AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway

 ,
,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. AICAR Inhibits Cell Growth in Prostate Cancer Cells, but Not in Non-Cancerous Prostate Cells
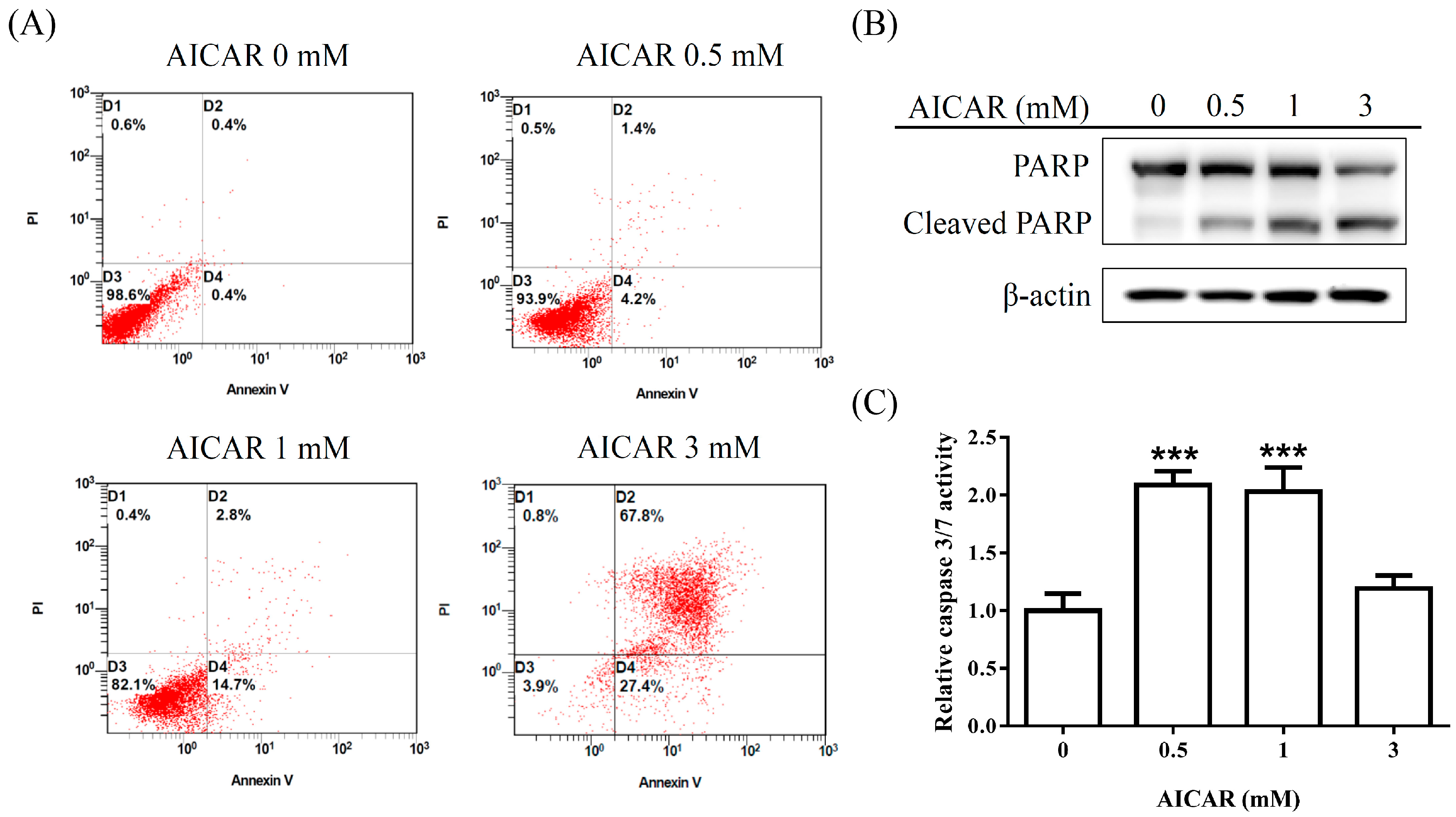
2.2. AICAR Induces Apoptosis in Prostate Cancer Cells
2.3. AICAR Inhibits Transforming Growth Factor-β (TGF-β)-Induced Epithelial to Mesenchymal Transition (EMT) and Attenuates TGF-β-Induced Migration and Invasion Activities
2.4. AICAR Exhibits Synergistic Effect with Docetaxel Treatment
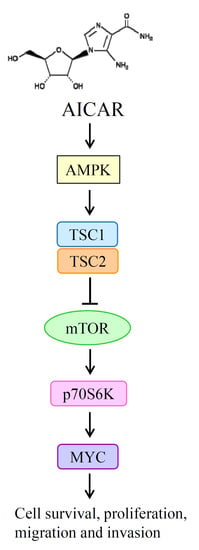
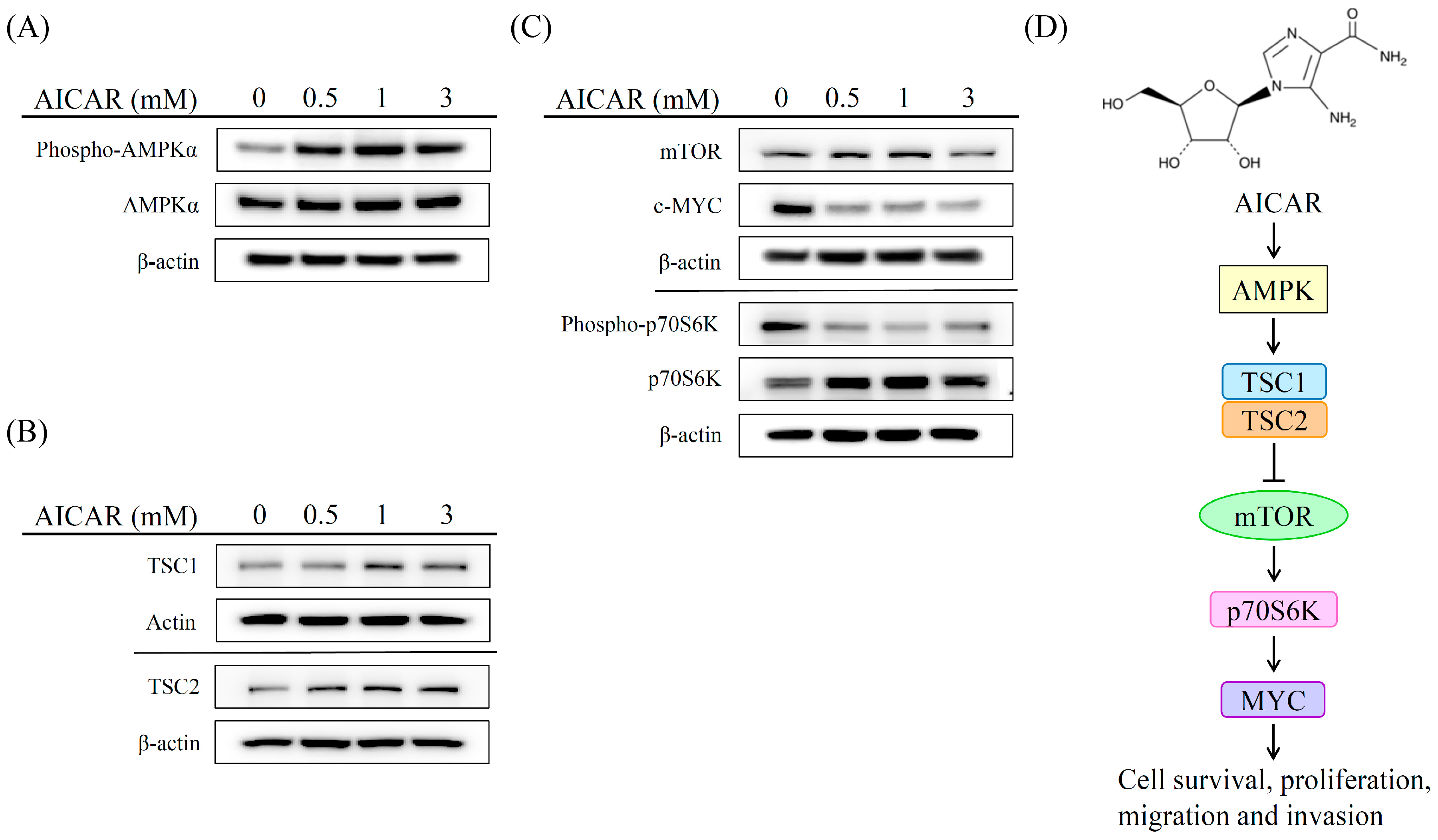
2.5. AICAR Inhibits the Growth of Prostate Cancer Cells Through Activating an Ampk/Mtor-Dependent Pathway
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. MTT Assay
4.4. Soft Agar Colony Formation Assay
4.5. Apoptosis Assay
4.6. Western Blot
4.7. Caspase 3/7 Activity Assay
4.8. Migration Assay
4.9. Invasion Assay
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AICAR | 5-aminoimidazole-4-carbox-amide-1-β-d-ribofuranoside |
| AMPK | AMP-activated protein kinase |
| ADT | Androgen deprivation therapy |
| CRPC | Castration-resistant prostate cancer |
| AR | Androgen receptor |
| ER | Estrogen receptor |
| FBS | Fetal bovine serum |
| BSA | Bovine serum albumin |
| PBS | Phosphate-buffered saline |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| PI | Propidium iodide |
| TBST | Tris-buffered saline/Tween 20 |
| PARP | Poly(ADP-ribose) polymerase |
| TGF-β | Transforming growth factor-beta |
| EMT | Epithelial to mesenchymal transition |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Litwin, M.S.; Tan, H.J. The diagnosis and treatment of prostate cancer: A review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.; Taneja, S. Focal therapy for prostate cancer: The current status. Prostate Int. 2015, 3, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Asmane, I.; Ceraline, J.; Duclos, B.; Rob, L.; Litique, V.; Barthelemy, P.; Bergerat, J.P.; Dufour, P.; Kurtz, J.E. New strategies for medical management of castration-resistant prostate cancer. Oncology 2011, 80, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Théodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Gao, A.C. Mechanisms of persistent activation of the androgen receptor in CRPC: Recent advances and future perspectives. World J. Urol. 2012, 30, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and their receptors in prostate cancer: Therapeutic implications. Front. Oncol. 2018, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; di Donato, M.; di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Grahame Hardie, D. Regulation of AMP-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Nagy, L.; Fodor, T.; Liaudet, L.; Pacher, P. Poly(ADP-ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol. Metab. 2015, 26, 75–83. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell. Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Icard, P.; Lincet, H. A global view of the biochemical pathways involved in the regulation of the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1826, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Mitochondria in cancer. Oncogene 2006, 25, 4630–4632. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell. Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef]
- Fodor, T.; Szanto, M.; Abdul-Rahman, O.; Nagy, L.; Der, A.; Kiss, B.; Bai, P. Combined treatment of MCF-7 cells with AICAR and methotrexate, arrests cell cycle and reverses Warburg metabolism through AMP-activated protein kinase (AMPK) and FOXO1. PLoS ONE 2016, 11, e0150232. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Kim, J.Y.; Ghafoory, S.; Duvaci, T.; Rafiee, R.; Theobald, J.; Alborzinia, H.; Holenya, P.; Fredebohm, J.; Merz, K.H.; et al. Methylisoindigo preferentially kills cancer stem cells by interfering cell metabolism via inhibition of LKB1 and activation of AMPK in PDACs. Mol. Oncol. 2016, 10, 806–824. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, Y.K.; Kim, H.J.; Park, O.J.; Kim, Y.M. AMPK interacts with beta-catenin in the regulation of hepatocellular carcinoma cell proliferation and survival with selenium treatment. Oncol. Rep. 2016, 35, 1566–1572. [Google Scholar] [CrossRef]
- Choudhury, Y.; Yang, Z.; Ahmad, I.; Nixon, C.; Salt, I.P.; Leung, H.Y. AMP-activated protein kinase (AMPK) as a potential therapeutic target independent of PI3K/Akt signaling in prostate cancer. Oncoscience 2014, 1, 446–456. [Google Scholar] [CrossRef]
- Sauer, H.; Engel, S.; Milosevic, N.; Sharifpanah, F.; Wartenberg, M. Activation of AMP-kinase by AICAR induces apoptosis of DU-145 prostate cancer cells through generation of reactive oxygen species and activation of c-Jun N-terminal kinase. Int. J. Oncol. 2012, 40, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, S.; Hardie, D.G. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim. Biophys. Acta 2010, 1804, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.L.; Lingrell, S.; Dyck, J.R.; Vance, D.E. Inhibition of hepatic phosphatidylcholine synthesis by 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside is independent of AMP-activated protein kinase activation. J. Biol. Chem. 2007, 282, 4516–4523. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.M.; Santidrian, A.F.; Campas, C.; Gil, J. 5-Aminoimidazole-4-carboxamide riboside induces apoptosis in Jurkat cells, but the AMP-activated protein kinase is not involved. Biochem. J. 2003, 370, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Saha, A.K.; Wen, R.; Ruderman, N.B.; Luo, Z. AMP-activated protein kinase activators can inhibit the growth of prostate cancer cells by multiple mechanisms. Biochem. Biophys. Res. Commun. 2004, 321, 161–167. [Google Scholar] [CrossRef]
- Guo, F.; Liu, S.Q.; Gao, X.H.; Zhang, L.Y. AICAR induces AMPK-independent programmed necrosis in prostate cancer cells. Biochem. Biophys. Res. Commun. 2016, 474, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.S.; Millena, A.C.; Khan, S.A. TGF-beta effects on prostate cancer cell migration and invasion require FosB. Prostate 2017, 77, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Courchet, J.; Viollet, B.; Brenman, J.E.; Polleux, F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc. Natl. Acad. Sci. USA 2011, 108, 5849–5854. [Google Scholar] [CrossRef]
- Zhou, W.; Marcus, A.I.; Vertino, P.M. Dysregulation of mTOR activity through LKB1 inactivation. Chin. J. Cancer 2013, 32, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Frigo, D.E. A spatiotemporal hypothesis for the regulation, role, and targeting of AMPK in prostate cancer. Nat. Rev. Urol. 2017, 14, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell. Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef]
- Chen, L.; Han, F.; Qu, H.; Yan, H.; Ren, L.; Yang, S. Combination therapy with 5-amino-4-imidazolecarboxamide riboside and arsenic trioxide in acute myeloid leukemia cells involving AMPK/TSC2/mTOR pathway. Pharmazie 2013, 68, 117–123. [Google Scholar] [PubMed]
- Lee, K.C.; Lin, C.T.; Chang, S.F.; Chen, C.N.; Liu, J.L.; Huang, W.S. Effect of AICAR and 5-Fluorouracil on X-ray Repair, Cross-Complementing Group 1 Expression, and Consequent Cytotoxicity Regulation in Human HCT-116 Colorectal Cancer Cells. Int. J. Mol. Sci. 2017, 18, 2363. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; Gonzalez-Girones, D.M.; Iglesias-Serret, D.; Coll-Mulet, L.; Cosialls, A.M.; de Frias, M.; Campàs, C.; González-Barca, E.; Alonso, E.; Labi, V.; et al. AICAR induces apoptosis independently of AMPK and p53 through up-regulation of the BH3-only proteins BIM and NOXA in chronic lymphocytic leukemia cells. Blood 2010, 116, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Su, R.Y.; Chao, Y.; Chen, T.Y.; Huang, D.Y.; Lin, W.W. 5-Aminoimidazole-4-carboxamide riboside sensitizes TRAIL- and TNFα-induced cytotoxicity in colon cancer cells through AMP-activated protein kinase signaling. Mol. Cancer Ther. 2007, 6, 1562–1571. [Google Scholar] [CrossRef] [PubMed]
- Bubendorf, L.; Schopfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Guo, Y.; Chen, S.; Zhong, C.; Xue, Y.; Zhang, Y.; Lai, X.; Wei, Y.; Yu, S.; Zhang, J.; et al. Metformin enhances tamoxifen-mediated tumor growth inhibition in ER-positive breast carcinoma. BMC Cancer 2014, 11, 172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, H.; Zhou, X.; Li, Y.; Liu, T.; Yin, X.; Zhang, B. Role of metformin in inhibiting estrogen-induced proliferation and regulating ERα and ERβ expression in human endometrial cancer cells. Oncol. Lett. 2017, 14, 4949–4956. [Google Scholar] [CrossRef] [PubMed]
- Poorthuis, M.H.F.; Vernooij, R.W.M.; van Moorselaar, R.J.A.; de Reijke, T.M. First-line non-cytotoxic therapy in chemotherapy-naive patients with metastatic castration-resistant prostate cancer: A systematic review of 10 randomised clinical trials. BJU Int. 2017, 119, 831–845. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.-C.; Hsieh, K.-L.; Liu, P.-L.; Yeh, H.-C.; Huang, S.-P.; Fang, S.-H.; Cheng, W.-C.; Huang, K.-H.; Chiu, F.-Y.; Lin, I.-L.; et al. AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway. Int. J. Mol. Sci. 2019, 20, 1647. https://doi.org/10.3390/ijms20071647
Su C-C, Hsieh K-L, Liu P-L, Yeh H-C, Huang S-P, Fang S-H, Cheng W-C, Huang K-H, Chiu F-Y, Lin I-L, et al. AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway. International Journal of Molecular Sciences. 2019; 20(7):1647. https://doi.org/10.3390/ijms20071647
Chicago/Turabian StyleSu, Chia-Cheng, Kun-Lin Hsieh, Po-Len Liu, Hsin-Chih Yeh, Shu-Pin Huang, Shih-Hua Fang, Wei-Chung Cheng, Kuan-Hua Huang, Fang-Yen Chiu, I-Ling Lin, and et al. 2019. "AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway" International Journal of Molecular Sciences 20, no. 7: 1647. https://doi.org/10.3390/ijms20071647
APA StyleSu, C.-C., Hsieh, K.-L., Liu, P.-L., Yeh, H.-C., Huang, S.-P., Fang, S.-H., Cheng, W.-C., Huang, K.-H., Chiu, F.-Y., Lin, I.-L., Huang, M.-Y., & Li, C.-Y. (2019). AICAR Induces Apoptosis and Inhibits Migration and Invasion in Prostate Cancer Cells Through an AMPK/mTOR-Dependent Pathway. International Journal of Molecular Sciences, 20(7), 1647. https://doi.org/10.3390/ijms20071647