Preliminary Study on Clusterin Protein (sCLU) Expression in PC-12 Cells Overexpressing Wild-Type and Mutated (Swedish) AβPP genes Affected by Non-Steroid Isoprenoids and Water-Soluble Cholesterol




Abstract
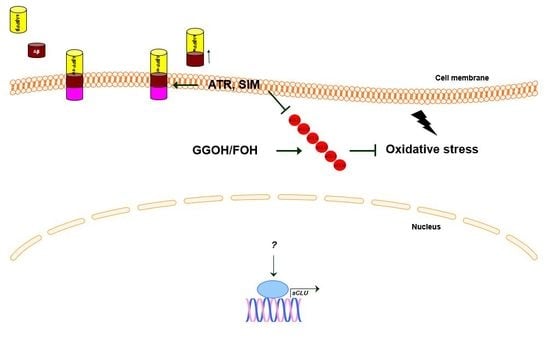
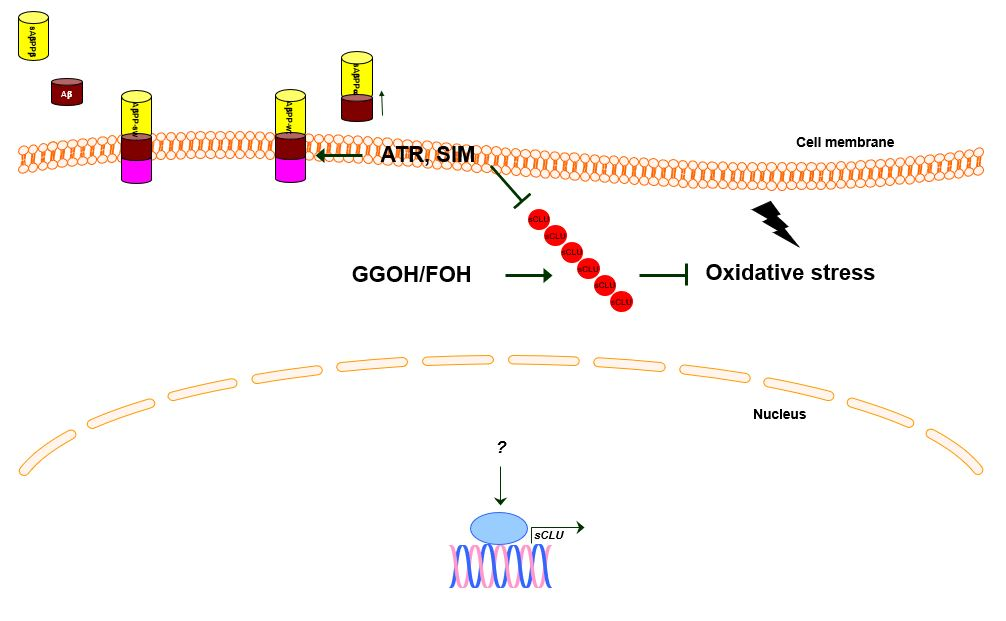
1. Introduction
2. Results
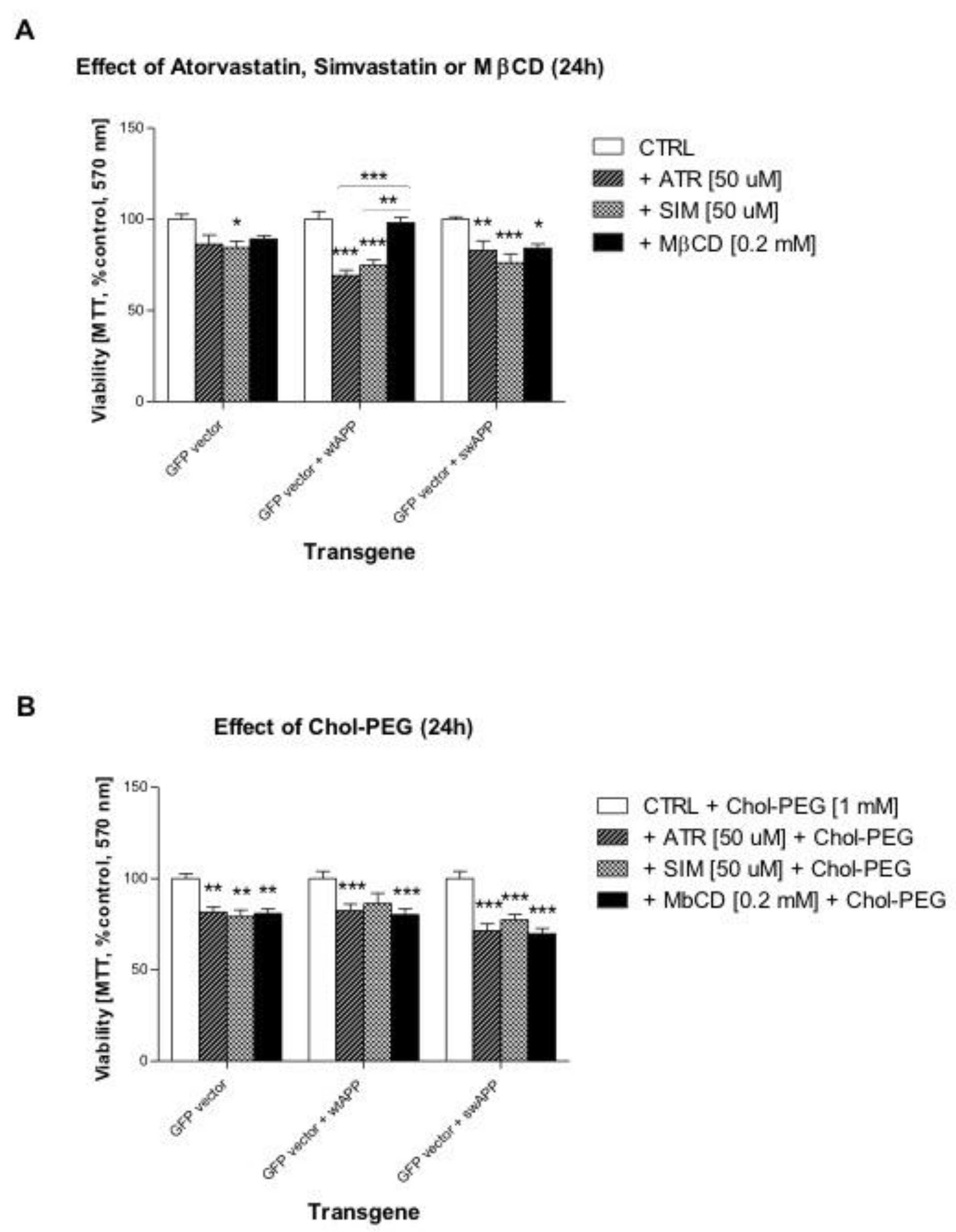
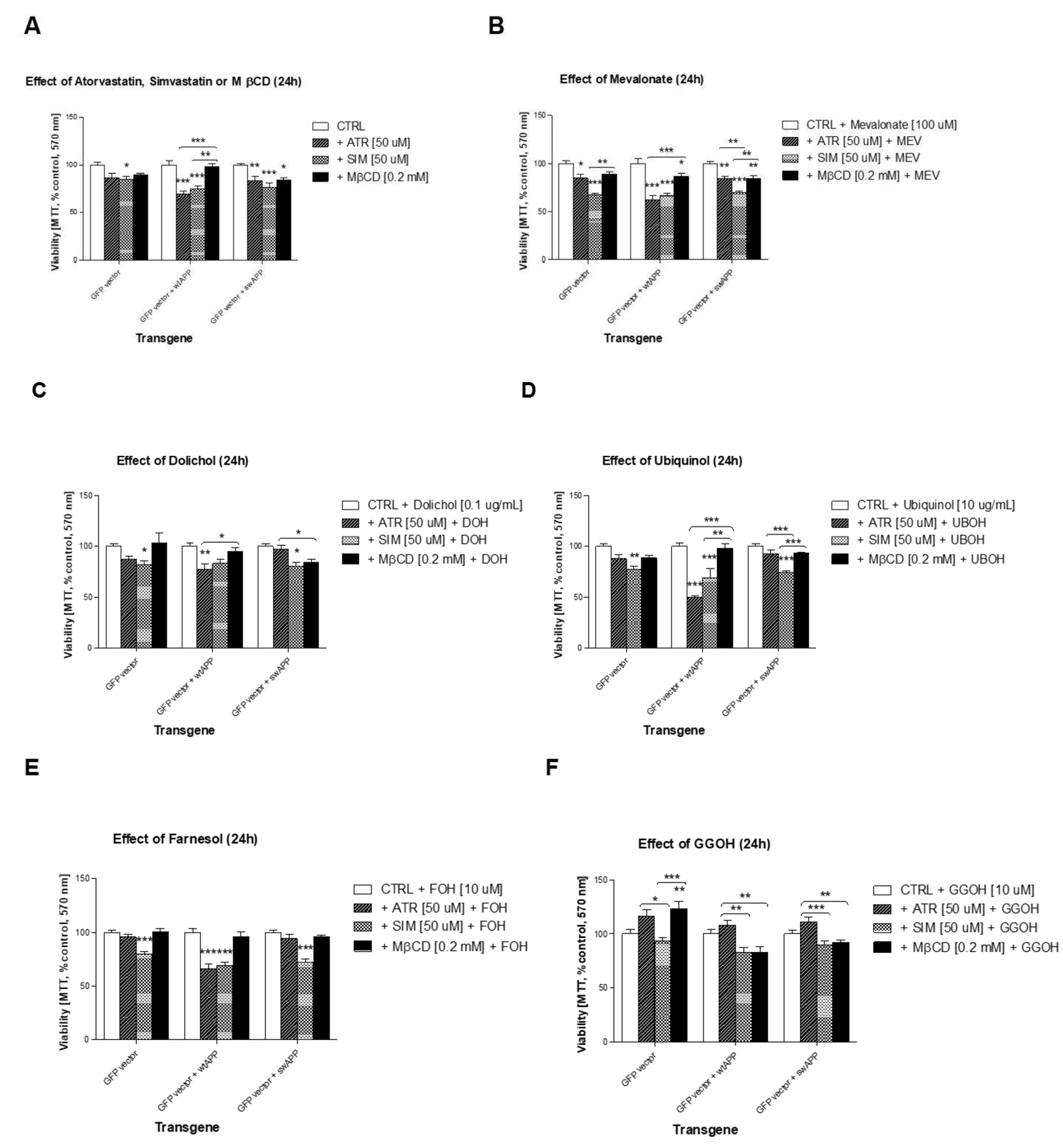
2.1. Effects of Selected Statins or MβCD on Cell Viability of Neuronal PC-12 Cells Overexpressing Wild-Type or Mutated (Swedish Mutation) AβPP Gene
2.2. Effect of Some Non-Sterol Isoprenoids on Cell Viability of Neuronal PC-12 Cells Overexpressing Wild-Type or Mutated (Swedish Mutation) AβPP Gene Treated with Selected Statins or MβCD
2.3. Effects of Selected MEV Pathway Enzyme Inhibitors on Cell Viability of Neuronal PC-12 Cells Overexpressing Wild-Type and Mutated (Swedish Mutation) AβPP Gene
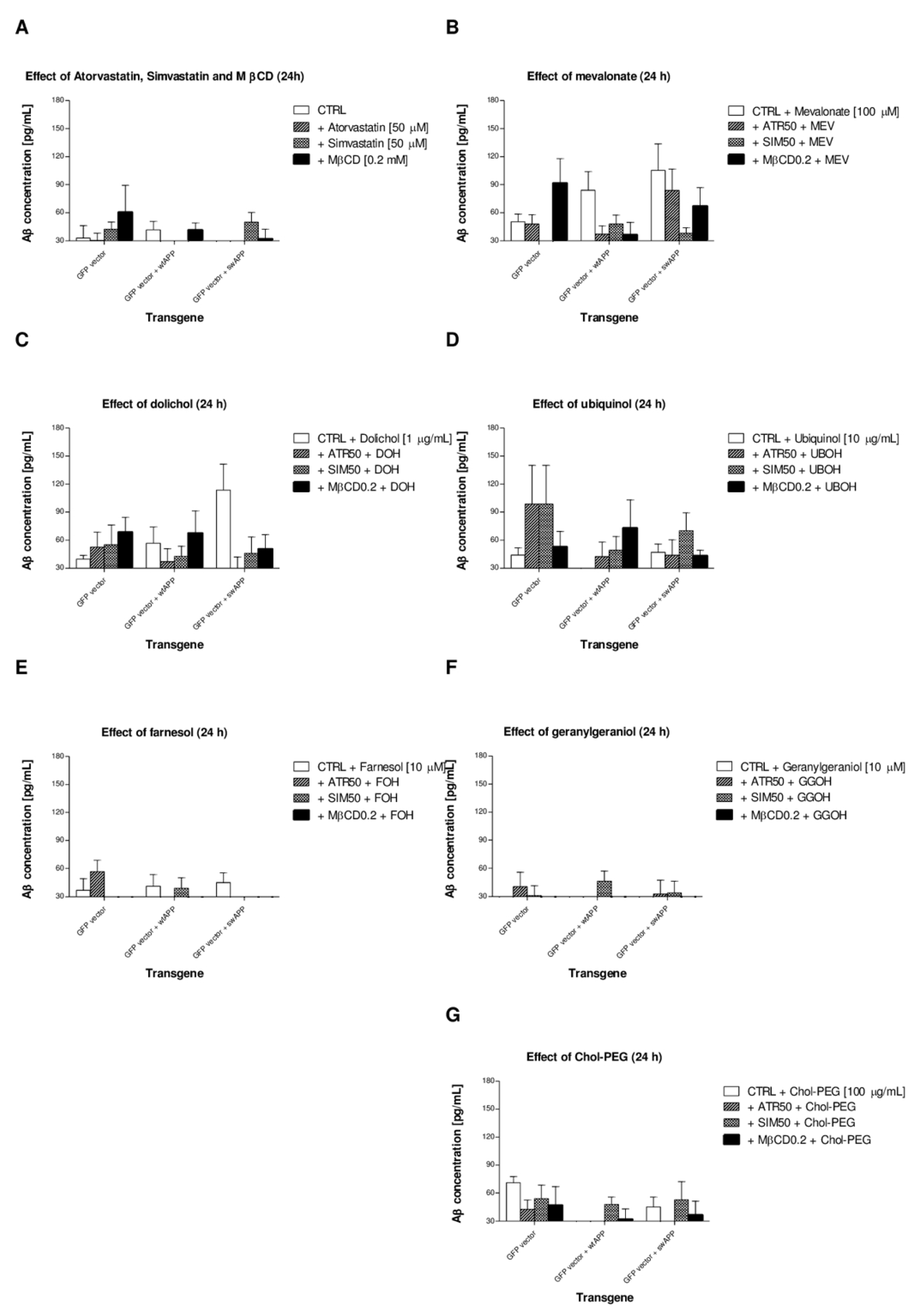
2.4. Effects of Non-Sterol Isoprenoids (MEV, DOH, UBOH, FOH, GGOH) vs. CHOL-PEG on β-Amyloid (Aβ1-40) Secretion by Neuronal PC-12 Cells Affected by MEV Pathway Modulators Statins or Cholesterol Chelator Methyl-β-Cyclodextrin (MβCD) in PC-12 Cells Overexpressing AβPP-wt or AβPP-sw Gene
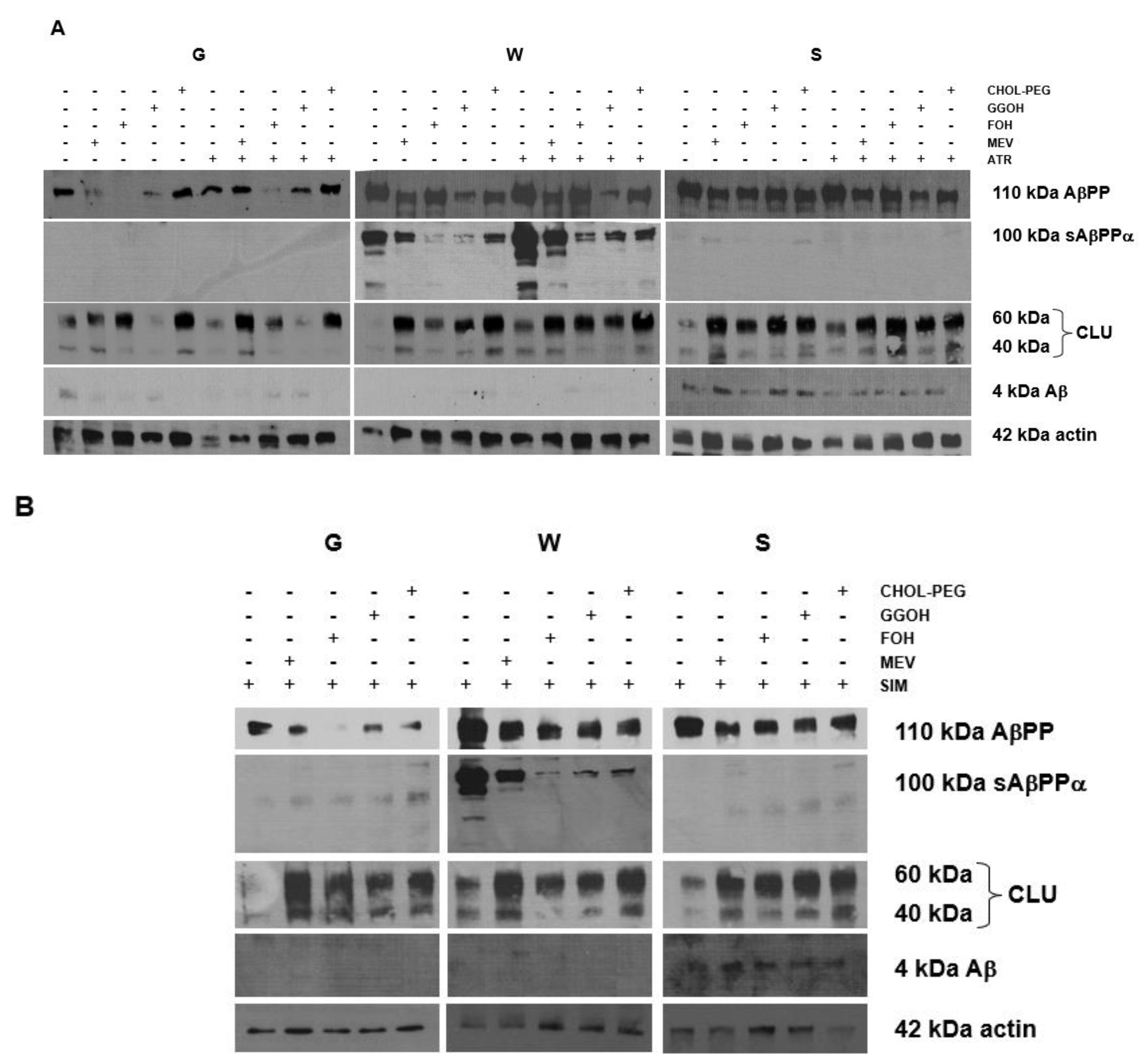
2.5. Effects of Mevalonate (MEV, FOH, GGOH) or Water-Soluble Cholesterol (Chol-PEG) on Selected Protein Expression Levels in Cells Affected by MEV Pathway Modulators (Atorvastatin – ATR, Simvastatin – SIM)
3. Discussion
4. Materials and Methods
4.1. PC-12 Cell Culture
4.2. Reagents
4.3. Cloning and Expression Vectors
4.4. Transfection of PC-12 Cells
4.5. Determination of Cell Viability
4.6. Cellular Aβ1-40 Assays
4.7. Detection of Vector Sequence in PC-12-Transfected Cells by PCR
4.8. Western Blot Analysis
5. Conclusions
- Statins, inhibitors of FTase, GGTase I, and SQS, as well as cholesterol chelator, reduce the viability of PC-12 neuronal cells.
- Statin-induced neurotoxicity is overruled by GGOH, but not cholesterol administration.
- sCLU expression is stimulated by isoprenoids and water-soluble cholesterol, however, this was not the case for GGOH administration in empty-vector cells.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α2M | α 2 macroglobulin |
| AβPP | Amyloid β precursor protein |
| AβPP-sw | Amyloid β precursor protein with Swedish mutation |
| AβPP-wt | Amyloid β precursor protein wild type |
| Aβ | Amyloid β |
| AD | Alzheimer’s disease |
| ANOVA | Analysis of variance |
| ApoE | Apolipoprotein E |
| ATR | Atorvastatin |
| BBB | Blood brain barrier |
| Chol-PEG | Water-soluble cholesterol |
| CLU | Clusterin |
| CSF | Cerebrospinal fluid |
| DMPP | Dimethylalkyl diphosphate |
| DOH | Dolichol |
| FOH | Farnesol |
| FPP | Farnesyl pyrophosphate |
| FTase | Farnesyl transferase |
| GGOH | Geranylgeraniol |
| GGPP | Geranylgeranyl pyrophosphate |
| GGTase | Geranylgeranyl transferase |
| GGTI | Geranylgeranyl transferase inhibitor |
| HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase |
| iCLU | Intracellular clusterin |
| IPP | Isopentyl pyrophosphate |
| LRP2 | Lipoprotein low density receptor related protein 2 |
| MβCD | Methyl-β-cyclodextrin |
| MEV | Mevalonate |
| MEVPP | Mevalonate pyrophosphate |
| NGF | Nerve growth factor |
| NOS | Nitrogen species |
| PC-12 | Rat neuronal pheochromocytoma PC-12 cells |
| RAGE | Advanced glycation end products |
| ROS | Reactive oxygen species |
| sCLU | Secretory clusterin |
| sGTPase | Small GTP-binding protein |
| S.E.M. | Standard Error of Mean |
| SIM | Simvastatin |
| SQS | Squalene synthase |
| TLR3 | Toll like receptor 3 |
| UBOH | Ubiquinol |
| WB | Western blot |
References
- Dietschy, J.M.; Turley, S.D. Thematic review series: Brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 2004, 45, 1375–1397. [Google Scholar] [CrossRef]
- Cartocci, V.; Segatto, M.; di Tunno, I.; Leone, S.; Pfrieger, F.W.; Pallottini, V. Modulation of the Isoprenoid/Cholesterol biosynthetic pathway during neuronal differentiation in vitro. J. Cell. Biochem. 2016, 117, 2036–2044. [Google Scholar] [CrossRef]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef]
- Moutinho, M.; Nunes, M.J.; Rodrigues, E. The mevalonate pathway in neurons: It’s not just about cholesterol. Exp. Cell Res. 2017, 360, 55–60. [Google Scholar] [CrossRef]
- Fracassi, A.; Maragnoni, M.; Rosso, P.; Pallottini, V.; Fioramonti, M.; Siteni, S.; Segatto, M. Statins and the Brain: More than Lipid Lowering Drugs? Curr. Neuropharmaol. 2019, 17, 59–93. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Dallas, S.; Hong, M.; Bendayan, R. Drug transporters in the central nervous system: Brain barriers and brain parenchyma considerations. Pharmaol. Rev. 2001, 53, 569–596. [Google Scholar] [CrossRef] [PubMed]
- Hooff, G.P.; Peters, I.; Muller, W.E.; Wood, W.G.; Eckert, G.P. Modulation of Cholesterol, Farnesyl- and Geranylgeranylpyrophosphate in neuronal SH-SY5Y APP965 cells—Impact for Amyloid-β production. Mol. Neurobiol. 2010a, 41, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Pierrot, N.; Tytea, D.; D’Auria, L.; Dewachter, I.; Gailly, P.; Hendrickx, A.; Tasiaux, B.; Haylani, L.E.; Muls, N.; N’kuli, F.; et al. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol. Med. 2013, 5, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.C.; Yu, J.T.; Li, Y.; Tan, L. Clusterin in Alzheimer’s disease. Adv. Clin. Chem. 2012, 56, 155–173. [Google Scholar]
- Duguid, J.R.; Bohmont, C.W.; Liu, N.G.; Tourtelotte, W.W. Changes in brain gene expression shared by scrapie and Alzheimer disease. Proc. Nat. Acad. Sci. USA 1989, 86, 7260–7264. [Google Scholar] [CrossRef]
- De Silva, H.V.; Harmony, J.A.; Stuart, W.D.; Gil, C.M.; Robbins, J. Apolipoprotein J: Structure and tissue distribution. Biochem. 1990, 29, 5380–5389. [Google Scholar] [CrossRef]
- Bell, R.D.; Sagare, A.P.; Friedman, A.E.; Bedi, G.S.; Holztman, D.M.; Deane, R.; Zlokovic, B.V. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoprotein E and J in the mouse central nervous system. J. Cerebral Blood Flow Metab. 2007, 27, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Wilson, M.R. Extraellular chaperones modulate the effects of Alzheimer’s patient cerebrospinal fluid on Abeta(1-42) toxicity and uptake. Cell Stress Chaperones 2010, 15, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle, V.; Essabbani, A.; De Pinieux, G.; Geramin, S.; Tourner, L.; Mistou, S.; Margottin-Gouget, F.; Anract, P.; Migaud, H.; Le Nen, D.; et al. Characterization and functional consequences of underexpression of clusterin in rheumathoid arthritis. J. Immunol. 2006, 177, 6471–6479. [Google Scholar] [CrossRef]
- McLoughlin, L.; Zhu, G.; Mistry, M.; Ley-Ebert, C.; Stuart, W.D.; Florio, C.J.; Groen, P.A.; Witt, S.A.; Kimball, T.R.; Witte, D.P.; et al. Apolipoprotein J/clusterin limits the severity of murine autoimmune myocarditis. J. Clin. Investig. 2000, 106, 1105–1113. [Google Scholar] [CrossRef]
- Bach, U.C.; Beiersdorfer, M.; Klock, G.; Cattarruzza, M.; Post, A.; Koch-Brandt, C. Apoptotic cell debris and phosphatidylserine-containing lipid vesicles induce apolipoprotein J (clusterin) gene expression in vital fibroblasts. Exp. Cell Res. 2001, 265, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Danik, M.; Chabot, J.G.; Hassan-Gonzalez, D.; Suh, M.; Quirion, R. Localization of sulfated glycoprotein-2/clusterin mRNA in the rat brain by in situ hybridization. J. Comp. Neurol. 1993, 334, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Charnay, Y.; Imhof, A.; Vallet, P.G.; Kovari, E.; Bouras, C.; Giannakopoulos, P. Clusterin in neurological disorders: Molecular perspectives and clinical relevance. Brain Res. Bull. 2012, 88, 434–443. [Google Scholar] [CrossRef]
- Bailey, R.W.; Aronow, B.; Harmony, J.A.; Griswold, M.D. Heat shock-initiated apoptosis is accelerated and removal of damaged cells is delayed in the testis of clusterin/ApoJ knock-out mice. Biol. Reprod. 2002, 66, 1042–1053. [Google Scholar] [CrossRef] [PubMed]
- Klock, G.; Baiersdorfer, M.; Koch-Brandt, C. Cell protective functions of secretory clusterin (sCLU). Adv. Cancer Res. 2009, 104, 115–135. [Google Scholar]
- Ling, I.-F.; Bhongsatiern, J.; Simpson, J.F.; Fardo, D.W.; Estus, S. Genetics of clusterin isoform expression and Alzheimer’s disease risk. PLoS ONE. 2012, 7, e33923. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, H.; Liu, J.; Li, J.; Li, H.; Ma, G.; Jiang, Y.; Chen, Z.; Zhao, B.; Li, K. The CLU Gene rs11136000 variant is significantly associated with Alzheimer’s disease in caucasian and asian populations. Neuromol. Med. 2014, 16, 52–60. [Google Scholar] [CrossRef]
- Zhu, R.; Liu, X.; He, Z. Association between CLU gene rs11136000 polymorphism and Alzheimer’s disease: An updated meta-analysis. Neurol. Sci. 2018, 39, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Lidstrom, A.M.; Bogdanovic, N.; Hesse, C.; Volkman, I.; Davidsson, P.; Blennov, K. Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer’s disease. Exp. Neurol. 1998, 154, 511–521. [Google Scholar] [CrossRef]
- Calero, M.; Rostagno, A.; Matsubara, E.; Zlokovic, B.; Frangione, B.; Ghiso, J. Apolipoprotein J (clusterin) and Alzheimer’s disease. Microsc. Res. Tech. 2000, 50, 305–315. [Google Scholar] [CrossRef]
- Miners, J.S.; Clarke, P.; Love, S. Clusterin levels are increased in Alzheimer’s disease and influence the regional distribution of Aβ. Brain Pathol. 2017, 27, 305–313. [Google Scholar] [CrossRef]
- Li, X.; Ma, Y.; Wei, X.; Li, Y.; Wu, H.; Zhuang, J.; Zhao, Z. Clusterin in Alzheimer’s disease: A player in the biological behavior of amyloid-beta. Neurosci. Bull. 2014, 30, 162–168. [Google Scholar] [CrossRef]
- Choi Miura, N.-C.; Oda, T. Relationship between multifunctional protein “clusterin” and Alzheimer disease. Neurobiol. Aging 1996, 17, 717–722. [Google Scholar] [PubMed]
- Baig, S.; Palmer, L.E.; Owen, M.J.; Williams, J.; Kehoe, P.G.; Love, S. Clusterin mRNA and protein in Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 28, 337–344. [Google Scholar] [CrossRef]
- Nuutinen, T.; Huuskonen, J.; Suuronen, T.; Ojala, J.; Miettinen, R.; Salminen, A. Amyloid-beta-1-42 induced endocytosis and clusterin/apoJ protein accumulation in cultured human astrocytes. Neurochem. Int. 2007, 50, 540–547. [Google Scholar] [CrossRef]
- Nuutinen, T.; Suuronen, T.; Kauppinen, A.; Salminen, A. Valproic acid stimulates clusterin expression in human astrocytes: Implications for Alzheimer’s disease. Neurosci. Lett. 2010, 475, 64–68. [Google Scholar] [CrossRef]
- Zhou, Y.; Hayashi, I.; Womg, J.; Tugusheva, K.; Renger, J.J.; Zerbinatti, C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein tau in Alzheimer’s disease. PLoS ONE 2014, e103187. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Poon, S.; Meechan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef]
- Wilson, M.; Yerbury, J.J.; Poon, S. Potential roles of abundant extracellular chaperones in the control of amyloid formation and toxicity. Mol. Biosyst. 2008, 4, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Petropoulou, C.; Trougakos, I.P.; Kolettas, E.; Toussaint, O.; Gonos, E.S. Clusterin/apolipoprotein J is a novel biomarker of cellular senescence that does not affect the proliferative capacity of human diploid fibroblasts. FEBS Lett. 2001, 509, 287–297. [Google Scholar] [CrossRef]
- Trougakos, I.P.; Gonos, E.S. Clusterin/apolipoprotein J in Human aging and cancer. Int. J. Biochem. Cell Biol. 2002, 34, 1430–1448. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. ApoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2007, 118, 4002–4013. [Google Scholar] [CrossRef]
- Pająk, B.; Kania, E.; Orzechowski, A. Nucleofection of rat pheochromocytoma PC-12 cells with human mutated β-amyloid precursor protein gene (APP-sw) leads to reduced viability, autophagy-like process and increased expression and secretion of β amyloid. BioMed Res. Int. 2015, 2015, 746092. [Google Scholar] [CrossRef]
- Moutinho, M.; Nunes, M.J.; Correira, J.C.; Gama, M.J.; Castro-Caldas, M.; Cedazo-Minguea, A.; Rodrigues, C.M.; Bjorkhem, I.; Ruas, J.L.; Rodrigues, E. Neuronal cholesterol metabolism increases dendritic outgrowth and synaptic markers via concerted action of GGTase-I and Trk. Sci. Rep. 2016, 6, 30298. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Phys. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Segatto, M.; Manduca, A.; Lecis, C.; Rosso, P.; Jozwiak, A.; Swiezewska, E.; Moreno, S.; Trezza, V.; Pallottini, V. Simvastatin treatment highlights a new role for the Isoprenoid/Cholesterol biosyntheti pathway in the modulation of emotional reactivity and cognitive performane in rats. Neuropsychopharmacol. 2014, 39, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.K.; Lerux, C.; Rosa-Neto, P.; Hamel, E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memry deficits. J. Neurochem. 2012, 32, 4705–4715. [Google Scholar]
- Leung, K.F.; Baron, R.; Seabra, M.C. Geranylgeranylation of Rab GTPases. J. Lipid Res. 2006, 47, 467–475. [Google Scholar] [CrossRef]
- Zhang, H.; Seabra, M.C.; Deisenhofer, J. Crystal structure of Rab geranylgeranyltransferase at 2.0 Å resolution. Structure 2000, 8, 241–251. [Google Scholar] [CrossRef]
- Jaśkiewicz, A.; Pająk, B.; Litwiniuk, A.; Urbańska, K.; Orzechowski, A. Geranylgeraniol prevents statin-dependent myotoxicity in C2C12 muscle cells through RAP1 GTPase prenylation and cytoprotective autophagy. Oxid. Med. Cell. Longev. 2018, 2018, 6463807. [Google Scholar] [CrossRef] [PubMed]
- Marz, P.; Otten, U.; Miserez, A.R. Statins induce differentiation and cell death in neurons and astroglia. Glia 2007, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.W.; Halford, R.W.; Ramirez, D.M.; Shah, R.; Kotti, T. Cholesterol 24-hydroxylase: An enzyme of cholesterol turnover in the brain. Ann. Rev. Biochem. 2009, 78, 1017–1040. [Google Scholar] [CrossRef]
- Schulz, J.G.; Bosel, J.; Stoeckel, M.; Megow, D.; Dirnagl, U.; Endres, M. HMG-CoA reductase inhibition causes neurite loss by interfering with geranylgeranylpyrophosphate synthesis. J. Neurochem. 2004, 89, 24–31. [Google Scholar] [CrossRef]
- Afshordel, S.; Wood, W.G.; Igbavboa, U.; Muller, W.E.; Eckert, G.P. Impaired geranylgeranyltransferase-I regulation reduces membrane-associated Rho protein levels in aged mouse brain. J. Neurochem. 2014, 129, 732–742. [Google Scholar] [CrossRef]
- Mohamed, A.; Smith, K.; Chaves, E.P. The mevalonate pathway in Alzheimer’s disease—Cholesterol and non-sterol isoprenoids. In Alzheimer’s Diseases Challenges for the Future; Zerr, I., Ed.; InTech: Rijeka, Croatia, 2015. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, W.G.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnami, H.P.; Guarnieri, P.; Caneda, C.; Ruderish, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurochem. 2014, 129, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.M.; Fuentes, G.; Rausell, A.; Valencia, A. The Ras protein superfamily: Evolutionary tree and the role of conserved amino acids. J. Biol. Chem. 2012, 196, 189–201. [Google Scholar]
- Pike, L.J. Rafts defined: A report on the Keystone symposium on lipid rafts and cell function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef]
- Aguilar, B.J.; Zhu, Y.; Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimer’s Res. Ther. 2017, 9, 97. [Google Scholar] [CrossRef]
- Ostrowski, S.M.; Wilkinson, B.L.; Golde, T.E.; Landreth, G. Statins reduce Amyloid-production through inhibition of protein isoprenylation. J. Biol. Chem. 2007, 2007, 26832–26844. [Google Scholar] [CrossRef]
- Scheper, W.; Hoozemans, J.J.M.; Hoogenraad, C.C.; Rozemuller, A.J.M.; Eikelenboom, P.; Baas, F. Rab6 is increased in Alzheimer’s disease brain and correlates with endoplasmic reticulum stress. Neuropathol. Appl. Neurobiol. 2007, 33, 523–532. [Google Scholar] [CrossRef]
- Scheper, W.; Zwart, R.; Baas, F. Rab6 membrane association is dependent of Presenilin 1 and cellular phosphorylation events. Brain Res. Mol. Brain Res. 2004, 122, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kukar, T.; Murphy, M.P.; Eriksen, J.L.; Sagi, S.A.; Weggen, S.; Smith, T.E.; Ladd, T.; Khan, M.A.; Kache, R.; Beard, J.; et al. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat. Med. 2005, 11, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Hooff, G.P.; Wood, W.G.; Muller, W.E.; Eckert, G.P. Isoprenoids, small GTPases and Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1801, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.G.; Eckert, G.P.; Igbavboa, U.; Muller, W.E. Statins and neuroprotection. A prescription to move the field forward. Ann. N. Y. Acad. Sci. 2010, 1199, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Eckert, G.P.; Hooff, G.P.; Strandjord, D.M.; Igbavboa, U.; Volmer, D.A.; Müller, W.E.; Wood, G.P. Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol. Dis. 2009, 35, 251–257. [Google Scholar] [CrossRef]
- Ng Ying Kin, N.M.; Palo, J.; Haltia, M.; Wolfe, L.S. High levels of brain dolichols in neuronal ceroid-lipofuscinosis and senescence. J. Neurochem. 1983, 40, 1465–1473. [Google Scholar] [PubMed]
- Edlund, C.; Soderberg, M.; Kristensson, K.; Dallner, G. Ubiquinone, dolichol, and cholesterol metabolism in aging and Alzheimer’s disease. Biochem. Cell Biol. 1992, 70, 422–428. [Google Scholar] [CrossRef]
- Soderberg, M.; Edlund, C.; Alafuzoff, I.; Kristensson, K.; Dallner, G. Lipid composition in different regions of the brain in Alzheimer’s disease/senile dementia of Alzheimer’s type. J. Neurochem. 1992, 59, 1646–1653. [Google Scholar] [CrossRef]
- Riekse, R.G.; Li, G.; Petrie, E.C.; Leverenz, J.B.; Vavrek, D.; Vuletic, S.; Albers, J.J.; Montine, T.J.; Lee, V.M.; Lee, M.; et al. Effect of statins on Alzheimer’s disease biomarkers in cerebrospinal fluid. J. Alzheimer’s Dis. 2006, 10, 399–406. [Google Scholar] [CrossRef]
- Li, G.; Larson, E.B.; Sonnen, J.A.; Shofer, J.B.; Petrie, E.C.; Schantz, A.; Perskind, E.R.; Raskind, M.A.; Breitner, J.C.; Montine, T.J. Statin therapy is associated with reduced neuropathologic changes of Alzheimer’s disease. Neurology 2007, 69, 878–885. [Google Scholar] [CrossRef]
- Pelleieux, S.; Picard, S.; Lamarre-Theroux, L.; Dea, D.; Leduc, V.; Tsantrizos, Y.S.; Poirer, J. Isoprenoids and tau pathology in sporadic Alzheimer’s disease. Neurobiol. Aging 2018, 65, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Julien, C.; Frappier, J.; Miron, J.; Theroux, L.; Dea, D.; Bretner, J.C.S.; Poirer, J. Alterations in cholesterol metabolism–related genes in sporadic Alzheimer’s disease. Neurobiol. Aging 2018, 66, 180. [Google Scholar] [CrossRef]
- Lee, K.J.; Moussa, C.E.; Lee, Y.; Sung, Y.; Howell, B.W.; Turner, R.S.; Pak, D.T.; Hoe, H.S. Beta amyloid-independent role of amyloid precursor protein in generation and maintance of dendritic spines. Neuroscience 2010, 169, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Ramser, E.M.; Yamashita, M.; Nakajima, K.; Mori, H.; Silverman, M.A.; Tomiyama, T. Intracellular amyloid b oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol. Commun. 2015, 3, 51. [Google Scholar] [CrossRef]
- Wirths, O.; Multhaup, G.; Czech, C.; Blanchard, V.; Moussaoui, S.; Tremp, G.; Pradier, L.; Beyreuther, K.; Bayer, T.A. Intraneuronal Aβ accumulation precedes plaque formation in β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci. Lett. 2001, 306, 116–120. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, F.; Grrengard, P.; Gouras, G.K. Intraneuronal Alzheimer aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzhemer’s disease: An immunoelectron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Van der Burgh, R.; Nijhuis, L.; Pervolaraki, K.; Compeer, E.; Jongeneel, L.H.; van Gijn, M.; Coffer, P.J.; Murphy, M.P.; Mastroberrardino, P.G.; Frenkel, J.; et al. Defects in mitochondrial clearance predispose human monocytes to interleukin-1β hypersecretion. J. Biol. Chem. 2014, 289, 5000–5012. [Google Scholar] [CrossRef]
- Kuijk, L.M.; Beekman, J.M.; Koster, J.; Waterham, H.R.; Frenkel, J.; Coffer, P.J. HMG-CoA reductase inhibition induces IL-1β release through Rac1/PI3K/PKB-dependent caspase-1 activation. Blood 2008, 112, 3563–3573. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Tricario, P.M.; Piscianz, E.; Kleiner, G.; Brumatti, L.V.; Crovella, S. Lovastatin induces apoptosis through the mitochondrial pathway in an undifferentiated SH-SY5Y neuroblastoma cell line. Cell Death Dis. 2013, 4, e858. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Zanim, V.; Piscianz, E.; Tricario, P.M.; Vuch, J.; Girardelli, M.; Monasta, L.; Bianco, A.M.; Crovella, S. Lovastatin-induced apoptosis is modulated by geranylgeraniol in a neuroblastoma cell line. Int. J. Dev. Neurosci. 2012, 30, 451–456. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Marcuzzi, A.; Piscianz, E.; Monasta, L.; Crovella, S.; Kleiner, G. Mevalonate kinase deficiency and neuroinflammation: Balance between apoptosis and pyroptosis. Int. J. Mol. Sci. 2013, 14, 23274–23288. [Google Scholar] [CrossRef]
- Tricarico, P.M.; Kleiner, G.; Valencic, E.; Campisciano, G.; Girardelli, M.; Crovella, S.; Knowles, A.; Marcuzzi, A. Block of the mevalonate pathway triggers oxidative and inflammatory molecular mechanisms modulated by exogeneous isoprenoid compounds. Int. J. Mol. Sci. 2014, 15, 6843–6853. [Google Scholar] [CrossRef]
- Bodeman, B.O.; Orvedahl, A.; Cheng, T.; Ram, R.R.; Ou, Y.H.; Formstecher, E.; Maiti, M.; Hazelett, C.C.; Wauson, E.M.; Balakireva, M.; et al. RalB and exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 2011, 144, 253–267. [Google Scholar] [CrossRef]
- Bento, C.F.; Puri, C.; Moreau, K.; Rubinsztein, D.C. The role of membrane trafficking small GTPases in the regulation of autophagy. J. Cell Sci. 2013, 126, 1059–1069. [Google Scholar] [CrossRef]
- Sirvent, P.; Mercier, J.; Lacampagne, A. New insights into mechanisms of statin-associated myotoxicity. Curr. Opin. Pharmacol. 2008, 8, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Van der Burgh, R.; Pervolaraki, K.; Turkenburg, M.; Waterham, H.R.; Frenkel, J.; Boes, M. Unprenylated RhoA contributes to IL-1β hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity. J. Biol. Chem. 2014, 289, 27757–27765. [Google Scholar] [CrossRef]
- Jaśkiewicz, A.; Pająk, B.; Łabieniec-Watała, M.; de Palma, C.; Orzechowski, A. Diverse action of selected statins on skeletal muscle cells—An attempt to explain geranylgeraniol (GGOH) protective effect in statin-associated myopathy (SAM). J. Clin. Med. Under revision.
- Campia, I.; Lussiana, C.; Pescarmona, G.; Ghigo, D.; Bosia, A.; Riganti, C. Geranylgeraniol prevents the cytotoxic effects of mevastatin in THP-1 cells, without decreasing the beneficial effects on cholesterol synthesis. Br. J. Pharmacol. 2009, 158, 1777–1786. [Google Scholar] [CrossRef]
- Mandey, S.H.; Kuijk, L.M.; Frenkel, J.; Waterham, H.R. A role for geranylgeranylation in interleukin-1β secretion. Arthritis Rheumatol. 2006, 54, 3690–3695. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Tommasini, A.; Crovella, S.; Pontillo, A. Natural isoprenoids inhibit LPS-induced-production of cytokines and nitric oxide in aminobisphosphonate-treated monocytes. Int. Immunopharmacol. 2003, 10, 639–642. [Google Scholar] [CrossRef]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Nat. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Vince, J.E. Ambiguities in NLRP3 inflammasome regulation: Is there a role for mitochondria? Biochim. Biophys. Acta 2014, 1840, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Tatsuna, I.; Uchida, D.; Moroo, I.; Morio, H.; Nakamura, S.; Noguchi, Y.; Yasuda, T.; Kitagawa, M.; Saito, Y.; et al. Geranylgeranyl-pyrophosphate, an isoprenoid of mevalonate cascade, is a critical compound for rat primary cultured cortical neurons to protect the cell death induced by 3-hydroxy-3-methylglutaryl-CoA reductase inhibition. J. Neurochem. 2000, 20, 2852–2859. [Google Scholar] [CrossRef]
- Jacobson, M.D.; Burne, J.F.; Raff, M.C. Programmed cell death and Bcl-2 protection in the absence of a nucleus. EMBO J. 1994, 13, 1899–1910. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Flanking Region | Primers and their Sequences |
|---|---|
| VP1.5 (forward seq primer) | 5′ GGACTTTCCAAAATGTCG 3′ Tm = 51 oC |
| XL39 (reverse seq primer) | 5′ ATTAGGACAAGGCTGGTGGG 3′ Tm = 60 oC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pająk, B.; Kania, E.; Gołaszewska, A.; Orzechowski, A. Preliminary Study on Clusterin Protein (sCLU) Expression in PC-12 Cells Overexpressing Wild-Type and Mutated (Swedish) AβPP genes Affected by Non-Steroid Isoprenoids and Water-Soluble Cholesterol. Int. J. Mol. Sci. 2019, 20, 1481. https://doi.org/10.3390/ijms20061481
Pająk B, Kania E, Gołaszewska A, Orzechowski A. Preliminary Study on Clusterin Protein (sCLU) Expression in PC-12 Cells Overexpressing Wild-Type and Mutated (Swedish) AβPP genes Affected by Non-Steroid Isoprenoids and Water-Soluble Cholesterol. International Journal of Molecular Sciences. 2019; 20(6):1481. https://doi.org/10.3390/ijms20061481
Chicago/Turabian StylePająk, Beata, Elżbieta Kania, Anita Gołaszewska, and Arkadiusz Orzechowski. 2019. "Preliminary Study on Clusterin Protein (sCLU) Expression in PC-12 Cells Overexpressing Wild-Type and Mutated (Swedish) AβPP genes Affected by Non-Steroid Isoprenoids and Water-Soluble Cholesterol" International Journal of Molecular Sciences 20, no. 6: 1481. https://doi.org/10.3390/ijms20061481
APA StylePająk, B., Kania, E., Gołaszewska, A., & Orzechowski, A. (2019). Preliminary Study on Clusterin Protein (sCLU) Expression in PC-12 Cells Overexpressing Wild-Type and Mutated (Swedish) AβPP genes Affected by Non-Steroid Isoprenoids and Water-Soluble Cholesterol. International Journal of Molecular Sciences, 20(6), 1481. https://doi.org/10.3390/ijms20061481