Plasma Rich in Growth Factors (PRGF) Disrupt the Blood-Brain Barrier Integrity and Elevate Amyloid Pathology in the Brains of 5XFAD Mice

 , and
, and 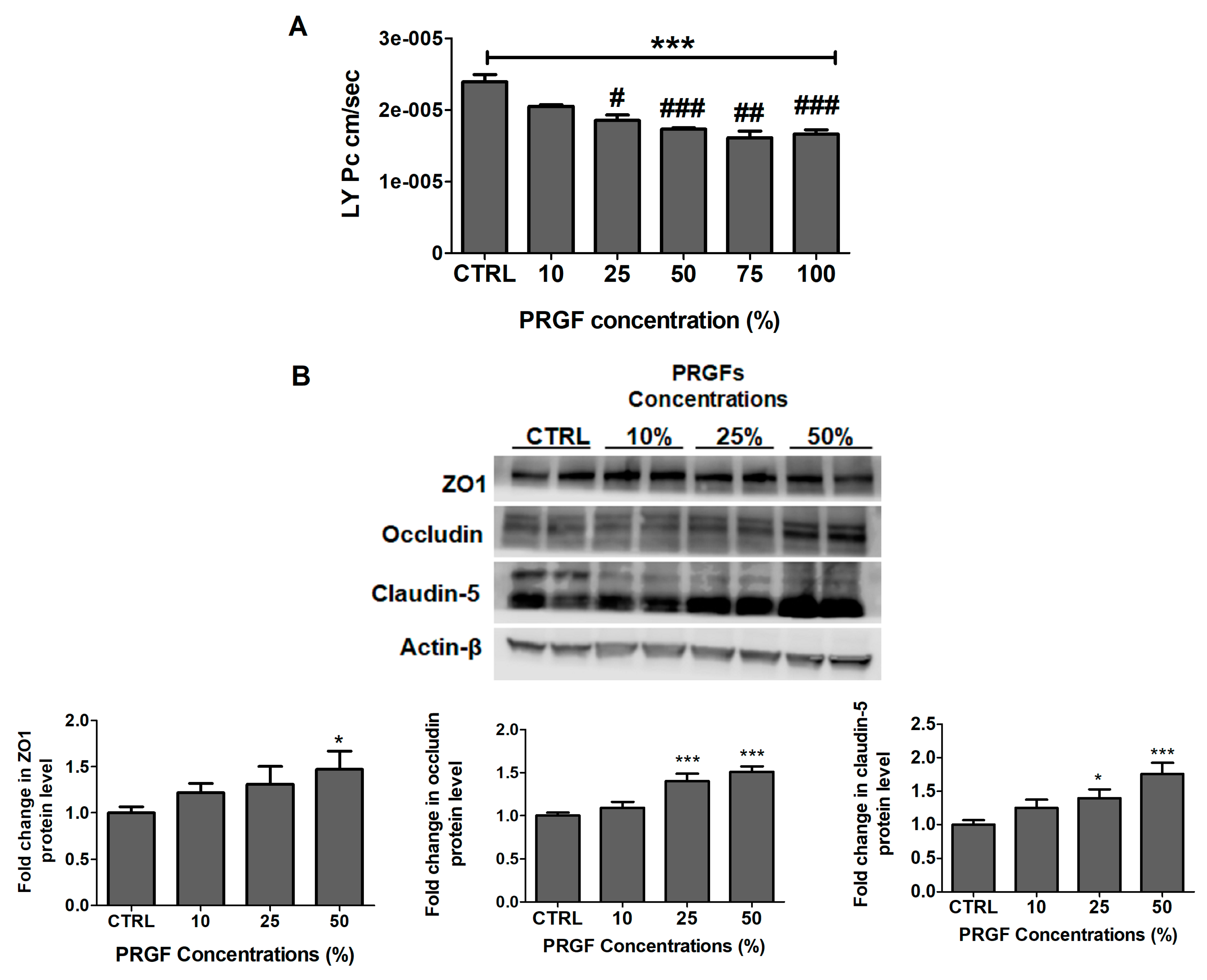{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Growth Factor Concentrations in Activated Plasma
2.2. PRGF Increased bEnd3 Monolayer Tightness and Aβ42 Transport Across the Cell-Based BBB Model
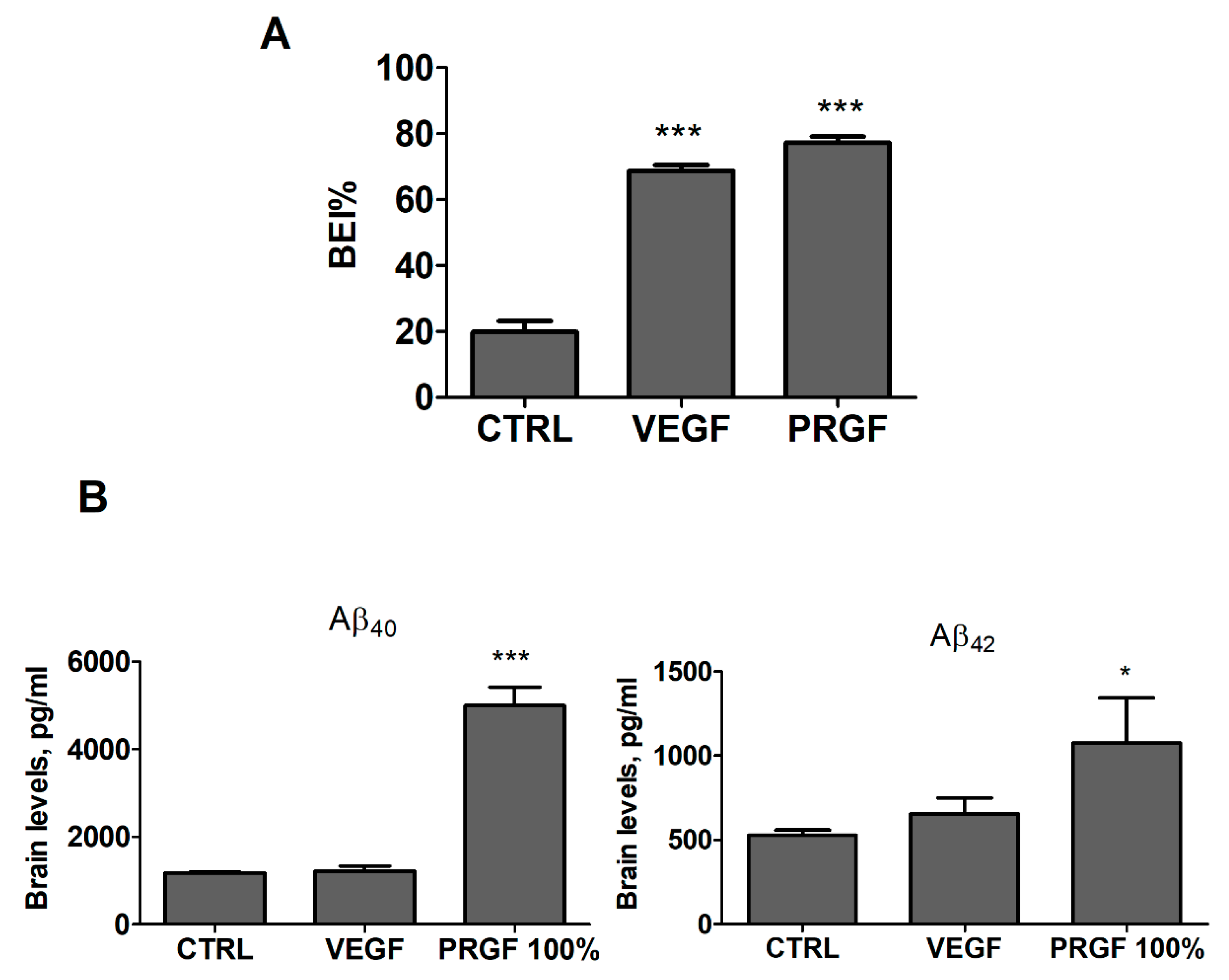
2.3. VEGF and PRGF Enhanced 125I-Aβ40 Clearance across Mice BBB with Differing Effect on Aβ Brain Load
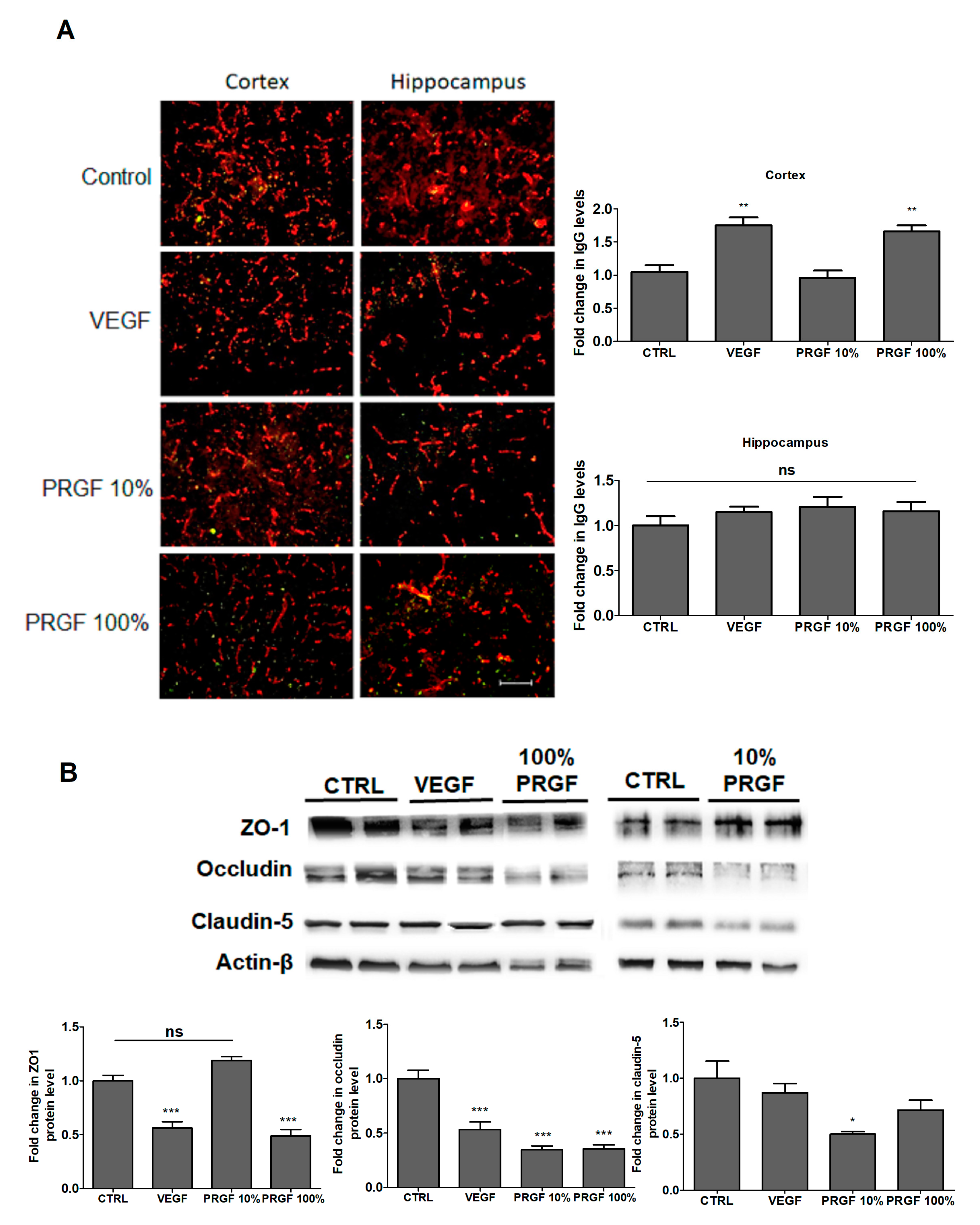
2.4. VEGF and PRGF modulated BBB tightness in 5xFAD mice brains
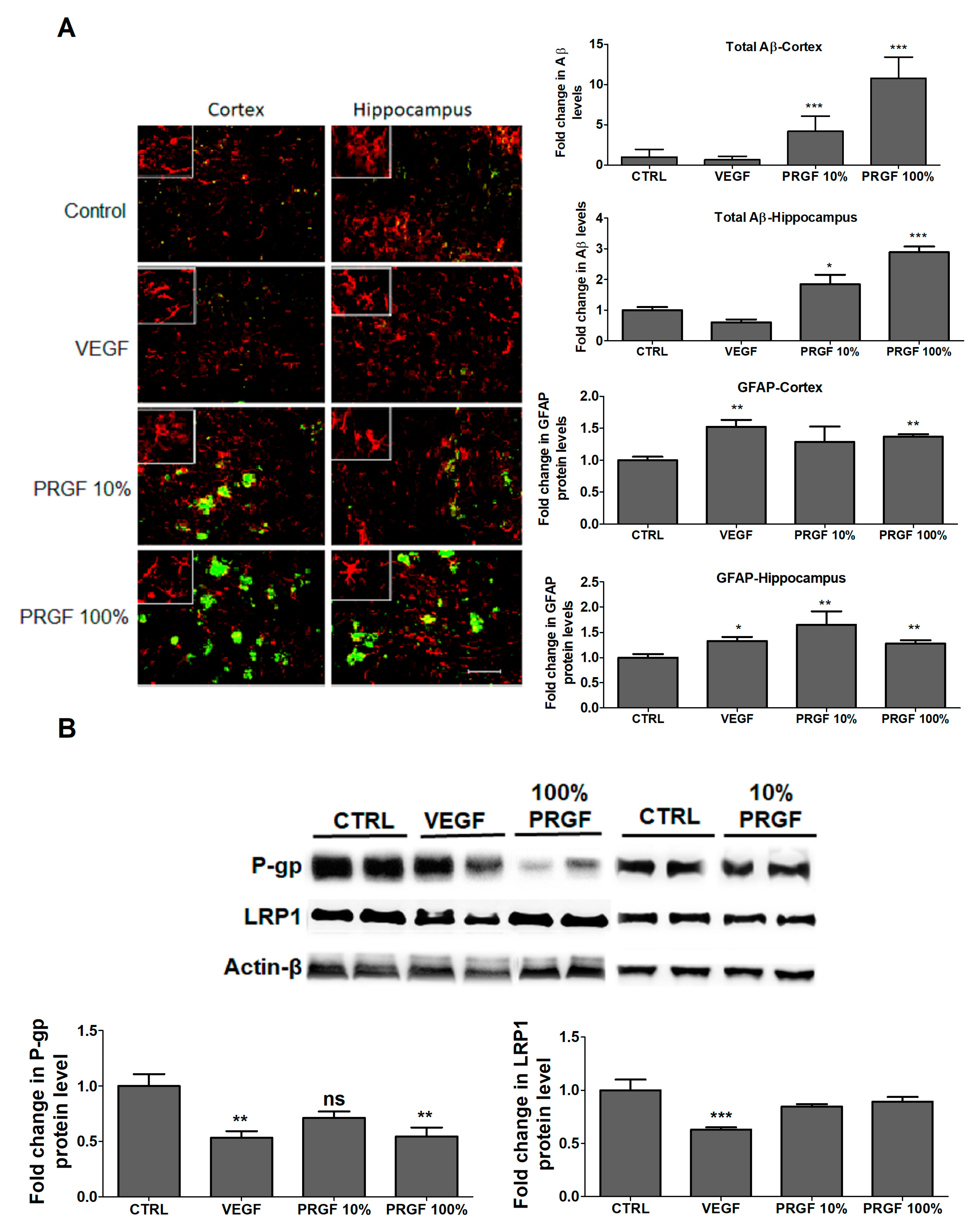
2.5. VEGF and PRGF Altered Aβ Levels, Increased Astrocytes Activation and Altered Aβ Major Transport Proteins in 5xFAD Mice Brains
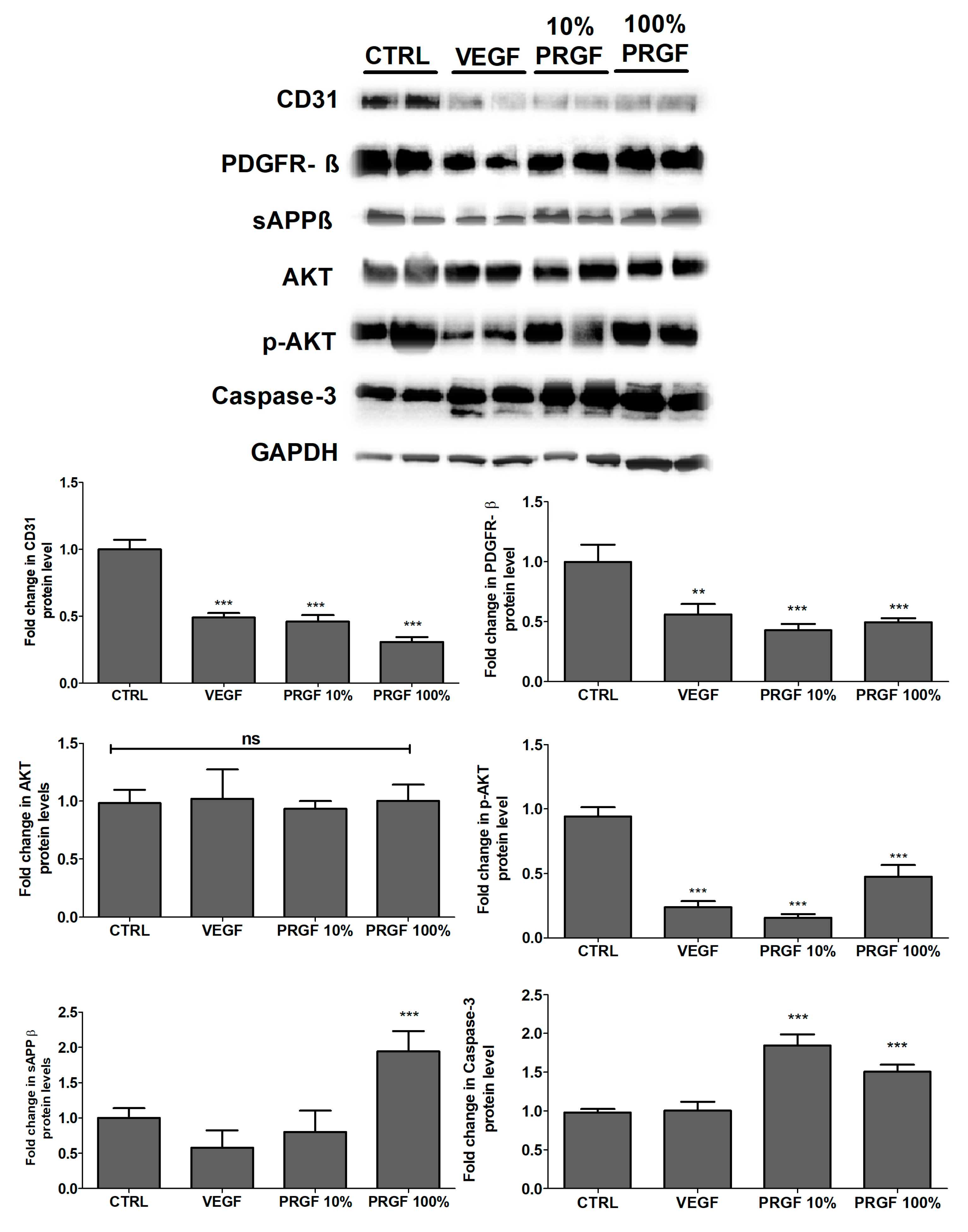
2.6. VEGF and PRGF Reduced Angiogenesis Markers, Increased Apoptosis, and Increased Amyloid Production
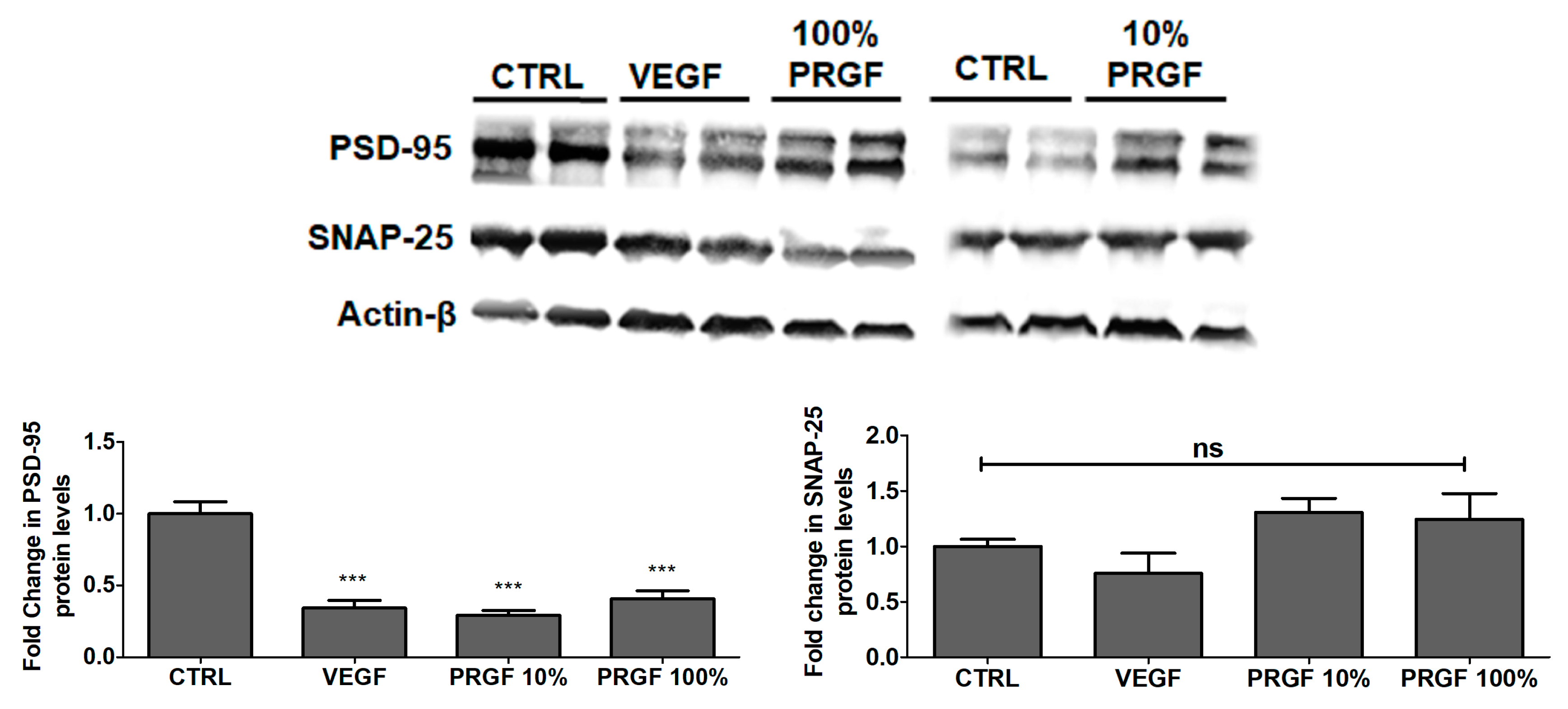
2.7. VEGF and PRGF Reduced the Expression of Postsynaptic Density-95 (PSD-95) in 5xFAD Mice Brains
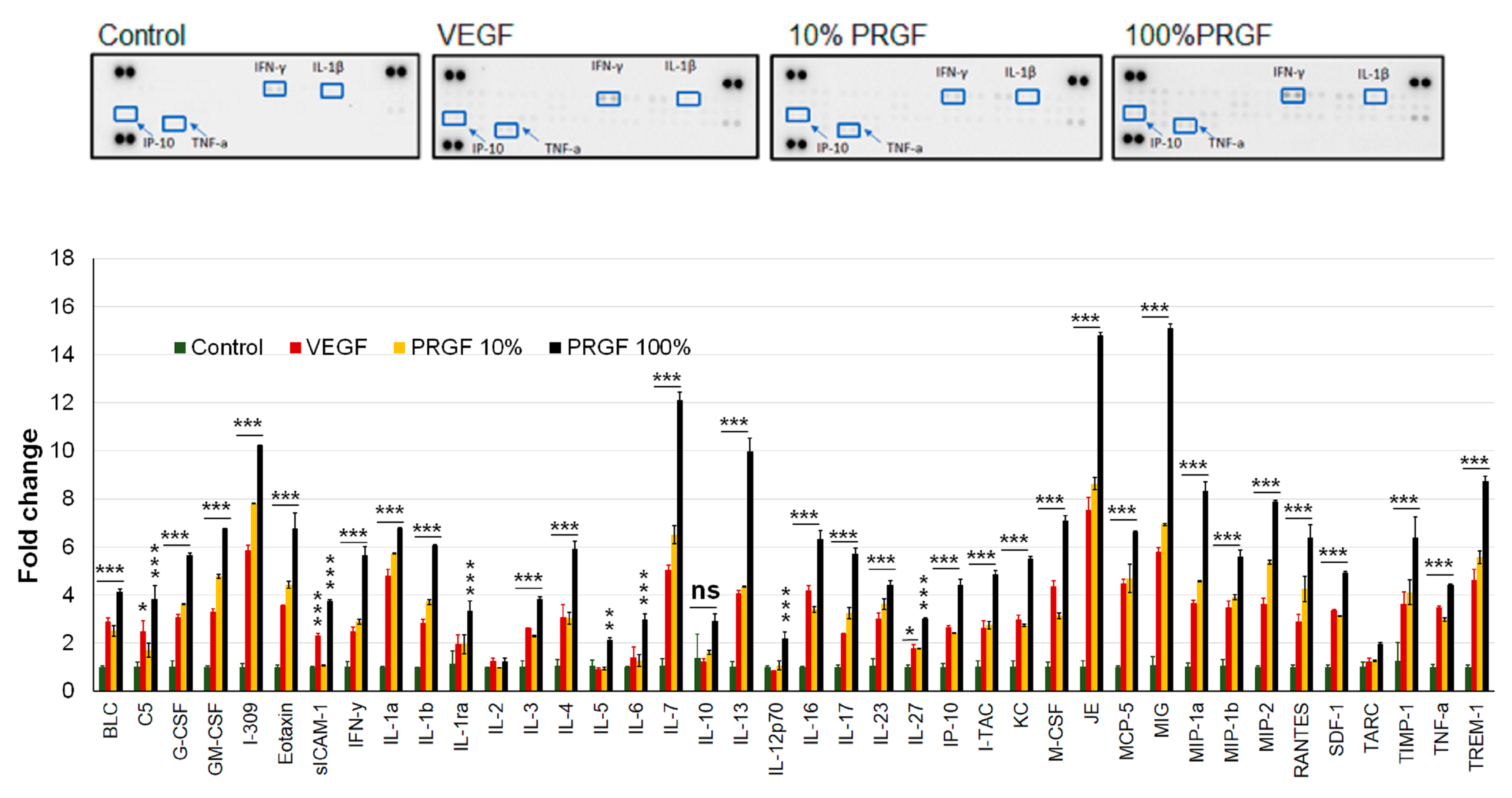
2.8. VEGF and PRGF Treatment Increased Cytokines and Neuroinflammatory Markers in 5xFAD Mice Brains
3. Discussion
4. Materials and Methods
4.1. Plasma Rich Derived Growth Factors Extraction and Quantification
4.2. In-Vitro Cell Culture
4.3. Lucifer Yellow Permeability Assay
4.4. Amyloid-β Transport Across bEnd3 Cell Monolayer
4.5. Animals
4.6. Animal Treatment
4.7. 5xFAD Brain Efflux Index Study
4.8. Isolation of Mouse Brain Microvessels
4.9. Western Blot Analysis
4.10. Immunohistochemistry Analysis
4.11. Amyloid-β Quantification by ELISA
4.12. Cytokine Array
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| BBB | Blood-Brain Barrier |
| Aβ | Amyloid- β |
| P-gp | P-glycoprotein |
| LRP1 | Low-Density Lipoprotein Receptor Related Protein-1 |
| TJs | Tight Junction Proteins |
| ZO | Zonula-occludin |
| PRGF | Plasma Rich in Growth Factors |
| VEGF | Vascular Endothelial Growth Factor |
| TGF-β1 | Transforming Growth Factor |
| CD | Cluster of Differentiation |
| PDGFR | Platelet-Derived Growth Factor Receptor |
| APP | Amyloid Precursor Protein |
| AKT | Protein Kinase B |
| p-AKT | Phosphorylated AKT |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| TNF | Tumor Necrosis Factor |
| INF | Interferon |
| IL | Interleukin |
| IP | Interferon Gamma-Induced Protein 10 |
References
- Citron, M. Alzheimer’s Disease: Strategies for Disease Modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar]
- Alzheimer’s Association. 2018 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s Disease: Genes, Proteins, and Therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Tau in Alzheimer’s Disease. Trends Cell Biol. 1998, 8, 425–427. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Lee, V.M. Fatal Attractions’ of Proteins: A Comprehensive Hypothetical Mechanism Underlying Alzheimer’s Disease and Other Neurodegenerative Disorders. Ann. N. Y. Acad. Sci. 2000, 924, 62–67. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I. Neurofibrillary Pathology Leads to Synaptic Loss and Not the Other Way around in Alzheimer Disease. J. Alzheimers Dis. 2002, 4, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Fujiyoshi, M.; Tachikawa, M.; Ohtsuki, S.; Ito, S.; Uchida, Y.; Akanuma, S.I.; Kamiie, J.; Hashimoto, T.; Hosoya, K.I.; Iwatsubo, T.; et al. Amyloid-β Peptide(1-40) Elimination from Cerebrospinal Fluid Involves Low-Density Lipoprotein Receptor-Related Protein 1 at the Blood-Cerebrospinal Fluid Barrier. J. Neurochem. 2011, 118, 407–415. [Google Scholar] [CrossRef]
- Ghersi-Egea, J.F.; Gorevic, P.D.; Ghiso, J.; Frangione, B.; Patlak, C.S.; Fenstermacher, J.D. Fate of Cerebrospinal Fluid-Borne Amyloid β-Peptide: Rapid Clearance into Blood and Appreciable Accumulation by Cerebral Arteries. J. Neurochem. 1996, 67, 880–883. [Google Scholar] [CrossRef]
- Lam, F.C.; Liu, R.; Lu, P.; Shapiro, A.B.; Renoir, J.M.; Sharom, F.J.; Reiner, P.B. Beta-Amyloid Efflux Mediated by P-glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein Deficiency at the Blood-Brain Barrier Increases Amyloid-Beta Deposition in an Alzheimer Disease Mouse Model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef] [PubMed]
- Abuznait, A.H.; Cain, C.; Ingram, D.; Burk, D.; Kaddoumi, A. Up-Regulation of P-glycoprotein Reduces Intracellular Accumulation of Beta Amyloid: Investigation of P-glycoprotein as a Novel Therapeutic Target for Alzheimer’s Disease. J. Pharm. Pharmacol. 2011, 63, 1111–1118. [Google Scholar] [CrossRef]
- Kuhnke, D.; Jedlitschky, G.; Grube, M.; Krohn, M.; Jucker, M.; Mosyagin, I.; Cascorbi, I.; Walker, L.C.; Kroemer, H.K.; Warzok, R.W.; et al. MDR1-P-Glycoprotein (ABCB1) Mediates Transport of Alzheimer’s Amyloid-β Peptides-Implications for the Mechanisms of Aβ Clearance at the Blood–Brain Barrier. Brain Pathol. 2007, 17, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.A.; Keller, J.N.; Kaddoumi, A. Role of P-Glycoprotein in Mediating Rivastigmine Effect on Amyloid-β Brain Load and Related Pathology in Alzheimer’s Disease Mouse Model. Biochim. Biophys. Acta 2016, 1862, 778–787. [Google Scholar] [CrossRef]
- Batarseh, Y.S.; Bharate, S.S.; Kumar, V.; Kumar, A.; Vishwakarma, R.A.; Bharate, S.B.; Kaddoumi, A. Crocus Sativus Extract Tightens the Blood-Brain Barrier, Reduces Amyloid β Load and Related Toxicity in 5XFAD Mice. ACS Chem. Neurosci. 2017, 8, 1756–1766. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Mohamed, L.A.; Al Rihani, S.B.; Batarseh, Y.S.; Duong, Q.V.; Keller, J.N.; Kaddoumi, A. High-Throughput Screening for Identification of Blood-Brain Barrier Integrity Enhancers: A Drug Repurposing Opportunity to Rectify Vascular Amyloid Toxicity. J. Alzheimers Dis. 2016, 53, 1499–1516. [Google Scholar] [CrossRef] [PubMed]
- Qosa, H.; Batarseh, Y.S.; Mohyeldin, M.; El Sayed, K.A.; Keller, J.N.; Kaddoumi, A. Oleocanthal Enhances Amyloid-β Clearance from the Brains of TgSwDI Mice and in Vitro across a Human Blood-Brain Barrier Model. ACS Chem. Neurosci. 2015, 6, 1849–1859. [Google Scholar] [CrossRef]
- Qosa, H.; Mohamed, L.A.; Batarseh, Y.S.; Alqahtani, S.; Ibrahim, B.; LeVine, H., III; Keller, J.N.; Kaddoumi, A. Extra-Virgin Olive Oil Attenuates Amyloid-β and Tau Pathologies in the Brains of TgSwDI Mice. J. Nutr. Biochem. 2015, 26, 1479–1490. [Google Scholar] [CrossRef]
- Qosa, H.; Abuznait, A.H.; Hill, R.A.; Kaddoumi, A. Enhanced Brain Amyloid-β Clearance by Rifampicin and Caffeine as a Possible Protective Mechanism against Alzheimer’s Disease. J. Alzheimers Dis. 2012, 31, 151–165. [Google Scholar] [CrossRef]
- Qosa, H.; Abuasal, B.S.; Romero, I.A.; Weksler, B.; Couraud, P.O.; Keller, J.N.; Kaddoumi, A. Differences in Amyloid-β Clearance across Mouse and Human Blood-Brain Barrier Models: Kinetic Analysis and Mechanistic Modeling. Neuropharmacology 2014, 79, 668–678. [Google Scholar] [CrossRef]
- Greene, C.; Campbell, M. Tight Junction Modulation of the Blood Brain Barrier: CNS Delivery of Small Molecules. Tissue Barriers 2016, 4, e1138017. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain Endothelial Cell-Cell Junctions: How to ‘Open’ the Blood Brain Barrier. Curr. Neuropharmacol. 2008, 6, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.S.; Monahan, A.J.; Carvey, P.M.; Hendey, B. Blood-Brain Barrier Pathology in Alzheimer’s and Parkinson’s Disease: Implications for Drug Therapy. Cell Transplant. 2007, 16, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.A.; Antimisiaris, D.; O’Brien, J. Drugs for Alzheimer’s Disease: Are They Effective? Pharmacol. Ther. 2010, 35, 208–211. [Google Scholar]
- Anitua, E.; Pascual, C.; Antequera, D.; Bolos, M.; Padilla, S.; Orive, G.; Carro, E. Plasma Rich in Growth Factors (PRGF-Endoret) Reduces Neuropathologic Hallmarks and Improves Cognitive Functions in an Alzheimer’s Disease Mouse Model. Neurobiol. Aging 2014, 35, 1582–1595. [Google Scholar] [CrossRef]
- Anitua, E.; Pascual, C.; Pérez-Gonzalez, R.; Antequera, D.; Padilla, S.; Orive, G.; Carro, E. Intranasal Delivery of Plasma and Platelet Growth Factors Using PRGF-Endoret System Enhances Neurogenesis in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2013, 8, e73118. [Google Scholar] [CrossRef] [PubMed]
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF Significantly Restores Impaired Memory Behavior in Alzheimer’s Mice by Improvement of Vascular Survival. Sci. Rep. 2013, 3, 2053. [Google Scholar] [CrossRef] [PubMed]
- McGinley, L.M.; Sims, E.; Lunn, J.S.; Kashlan, O.N.; Chen, K.S.; Bruno, E.S.; Pacut, C.M.; Hazel, T.; Johe, K.; Sakowski, S.A.; et al. Human Cortical Neural Stem Cells Expressing Insulin- like Growth Factor-I: A Novel Cellular Therapy for Alzheimer’s Disease. Stem Cells Transl. Med. 2016, 5, 379–391. [Google Scholar] [CrossRef]
- Chen, J.H.; Ke, K.F.; Lu, J.H.; Qiu, Y.H.; Peng, Y.P. Protection of TGF-β1 against Neuroinflammation and Neurodegeneration in Aβ1-42-Induced Alzheimer’s Disease Model Rats. PLoS ONE 2015, 10, e0116549. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular Dysfunction and Faulty Amyloid β-Peptide Clearance in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef]
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal Administration of CNS Therapeutics to Awake Mice. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Erdő, F.; Denes, L.; de Lange, E. Age-Associated Physiological and Pathological Changes at the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2017, 37, 4–24. [Google Scholar] [CrossRef] [PubMed]
- Evanson, J.R.; Guyton, M.K.; Oliver, D.L.; Hire, J.M.; Topolski, R.L.; Zumbrun, S.D.; McPherson, J.C.; Bojescul, J.A. Gender and Age Differences in Growth Factor Concentrations from Platelet-Rich Plasma in Adults. Mil. Med. 2014, 179, 799–805. [Google Scholar] [CrossRef]
- Mooren, R.E.; Hendriks, E.J.; van den Beucken, J.J.; Merkx, M.A.; Meijer, G.J.; Jansen, J.A.; Stoelinga, P.J. The Effect of Platelet-Rich Plasma in Vitro on Primary Cells: Rat Osteoblast-like Cells and Human Endothelial Cells. Tissue Eng. Part A 2010, 16, 3159–3172. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Merlo, S.; Sano, Y.; Kanda, T.; Sortino, M.A. Astrocytes Contribute to Aβ-Induced Blood–Brain Barrier Damage through Activation of Endothelial MMP9. J. Neurochem. 2017, 142, 464–477. [Google Scholar] [CrossRef]
- Zhang, J.B.; Li, M.F.; Zhang, H.X.; Li, Z.G.; Sun, H.R.; Zhang, J.S.; Wang, P.F. Association of Serum Vascular Endothelial Growth Factor Levels and Cerebral Microbleeds in Patients with Alzheimer’s Disease. Eur. J. Neurol. 2016, 23, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, Y.P.; Liu, Y.H.; Jiao, S.S.; Liu, L.; Wang, Y.J.; Fu, W.L. Diagnostic Utility of VEGF and Soluble CD40L Levels in Serum of Alzheimer’s Patients. Clin. Chim. Acta 2016, 453, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Herran, E.; Perez-Gonzalez, R.; Igartua, M.; Pedraz, J.L.; Carro, E.; Hernandez, R.M. Enhanced Hippocampal Neurogenesis in APP/Ps1 Mouse Model of Alzheimer’s Disease After Implantation of VEGF-Loaded PLGA Nanospheres. Curr. Alzheimer Res. 2015, 12, 932–940. [Google Scholar] [CrossRef]
- Nurden, A.T. Platelets, Inflammation and Tissue Regeneration. Thromb. Haemost. 2011, 105, S13–S33. [Google Scholar] [CrossRef]
- Trinchese, F.; Liu, S.; Battaglia, F.; Walter, S.; Mathews, P.M.; Arancio, O. Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann. Neurol. 2004, 55, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial Functions of Platelet/Endothelial Cell Adhesion Molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes Are Required for Blood-Brain Barrier Integrity during Embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Cockerill, I.; Oliver, J.A.; Xu, H.; Fu, B.M.; Zhu, D. Blood-Brain Barrier Integrity and Clearance of Amyloid-β from the BBB. Adv. Exp. Med. Biol. 2018, 1097, 261–278. [Google Scholar]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-β(1-42) across the blood-brain barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Baek, S.J.; Heo, S.; Park, H.J.; Kwon, H.; Lee, S.; Jung, J.; Park, S.J.; Kim, B.C.; Lee, Y.C.; et al. Direct Pharmacological Akt Activation Rescues Alzheimer’s Disease like Memory Impairments and Aberrant Synaptic Plasticity. Neuropharmacology 2018, 128, 282–292. [Google Scholar] [CrossRef]
- Wang, P.; Xie, Z.H.; Guo, Y.J.; Zhao, C.P.; Jiang, H.; Song, Y.; Zhu, Z.Y.; Lai, C.; Xu, S.L.; Bi, J.Z. VEGF-induced angiogenesis ameliorates the memory impairment in APP transgenic mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2011, 411, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Mao, X.; Xie, L.; Greenberg, D.A.; Jin, K. Expression Level of Vascular Endothelial Growth Factor in Hippocampus Is Associated with Cognitive Impairment in Patients with Alzheimer’s Disease. Neurobiol. Aging 2013, 34, 1412–1415. [Google Scholar] [CrossRef]
- Thomas, T.; Miners, S.; Love, S. Post-mortem assessment of hypoperfusion of cerebral cortex in Alzheimer’s disease and vascular dementia. Brain 2015, 138, 1059–1069. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.; Miners, J.S.; Allen, S.; Love, S. VEGFR1 and VEGFR2 in Alzheimer’s Disease. J. Alzheimer Dis. 2018, 61, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Giannoni, P.; Arango-Lievano, M.; Neves, I.D.; Rousset, M.C.; Baranger, K.; Rivera, S.; Jeanneteau, F.; Claeysen, S.; Marchi, N. Cerebrovascular Pathology during the Progression of Experimental Alzheimer’s Disease. Neurobiol. Dis. 2016, 88, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Romer, L.H.; McLean, N.V.; Yan, H.C.; Daise, M.; Sun, J.; DeLisser, H.M. IFN-Gamma and TNF-Alpha Induce Redistribution of PECAM-1 (CD31) on Human Endothelial Cells. J. Immunol. 1995, 154, 6582–6592. [Google Scholar]
- Zhang, F.; Jiang, L. Neuroinflammation in Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2015, 11, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.; Miler, C.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-β1-40 peptide from brain by LDL receptor-related protein-1 at the blood brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duong, Q.-V.; Kintzing, M.L.; Kintzing, W.E.; Abdallah, I.M.; Brannen, A.D.; Kaddoumi, A. Plasma Rich in Growth Factors (PRGF) Disrupt the Blood-Brain Barrier Integrity and Elevate Amyloid Pathology in the Brains of 5XFAD Mice. Int. J. Mol. Sci. 2019, 20, 1489. https://doi.org/10.3390/ijms20061489
Duong Q-V, Kintzing ML, Kintzing WE, Abdallah IM, Brannen AD, Kaddoumi A. Plasma Rich in Growth Factors (PRGF) Disrupt the Blood-Brain Barrier Integrity and Elevate Amyloid Pathology in the Brains of 5XFAD Mice. International Journal of Molecular Sciences. 2019; 20(6):1489. https://doi.org/10.3390/ijms20061489
Chicago/Turabian StyleDuong, Quoc-Viet, Margia L. Kintzing, William E. Kintzing, Ihab M. Abdallah, Andrew D. Brannen, and Amal Kaddoumi. 2019. "Plasma Rich in Growth Factors (PRGF) Disrupt the Blood-Brain Barrier Integrity and Elevate Amyloid Pathology in the Brains of 5XFAD Mice" International Journal of Molecular Sciences 20, no. 6: 1489. https://doi.org/10.3390/ijms20061489
APA StyleDuong, Q.-V., Kintzing, M. L., Kintzing, W. E., Abdallah, I. M., Brannen, A. D., & Kaddoumi, A. (2019). Plasma Rich in Growth Factors (PRGF) Disrupt the Blood-Brain Barrier Integrity and Elevate Amyloid Pathology in the Brains of 5XFAD Mice. International Journal of Molecular Sciences, 20(6), 1489. https://doi.org/10.3390/ijms20061489