing and cell death. Dev. Cell 2012, 23, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Hubbard, S.R. How IRE1 reacts to ER stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.-G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef]
- Kim, S.; Woo, C.-H. Laminar Flow Inhibits ER Stress-Induced Endothelial Apoptosis through PI3K/Akt-Dependent Signaling Pathway. Mol. Cells 2018, 41, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, J.; Danniel, M.; Wang, X.; Wang, W.; Zeng, L.; Shen, L. The interaction between XBP1 and eNOS contributes to endothelial cell migration. Exp. Cell Res. 2018, 363, 262–270. [Google Scholar] [CrossRef]
- Frakes, A.E.; Dillin, A. The UPR(ER): Sensor and Coordinator of Organismal Homeostasis. Mol. Cell 2017, 66, 761–771. [Google Scholar] [CrossRef]
- Lenna, S.; Townsend, D.M.; Tan, F.K.; Kapanadze, B.; Markiewicz, M.; Trojanowska, M.; Scorza, R. HLA-B35 upregulates endothelin-1 and downregulates endothelial nitric oxide synthase via endoplasmic reticulum stress response in endothelial cells. J. Immunol. 2010, 184, 4654–4661. [Google Scholar] [CrossRef]
- Spitler, K.M.; Matsumoto, T.; Webb, R.C. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am. J. Physiol.-Heart Circ. Physiol. 2013, 305, H344–H353. [Google Scholar] [CrossRef]
- Galán, M.; Kassan, M.; Kadowitz, P.J.; Trebak, M.; Belmadani, S.; Matrougui, K. Mechanism of endoplasmic reticulum stress-induced vascular endothelial dysfunction. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2014, 1843, 1063–1075. [Google Scholar] [CrossRef]
- Walsh, L.K.; Restaino, R.M.; Neuringer, M.; Manrique, C.; Padilla, J. Administration of tauroursodeoxycholic acid prevents endothelial dysfunction caused by an oral glucose load. Clin. Sci. 2016, 130, 1881–1888. [Google Scholar] [CrossRef]
- Maamoun, H.; Zachariah, M.; McVey, J.H.; Green, F.R.; Agouni, A. Heme oxygenase (HO)-1 induction prevents Endoplasmic Reticulum stress-mediated endothelial cell death and impaired angiogenic capacity. Biochem. Pharmacol. 2017, 127, 46–59. [Google Scholar] [CrossRef]
- Yu, W.; Liu, X.; Feng, L.; Yang, H.; Yu, W.; Feng, T.; Wang, S.; Wang, J.; Liu, N. Glycation of paraoxonase 1 by high glucose instigates endoplasmic reticulum stress to induce endothelial dysfunction in vivo. Sci. Rep. 2017, 7, 45827. [Google Scholar] [CrossRef]
- Choi, S.-K.; Lim, M.; Byeon, S.-H.; Lee, Y.-H. Inhibition of endoplasmic reticulum stress improves coronary artery function in the spontaneously hypertensive rats. Sci. Rep. 2016, 6, 31925. [Google Scholar] [CrossRef]
- Dong, Y.; Fernandes, C.; Liu, Y.; Wu, Y.; Wu, H.; Brophy, M.L.; Deng, L.; Song, K.; Wen, A.; Wong, S.; et al. Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diabetes Vasc. Dis. Res. 2017, 14, 14–23. [Google Scholar] [CrossRef]
- Tsukano, H.; Gotoh, T.; Endo, M.; Miyata, K.; Tazume, H.; Kadomatsu, T.; Yano, M.; Iwawaki, T.; Kohno, K.; Araki, K.; et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1925–1932. [Google Scholar] [CrossRef]
- Zhou, J.; Lhotak, S.; Hilditch, B.A.; Austin, R.C. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2005, 111, 1814–1821. [Google Scholar] [CrossRef]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef]
- Theodorou, K.; Boon, R.A. Endothelial Cell Metabolism in Atherosclerosis. Front. Cell Dev. Biol. 2018, 6, 82. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, D.; Liu, X.; Li, X.; Liu, F.; Yu, Y.; Jia, S.; Zhou, Y.; Zhao, Y. Endoplasmic Reticulum Stress Affects Lipid Metabolism in Atherosclerosis Via CHOP Activation and Over-Expression of miR-33. Cell. Physiol. Biochem. 2018, 48, 1995–2010. [Google Scholar] [CrossRef]
- Tian, H.; Li, Y.; Kang, P.; Wang, Z.; Yue, F.; Jiao, P.; Yang, N.; Qin, S.; Yao, S. Endoplasmic reticulum stress-dependent autophagy inhibits glycated high-density lipoprotein-induced macrophage apoptosis by inhibiting CHOP pathway. J. Cell. Mol. Med. 2019. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Qi, R.; Wang, Y.; Zhang, X.; Yu, M.; Tang, Y.; Wang, M.; Shu, Y.N.; Huang, W.; et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: The effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc. Res. 2015, 107, 579–589. [Google Scholar] [CrossRef]
- Yingchun, H.; Yahong, M.; Jiangping, W.; Xiaokui, H.; Xiaohong, Z. Increased inflammation, endoplasmic reticulum stress and oxidative stress in endothelial and macrophage cells exacerbate atherosclerosis in ApoCIII transgenic mice. Lipids Health Dis. 2018, 17, 220. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Panzhinskiy, E.; Hua, Y.; Culver, B.; Ren, J.; Nair, S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia 2013, 56, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; Laratta, E.; Procopio, C.; Hribal, M.L.; Sciacqua, A.; Perticone, M.; Miele, C.; Perticone, F.; Sesti, G. Interleukin-6 impairs the insulin signaling pathway, promoting production of nitric oxide in human umbilical vein endothelial cells. Mol. Cell. Biol. 2007, 27, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Colgan, S.M.; Tang, D.; Werstuck, G.H.; Austin, R.C. Endoplasmic reticulum stress causes the activation of sterol regulatory element binding protein-2. Int. J. Biochem. Cell Biol. 2007, 39, 1843–1851. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Maria Assunta, P.; Sara, G.; Carmela, N.; Maria Rosaria, C.; Monica, M. Endothelial Dysfunction in Diabetes: From Mechanisms to Therapeutic Targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef]
- Okon, E.B.; Chung, A.W.Y.; Rauniyar, P.; Padilla, E.; Tejerina, T.; McManus, B.M.; Luo, H.; van Breemen, C. Compromised Arterial Function in Human Type 2 Diabetic Patients. Diabetes 2005, 54, 2415–2423. [Google Scholar] [CrossRef]
- Duncan, E.R.; Crossey, P.A.; Walker, S.; Anilkumar, N.; Poston, L.; Douglas, G.; Ezzat, V.A.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.I. Effect of Endothelium-Specific Insulin Resistance on Endothelial Function In Vivo. Diabetes 2008, 57, 3307–3314. [Google Scholar] [CrossRef] [PubMed]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Asp. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Vander Mierde, D.; Song, B.; Flamez, D.; Creemers, J.W.; Tsukamoto, K.; Ribick, M.; Schuit, F.C.; Kaufman, R.J. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat. Med. 2005, 11, 757–764. [Google Scholar] [CrossRef]
- Yung, H.W.; Korolchuk, S.; Tolkovsky, A.M.; Charnock-Jones, D.S.; Burton, G.J. Endoplasmic reticulum stress exacerbates ischemia-reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J. 2007, 21, 872–884. [Google Scholar] [CrossRef]
- Villalobos-Labra, R.; Saez, P.J.; Subiabre, M.; Silva, L.; Toledo, F.; Westermeier, F.; Pardo, F.; Farias, M.; Sobrevia, L. Pre-pregnancy maternal obesity associates with endoplasmic reticulum stress in human umbilical vein endothelium. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3195–3210. [Google Scholar] [CrossRef]
- Di Pietro, N.; Marcovecchio, M.L.; Di Silvestre, S.; de Giorgis, T.; Cordone, V.G.P.; Lanuti, P.; Chiarelli, F.; Bologna, G.; Mohn, A.; Pandolfi, A. Plasma from pre-pubertal obese children impairs insulin stimulated Nitric Oxide (NO) bioavailability in endothelial cells: Role of ER stress. Mol. Cell. Endocrinol. 2017, 443, 52–62. [Google Scholar] [CrossRef]
- Seals, D.R.; Chung, E.; Kaplon, R.E.; Cox-York, K.; Reese, L.; Gentile, C.L. Activation of the Unfolded Protein Response in Vascular Endothelial Cells of Nondiabetic Obese Adults. J. Clin. Endocrinol. Metab. 2013, 98, E1505–E1509. [Google Scholar] [CrossRef]
- Kim, J.-A.; Jang, H.-J.; Hwang, D.H. Toll-like receptor 4-induced endoplasmic reticulum stress contributes to impairment of vasodilator action of insulin. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E767–E776. [Google Scholar] [CrossRef]
- Shah, D.; Romero, F.; Guo, Z.; Sun, J.; Li, J.; Kallen, C.B.; Naik, U.P.; Summer, R. Obesity-Induced Endoplasmic Reticulum Stress Causes Lung Endothelial Dysfunction and Promotes Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2017, 57, 204–215. [Google Scholar] [CrossRef]
- Agouni, A.; Tual-Chalot, S.; Chalopin, M.; Duluc, L.; Mody, N.; Martinez, M.C.; Andriantsitohaina, R.; Delibegovic, M. Hepatic protein tyrosine phosphatase 1B (PTP1B) deficiency protects against obesity-induced endothelial dysfunction. Biochem. Pharmacol. 2014, 92, 607–617. [Google Scholar] [CrossRef]
- Agouni, A.; Mody, N.; Owen, C.; Czopek, A.; Zimmer, D.; Bentires-Alj, M.; Bence, K.K.; Delibegovic, M. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem. J. 2011, 438, 369–378. [Google Scholar] [CrossRef]
- Owen, C.; Lees, E.K.; Grant, L.; Zimmer, D.J.; Mody, N.; Bence, K.K.; Delibegovic, M. Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia 2013, 56, 2286–2296. [Google Scholar] [CrossRef]
- Van der Vlies, D.; Makkinje, M.; Jansens, A.; Braakman, I.; Verkleij, A.J.; Wirtz, K.W.; Post, J.A. Oxidation of ER resident proteins upon oxidative stress: Effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid. Redox Signal. 2003, 5, 381–387. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Carreras-Sureda, A.; Pihan, P.; Hetz, C. Calcium signaling at the endoplasmic reticulum: Fine-tuning stress responses. Cell Calcium 2018, 70, 24–31. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Gorlach, A.; Klappa, P.; Kietzmann, T. The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.; Cui, S.; Wang, L.; Fu, B.; Ding, R.; Yang, J.; Chen, X.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 51, 402–410. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef]
- Suganya, N.; Dornadula, S.; Chatterjee, S.; Mohanram, R.K. Quercetin improves endothelial function in diabetic rats through inhibition of endoplasmic reticulum stress-mediated oxidative stress. Eur. J. Pharmacol. 2018, 819, 80–88. [Google Scholar] [CrossRef]
- Choy, K.W.; Lau, Y.S.; Murugan, D.; Mustafa, M.R. Chronic treatment with paeonol improves endothelial function in mice through inhibition of endoplasmic reticulum stress-mediated oxidative stress. PLoS ONE 2017, 12, e0178365. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.O.; Aw, T.Y.; Holbrook, N.J. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef]
- Wang, X.Z.; Ron, D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 1996, 272, 1347–1349. [Google Scholar] [CrossRef]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Struzik, J.; Szulc-Dabrowska, L. NF-kappaB Signaling in Targeting Tumor Cells by Oncolytic Viruses-Therapeutic Perspectives. Cancers 2018, 10, 426. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gokcen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-kappaB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef]
- Roy, P.; Sarkar, U.A.; Basak, S. The NF-kappaB Activating Pathways in Multiple Myeloma. Biomedicines 2018, 6, 59. [Google Scholar] [CrossRef]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.; Szabowski, A.; Schorpp-Kistner, M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene 2001, 20, 2413–2423. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef]
- Huang, J.; Wan, L.; Lu, H.; Li, X. High expression of active ATF6 aggravates endoplasmic reticulum stress-induced vascular endothelial cell apoptosis through the mitochondrial apoptotic pathway. Mol. Med. Rep. 2018, 17, 6483–6489. [Google Scholar] [CrossRef]
- Bhatta, M.; Ma, J.H.; Wang, J.J.; Sakowski, J.; Zhang, S.X. Enhanced endoplasmic reticulum stress in bone marrow angiogenic progenitor cells in a mouse model of long-term experimental type 2 diabetes. Diabetologia 2015, 58, 2181–2190. [Google Scholar] [CrossRef]
- Sarvani, C.; Sireesh, D.; Ramkumar, K.M. Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacol. Res 2017, 119, 412–421. [Google Scholar] [CrossRef]
- Zhang, Z.; Tong, N.; Gong, Y.; Qiu, Q.; Yin, L.; Lv, X.; Wu, X. Valproate protects the retina from endoplasmic reticulum stress-induced apoptosis after ischemia-reperfusion injury. Neurosci. Lett. 2011, 504, 88–92. [Google Scholar] [CrossRef]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef]
- Tong, Q.; Wu, L.; Jiang, T.; Ou, Z.; Zhang, Y.; Zhu, D. Inhibition of endoplasmic reticulum stress-activated IRE1alpha-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of Parkinson’s disease. Eur. J. Pharmacol. 2016, 776, 106–115. [Google Scholar] [CrossRef]
- Lakshmanan, A.P.; Thandavarayan, R.A.; Palaniyandi, S.S.; Sari, F.R.; Meilei, H.; Giridharan, V.V.; Soetikno, V.; Suzuki, K.; Kodama, M.; Watanabe, K. Modulation of AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates ER stress-induced renal apoptosis in streptozotocin-induced diabetic mice. Eur. J. Pharm. Sci. 2011, 44, 627–634. [Google Scholar] [CrossRef]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef]
- Lee, E.S.; Kim, H.M.; Kang, J.S.; Lee, E.Y.; Yadav, D.; Kwon, M.H.; Kim, Y.M.; Kim, H.S.; Chung, C.H. Oleanolic acid and N-acetylcysteine ameliorate diabetic nephropathy through reduction of oxidative stress and endoplasmic reticulum stress in a type 2 diabetic rat model. Nephrol. Dial. Transpl. 2016, 31, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Natsume, Y.; Ito, S.; Satsu, H.; Shimizu, M. Protective effect of quercetin on ER stress caused by calcium dynamics dysregulation in intestinal epithelial cells. Toxicology 2009, 258, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Suganya, N.; Bhakkiyalakshmi, E.; Suriyanarayanan, S.; Paulmurugan, R.; Ramkumar, K.M. Quercetin ameliorates tunicamycin-induced endoplasmic reticulum stress in endothelial cells. Cell Prolif. 2014, 47, 231–240. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. Int. J. Mol. Sci. 2019, 20, 1658. https://doi.org/10.3390/ijms20071658
Maamoun H, Abdelsalam SS, Zeidan A, Korashy HM, Agouni A. Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. International Journal of Molecular Sciences. 2019; 20(7):1658. https://doi.org/10.3390/ijms20071658
Chicago/Turabian StyleMaamoun, Hatem, Shahenda S. Abdelsalam, Asad Zeidan, Hesham M. Korashy, and Abdelali Agouni. 2019. "Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes" International Journal of Molecular Sciences 20, no. 7: 1658. https://doi.org/10.3390/ijms20071658
APA StyleMaamoun, H., Abdelsalam, S. S., Zeidan, A., Korashy, H. M., & Agouni, A. (2019). Endoplasmic Reticulum Stress: A Critical Molecular Driver of Endothelial Dysfunction and Cardiovascular Disturbances Associated with Diabetes. International Journal of Molecular Sciences, 20(7), 1658. https://doi.org/10.3390/ijms20071658