Functional and Structural Features of Disease-Related Protein Variants


Abstract
1. Introduction
2. Results and Discussion
2.1. HVAR3D: A Dataset of Protein Variants with Structural and Functional Information
2.2. Structural Characterization of HVAR3D
2.3. Functional Characterization of HVAR3D
2.4. Structural and Functional Characterization of HVAR3D
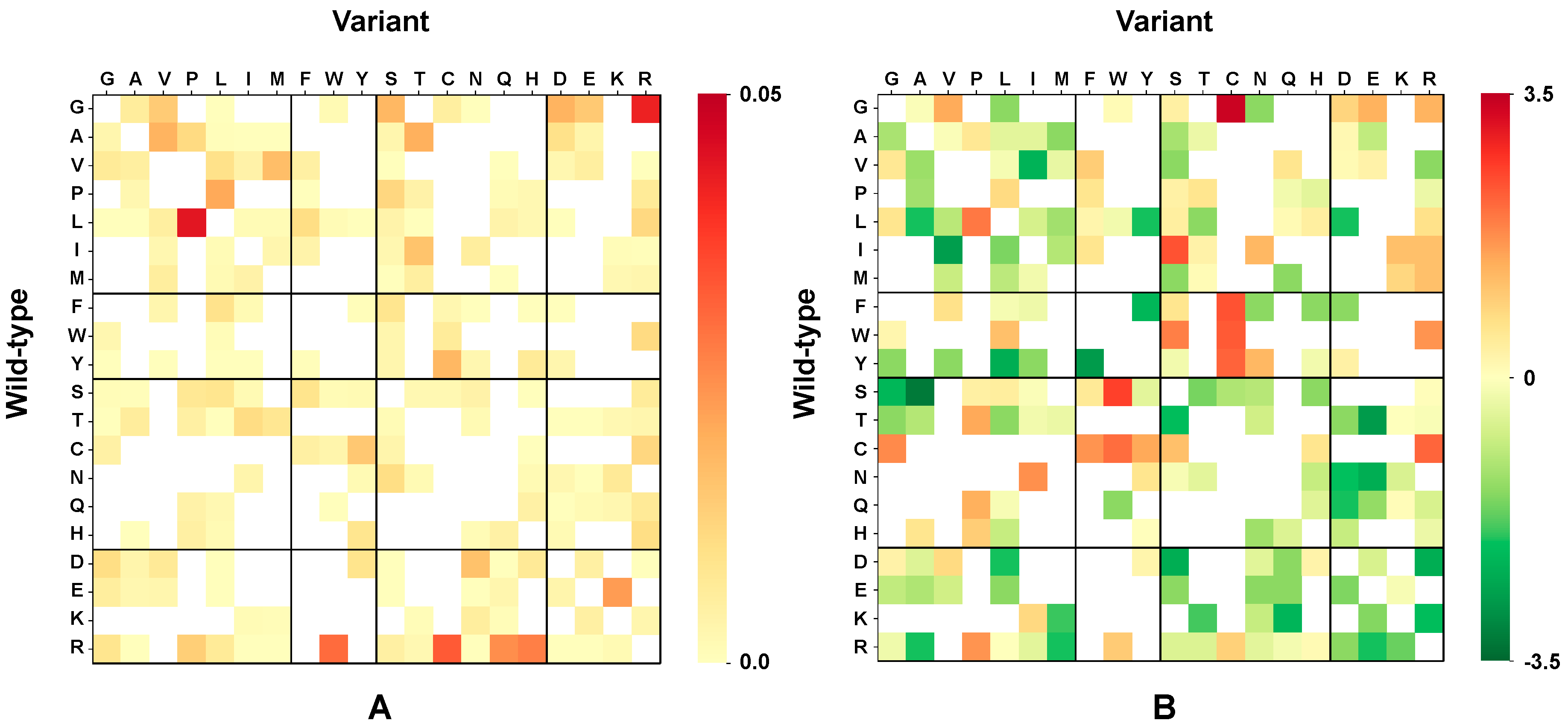
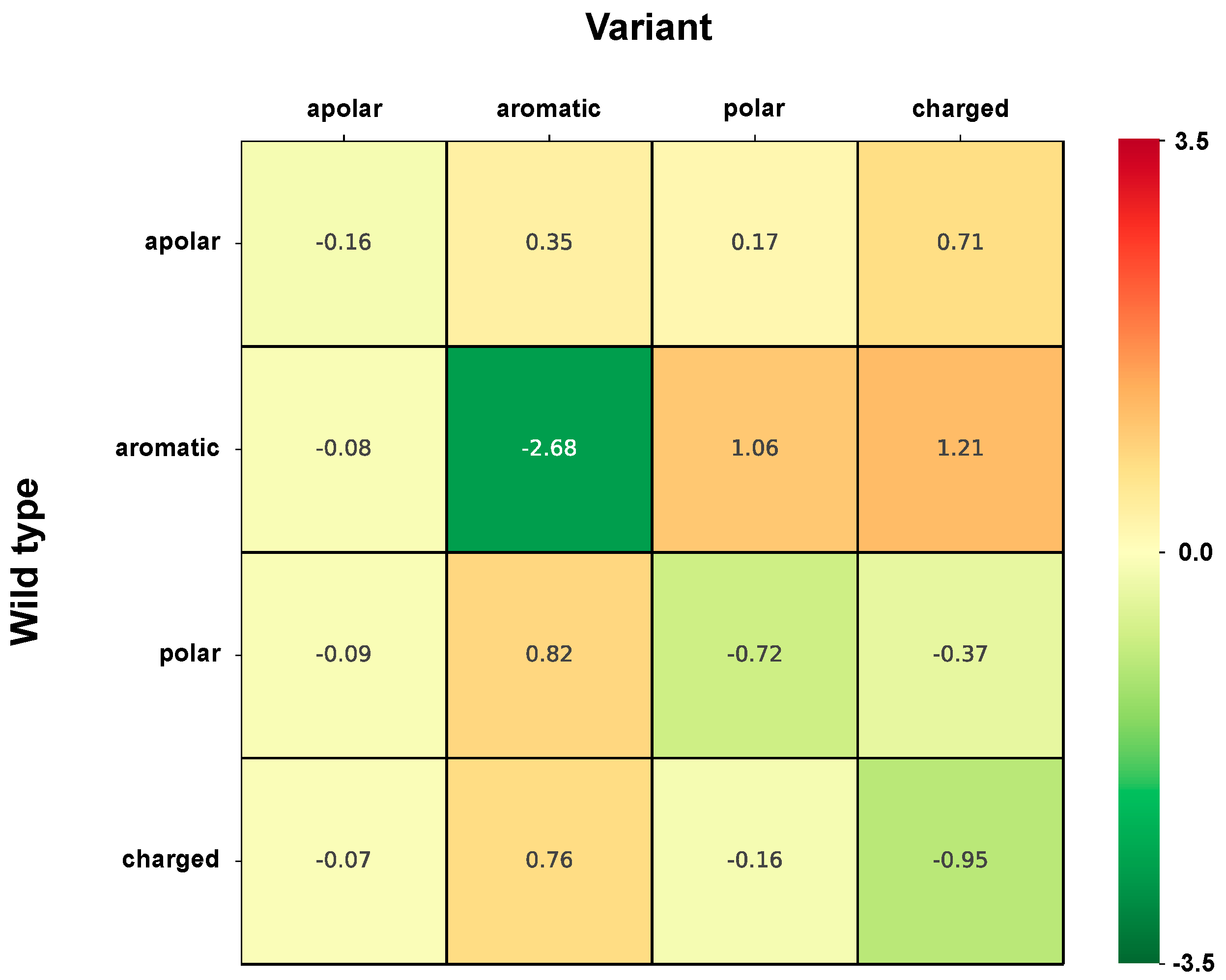
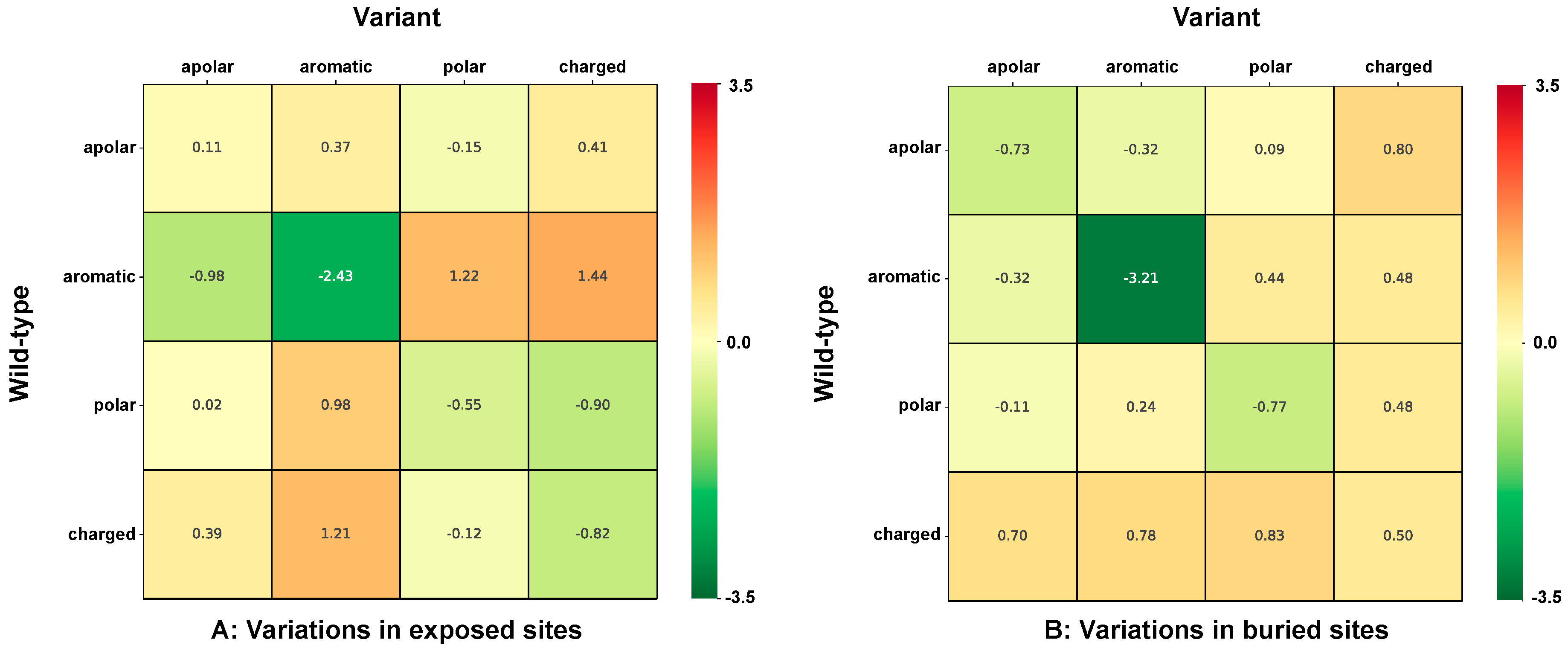
2.5. Chemico-Physical Characterization of Disease Related Variations in HVAR3D
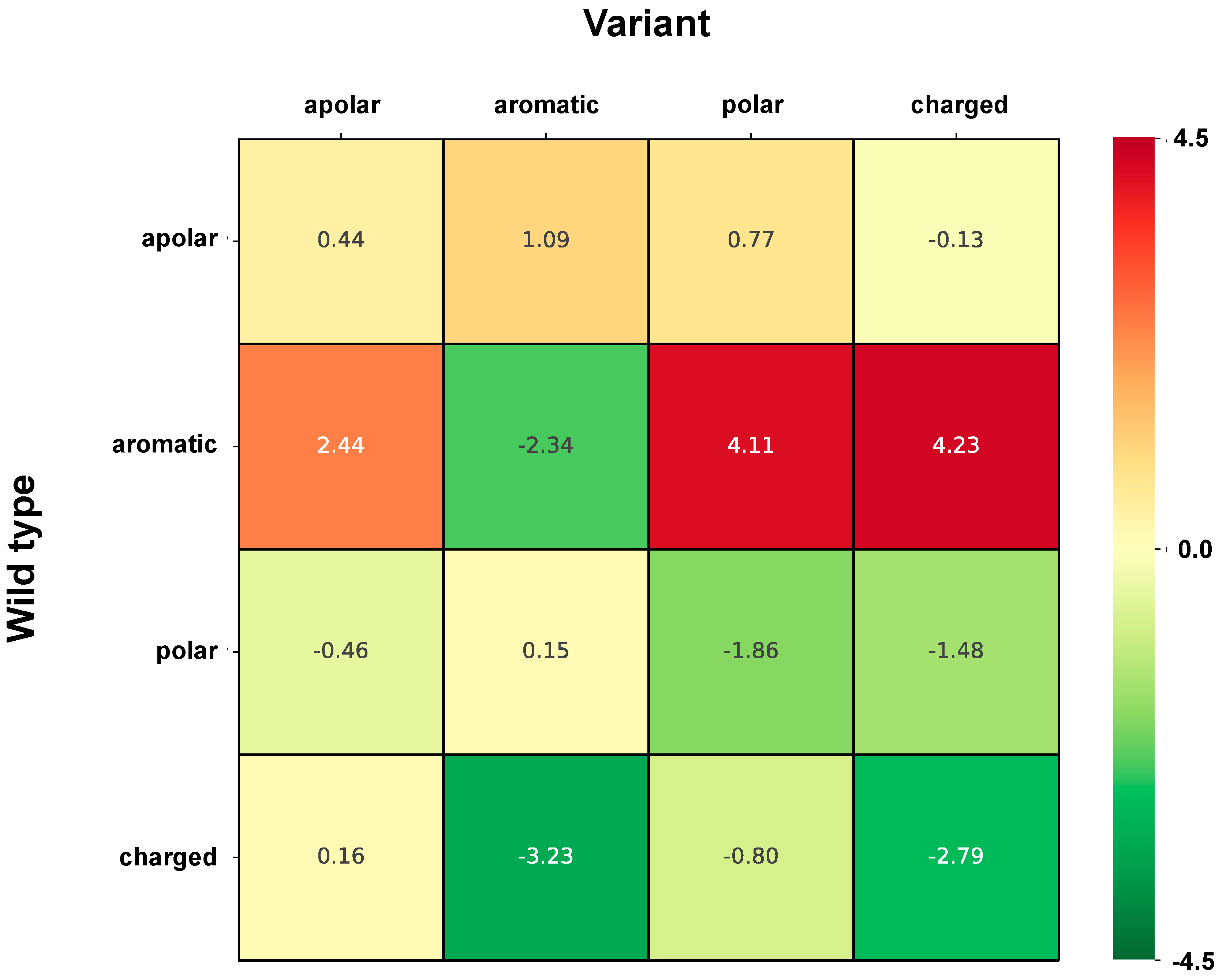
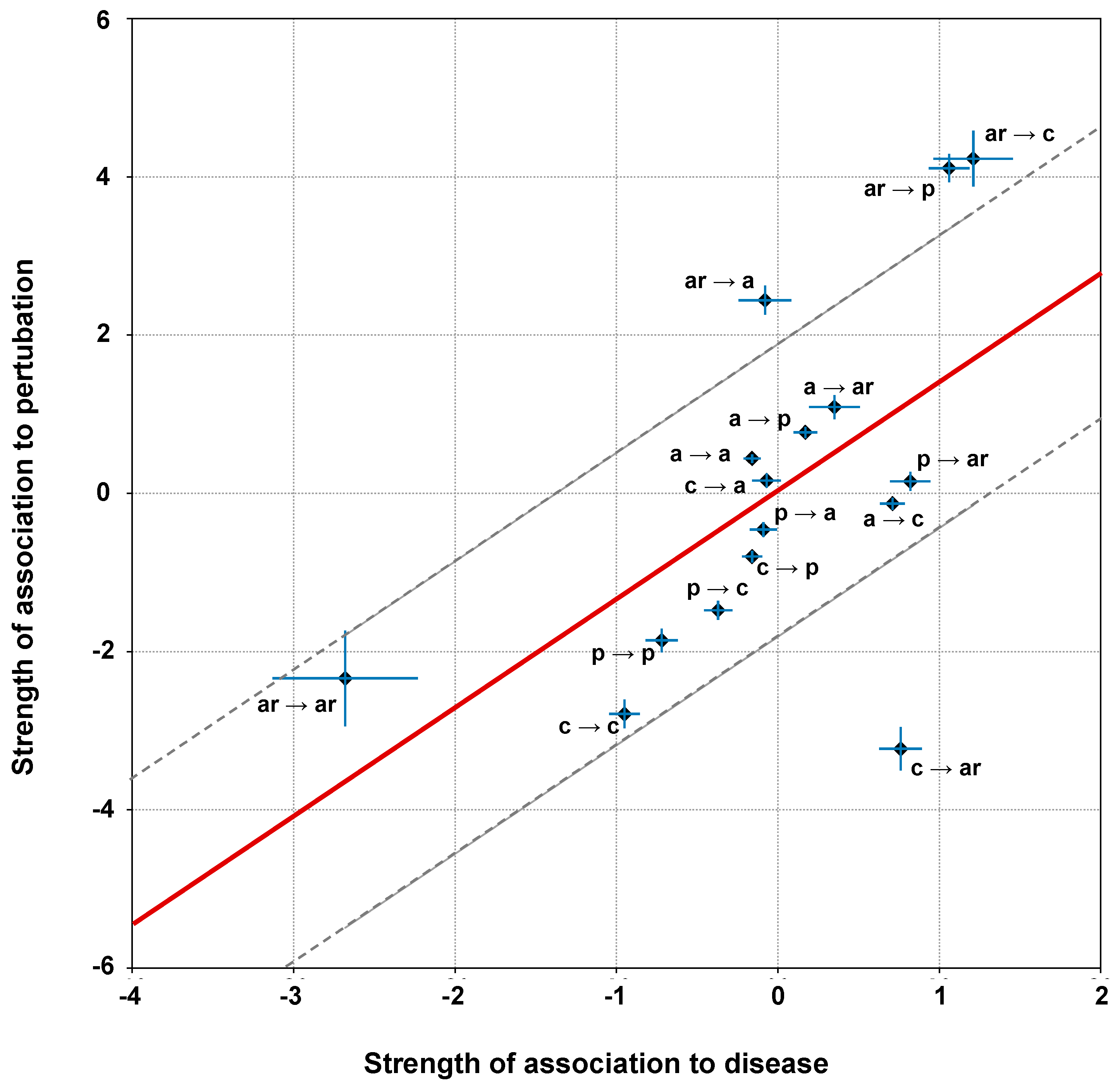
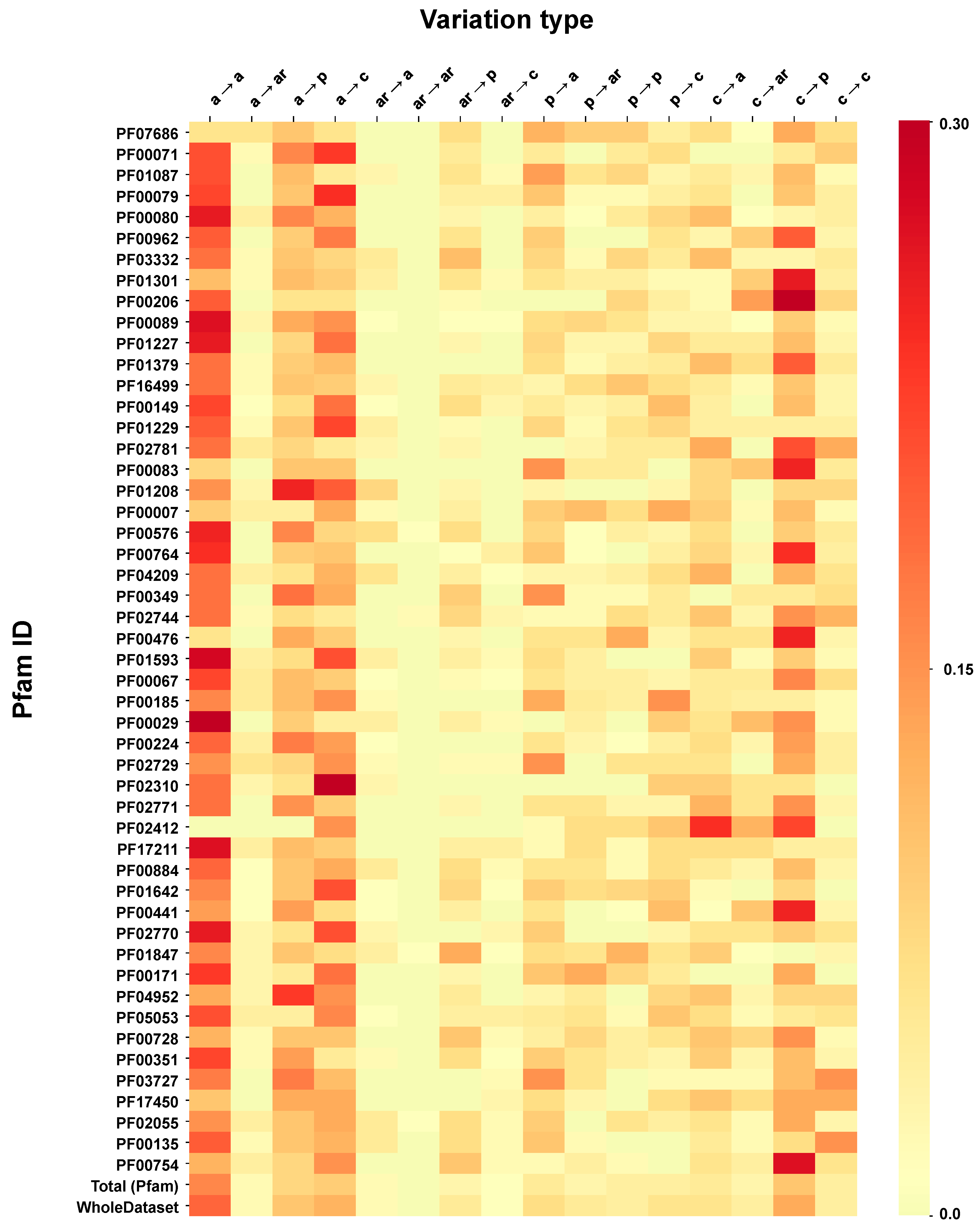
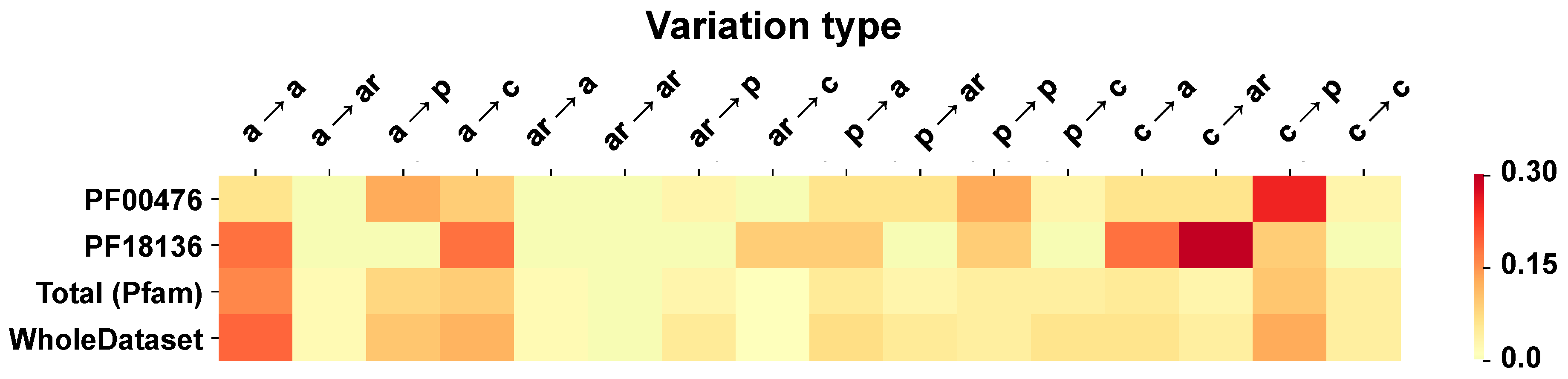
2.6. Correlation of Structural and Chemico-Physical Features of Disease SVRs in HVAR3D
2.6.1. Protein Stability Perturbartion and Chemico-Physical Features of Disease SVRs
2.6.2. Residue Solvent Exposure and Chemico-Physical Features of Disease SRVs
2.6.3. Pfam Protein Domains and Chemico-Physical Features of Disease SRVs
3. Materials and Methods
3.1. Variant Data Collection and Mapping to Protein Data Bank (PDB)
3.2. Retrieving Protein Annotations
3.3. Computing Residue Surface Exposure and Protein-Protein Interfaces
3.4. Prediction of the Impact of SRVs on Protein Stability
3.5. Computing the Association Strength of Each Variation Type to Disease and Pertubation
3.6. Computing Linear Regression and Residuals
3.7. Kullback–Lieber Divergence Between Distributions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SRV | Single Residue Variant |
| GO | Gene Ontology |
References
- Chakravorty, S.; Hegde, M. Gene and variant annotation for mendelian disorders in the era of advanced sequencing technologies. Annu. Rev. Genom. Hum. Genet. 2017, 31, 229–256. [Google Scholar] [CrossRef] [PubMed]
- Amberger, J.S.; Hamosh, A. Searching online mendelian inheritance in man (OMIM): A knowledgebase of human genes and genetic phenotypes. Curr. Protoc. Bioinform. 2017, 58, 1.2.1–1.2.12. [Google Scholar] [CrossRef]
- Babbi, G.; Martelli, P.L.; Profiti, G.; Bovo, S.; Savojardo, C.; Casadio, R. eDGAR: A database of Disease-Gene Associations with annotated Relationships among genes. BMC Genom. 2017, 18, 554. [Google Scholar] [CrossRef]
- Kroncke, B.M.; Vanoye, C.G.; Meiler, J.; George, A.L., Jr.; Sanders, C.R. Personalized biochemistry and biophysics. Biochemistry 2015, 54, 2551–2559. [Google Scholar] [CrossRef]
- Wang, Z.; Moult, J. SNPs, protein structure, and disease. Hum. Mutat. 2001, 17, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Steward, R.E.; MacArthur, M.W.; Laskowski, R.A.; Thornton, J.M. Molecular basis of inherited diseases: A structural perspective. Trends Genet. 2003, 19, 505–513. [Google Scholar] [CrossRef]
- Petukh, M.; Kucukkal, T.G.; Alexov, E. On human disease-causing amino acid variants: Statistical study of sequence and structural patterns. Hum. Mutat. 2015, 36, 524–534. [Google Scholar] [CrossRef]
- David, A.; Sternberg, M.J. The contribution of missense mutations in core and rim residues of protein-protein interfaces to human disease. J. Mol. Biol. 2015, 427, 2886–2898. [Google Scholar] [CrossRef]
- Gao, M.; Zhou, H.; Skolnick, J. Insights into disease-associated mutations in the human proteome through protein structural analysis. Structure 2015, 3, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Martelli, P.L.; Fariselli, P.; Savojardo, C.; Babbi, G.; Aggazio, F.; Casadio, R. Large scale analysis of protein stability in OMIM disease related human protein variants. BMC Genom. 2016, 17, 397. [Google Scholar] [CrossRef]
- Schaafsma, G.C.P.; Vihinen, M. Large differences in proportions of harmful and benign amino acid substitutions between proteins and diseases. Hum. Mutat. 2017, 38, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Schaafsma, G.C.P.; Vihinen, M. Representativeness of variation benchmark datasets. BMC Bioinform. 2018, 19, 461. [Google Scholar] [CrossRef] [PubMed]
- Medina-Carmona, E.; Fuchs, J.E.; Gavira, J.A.; Mesa-Torres, N.; Neira, J.L.; Salido, E.; Palomino-Morales, R.; Burgos, M.; Timson, D.J.; Pey, A.L. Enhanced vulnerability of human proteins towards disease-associated inactivation through divergent evolution. Hum. Mol. Genet. 2017, 26, 3531–3544. [Google Scholar] [CrossRef]
- Khoo, K.H.; Mayer, S.; Fersht, A.R. Effects of stability on the biological function of p53. J. Biol. Chem. 2009, 284, 30974–30980. [Google Scholar] [CrossRef]
- Khoo, K.H.; Andreeva, A.; Fersht, A.R. Adaptive evolution of p53 thermodynamic stability. J. Mol. Biol. 2009, 393, 161–175. [Google Scholar] [CrossRef]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. NAD(P)H quinone oxidoreductase (NQO1): An enzyme which needs just enough mobility, in just the right places. Biosci. Rep. 2019, 39, BSR20180459. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Li, Z.; Moult, J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005, 353, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO—Multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Costa, C.; Orozco, M.; de la Cruz, X. Characterization of disease-associated single amino acid polymorphisms in terms of sequence and structure properties. J. Mol. Biol. 2002, 315, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Casadio, R.; Vassura, M.; Tiwari, S.; Fariselli, P.; Martelli, P.L. Correlating disease-related mutations to their effect on protein stability: A large-scale analysis of the human proteome. Hum. Mutat. 2011, 32, 1161–1170. [Google Scholar] [CrossRef]
- Peng, Y.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins 2016, 84, 232–239. [Google Scholar] [CrossRef]
- Fariselli, P.; Martelli, P.L.; Savojardo, C.; Casadio, R. INPS: Predicting the impact of non-synonymous variations on protein stability from sequence. Bioinformatics 2015, 31, 2816–2821. [Google Scholar] [CrossRef]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef]
- Martin, A.C.R. Mapping PDB chains to UniProtKB entries. Bioinformatics 2005, 21, 4297–4301. [Google Scholar] [CrossRef]
- Velankar, S.; Dana, J.M.; Jacobsen, J.; van Ginkel, G.; Gane, P.J.; Luo, J.; Oldfield, T.J.; O’Donovan, C.; Martin, M.J.; Kleywegt, G.J. SIFTS: Structure Integration with Function, Taxonomy and Sequences resource. Nucleic Acids Res. 2013, 41, D483–D489. [Google Scholar] [CrossRef]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO::TermFinder—Open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Rost, B.; Sander, C. Conservation and prediction of solvent accessibility in protein families. Proteins 1994, 20, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Niroula, A.; Vihinen, M. Variation interpretation predictors: Principles, types, performance, and choice. Hum. Mutat. 2016, 37, 579–597. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| #PDB Entries | #SRVs | # SRVs Buried (b) | # SRVs Exposed (c) |
|---|---|---|---|
| 386 (D+D/N) (a) | 6621 | 3998 | 2623 |
| 241 (D/N) (a) | 5450 | 3325 | 2125 |
| 145 (D) (a) | 1171 | 673 | 498 |
| Biological Assembly | #PDB Entries | #D-SRVs (a) | #N-SRV (b) | #D-SRVs Interface (c) | #N-SRVs Interface (d) |
|---|---|---|---|---|---|
| Monomers | 146 | 2011 | 340 | - | - |
| Multimers | 240 | 3566 | 704 | 501 | 109 |
| EC(a) | #Protein Chains | #D-SRVs (b) | #N-SRVs (c) | #D-SRVs on: | #N-SRVs on: | ||
|---|---|---|---|---|---|---|---|
| ACTIVE SITE | BINDING SITE | ACTIVE SITE | BINDING SITE | ||||
| Oxidoreductases | 64 | 1045 | 107 | 2 | 20 | 0 | 0 |
| Transferases | 71 | 1058 | 122 | 5 | 20 | 0 | 0 |
| Hydrolases | 69 | 1342 | 143 | 7 | 10 | 0 | 1 |
| Lyases | 19 | 260 | 36 | 0 | 3 | 0 | 0 |
| Isomerases | 7 | 236 | 14 | 0 | 7 | 0 | 0 |
| Ligases | 11 | 138 | 15 | 0 | 7 | 0 | 2 |
| Multiple ECs | 7 | 179 | 18 | 0 | 0 | 0 | 0 |
| Total | 248 | 4258 | 455 | 14 | 67 | 0 | 3 |
| GO MF (a) | #Protein Chains | #D-SRVs | #N-SRVs | #D-SRVs on Binding Sites | #N-SRVs on Binding Sites |
|---|---|---|---|---|---|
| Antioxidant activity | 1 (1) (b) | 3 | 55 | 0 | 1 |
| Catalytic activity | 22 (20) | 291 | 52 | 0 | 0 |
| Molecular carrier activity | 2 (2) | 11 | 279 | 0 | 11 |
| Molecular function regulator | 25 (22) | 266 | 50 | 2 | 0 |
| Molecular transducer activity | 4 (3) | 10 | 13 | 0 | 0 |
| Structural molecule activity | 11 (4) | 167 | 19 | 0 | 0 |
| Transcription regulator activity | 4 (3) | 13 | 2 | 0 | 0 |
| Translation regulator activity | 1 (1) | 2 | 0 | 0 | 0 |
| Transporter activity | 10 (9) | 134 | 27 | 3 | 0 |
| Binding (c) | 29 | 297 | 29 | 0 | 0 |
| Multiple terms (d) | 31 (26) | 115 | 62 | 0 | 0 |
| Non annotated | 4 | 10 | 1 | 0 | 0 |
| Total | 144 (120) | 1319 | 589 | 5 | 12 |
| Pfam ID | Pfam Name | #Protein Chains | #D-SRVs | #N-SRVs |
|---|---|---|---|---|
| PF00351 | Biopterin-dependent aromatic amino acid hydroxylase | 3 | 213 | 5 |
| PF00884 | Sulfatase | 4 | 159 | 4 |
| PF16499 | Alpha galactosidase A | 2 | 143 | 15 |
| PF00023 | Ankyrin repeat | 1 | 1 | 2 |
| PF00050 | Kazal-type serine protease inhibitor domain | 1 | 1 | 2 |
| PF00070 | Pyridine nucleotide-disulphide oxidoreductase | 1 | 1 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savojardo, C.; Babbi, G.; Martelli, P.L.; Casadio, R. Functional and Structural Features of Disease-Related Protein Variants. Int. J. Mol. Sci. 2019, 20, 1530. https://doi.org/10.3390/ijms20071530
Savojardo C, Babbi G, Martelli PL, Casadio R. Functional and Structural Features of Disease-Related Protein Variants. International Journal of Molecular Sciences. 2019; 20(7):1530. https://doi.org/10.3390/ijms20071530
Chicago/Turabian StyleSavojardo, Castrense, Giulia Babbi, Pier Luigi Martelli, and Rita Casadio. 2019. "Functional and Structural Features of Disease-Related Protein Variants" International Journal of Molecular Sciences 20, no. 7: 1530. https://doi.org/10.3390/ijms20071530
APA StyleSavojardo, C., Babbi, G., Martelli, P. L., & Casadio, R. (2019). Functional and Structural Features of Disease-Related Protein Variants. International Journal of Molecular Sciences, 20(7), 1530. https://doi.org/10.3390/ijms20071530