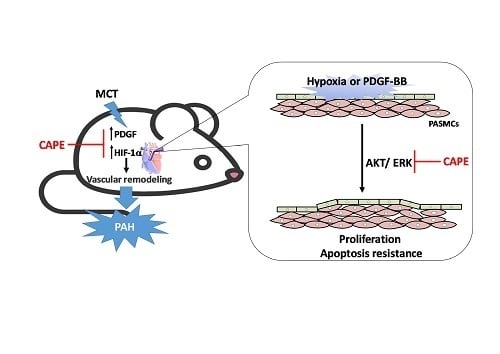
Caffeic Acid Phenethyl Ester Rescues Pulmonary Arterial Hypertension through the Inhibition of AKT/ERK-Dependent PDGF/HIF-1α In Vitro and In Vivo

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. CAPE Attenuates Pulmonary Hypertension and Right Ventricular Hypertrophy in MCT-Treated Rats
2.2. Caffeic Acid Phenethyl Ester (CAPE) Prevents Pulmonary Vascular Remodeling in Monocrotaline (MCT)-Induced Pulmonary Arterial Hypertension (PAH) Rat Model
2.3. CAPE Attenuates HIF-1α and PDGF-BB Expression in MCT-Treated Rats
2.4. CAPE Attenuates Hypoxia- and PDGF-BB-Induced HIF-1α Expression via the AKT/ERK Pathway in hPASMCs
2.5. CAPE Reduces Hypoxia- and PDGF-BB-Induced Proliferation via Inhibiting AKT/ERK Pathway in hPASMCs
2.6. CAPE Promotes the Apoptosis of hPASMCs
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. MCT Rat Studies
4.4. Hemodynamic Measurements
4.5. Histology and Immunohistochemical Analysis of Pulmonary Arteries
4.6. Cell Viability Assay
4.7. BrdU Proliferation Assay
4.8. Senescence-Associated β-Galactosidase (SA-β-gal) Staining
4.9. Transferase-Mediated Deoxyuridine Triphosphate-Biotin Nick End Labeling (TUNEL) Assays
4.10. Quantitative PCR (qPCR)
4.11. Western Blot
4.12. Measurement of PDGF-BB Generation
4.13. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| PAH | pulmonary arterial hypertension |
| hPASMCs | human pulmonary artery smooth muscle cells |
| HIF-1α | hypoxia-inducible factor-1α |
| CAPE | caffeic acid phenethyl ester |
| MCT | monocrotaline |
| RVSP | right ventricle systolic pressure |
| LVSP | systolic pressure of left ventricle |
| BrdU | bromodeoxyuridine |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PDGF-BB | platelet-derived growth factor |
| MMPs | matrix metalloproteinases |
| TIMPs | tissue inhibitors of metalloproteinases |
| VEGF | vascular endothelial growth factor |
References
- Hassoun, P.M.; Mouthon, L.; Barber, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.W.; et al. Inflammation, growth factors, and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 2009, 54 (Suppl. 1), S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Sadamura-Takenaka, Y.; Ito, T.; Noma, S.; Oyama, Y.; Yamada, S.; Kawahara, K.; Inoue, H.; Maruyama, I. HMGB1 promotes the development of pulmonary arterial hypertension in rats. PLoS ONE 2014, 9, e102482. [Google Scholar] [CrossRef]
- Cho, M.S.; Park, W.S.; Jung, W.K.; Qian, Z.J.; Lee, D.S.; Choi, J.S.; Lee, D.Y.; Park, S.G.; Seo, S.K.; Kim, H.J.; et al. Caffeic acid phenethyl ester promotes anti-inflammatory effects by inhibiting MAPK and NF-kappaB signaling in activated HMC-1 human mast cells. Pharm. Biol. 2014, 52, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Farkas, L.; Gauldie, J.; Voelkel, N.F.; Kolb, M. Pulmonary hypertension and idiopathic pulmonary fibrosis: A tale of angiogenesis, apoptosis, and growth factors. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1–15. [Google Scholar] [CrossRef]
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 2016, 47, 288–303. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoo, H.Y. Hypoxic pulmonary vasoconstriction and vascular contractility in monocrotaline-induced pulmonary arterial hypertensive rats. Korean J. Physiol. Pharmacol. 2016, 20, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Fandrey, J.; Gorr, T.A.; Gassmann, M. Regulating cellular oxygen sensing by hydroxylation. Cardiovasc. Res. 2006, 71, 642–651. [Google Scholar] [CrossRef]
- Rose, F.; Grimminger, F.; Appel, J.; Heller, M.; Pies, V.; Weissmann, N.; Fink, L.; Schmidt, S.; Krick, S.; Camenisch, G.; et al. Hypoxic pulmonary artery fibroblasts trigger proliferation of vascular smooth muscle cells: Role of hypoxia-inducible transcription factors. FASEB J. 2002, 16, 1660–1661. [Google Scholar] [CrossRef]
- Schultz, K.; Fanburg, B.L.; Beasley, D. Hypoxia and hypoxia-inducible factor-1alpha promote growth factor-induced proliferation of human vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2528–H2534. [Google Scholar] [CrossRef] [PubMed]
- Antoniu, S.A. Targeting PDGF pathway in pulmonary arterial hypertension. Expert Opin. Ther. Targets 2012, 16, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Belaiba, R.S.; Bonello, S.; Zahringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Gorlach, A. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef]
- Marquez, N.; Sancho, R.; Macho, A.; Calzado, M.A.; Fiebich, B.L.; Munoz, E. Caffeic acid phenethyl ester inhibits T-cell activation by targeting both nuclear factor of activated T-cells and NF-kappaB transcription factors. J. Pharmacol. Exp. Ther. 2004, 308, 993–1001. [Google Scholar] [CrossRef]
- Vilela, P.; de Oliveira, J.R.; de Barros, P.P.; Leao, M.V.; de Oliveira, L.D.; Jorge, A.O. In vitro effect of caffeic acid phenethyl ester on matrix metalloproteinases (MMP-1 and MMP-9) and their inhibitor (TIMP-1) in lipopolysaccharide-activated human monocytes. Arch. Oral. Biol. 2015, 60, 1196–1202. [Google Scholar] [CrossRef]
- Lin, H.P.; Lin, C.Y.; Huo, C.; Hsiao, P.H.; Su, L.C.; Jiang, S.S.; Chan, T.M.; Chang, C.H.; Chen, L.T.; Kung, H.J.; et al. Caffeic acid phenethyl ester induced cell cycle arrest and growth inhibition in androgen-independent prostate cancer cells via regulation of Skp2, p53, p21Cip1 and p27Kip1. Oncotarget 2015, 6, 6684–6707. [Google Scholar] [CrossRef]
- Tseng, T.H.; Shen, C.H.; Huang, W.S.; Chen, C.N.; Liang, W.H.; Lin, T.H.; Kuo, H.C. Activation of neutral-sphingomyelinase, MAPKs, and p75 NTR-mediating caffeic acid phenethyl ester-induced apoptosis in C6 glioma cells. J. Biomed. Sci. 2014, 21, 61. [Google Scholar] [CrossRef]
- Ho, H.C.; Chang, H.C.; Ting, C.T.; Kuo, C.Y.; Yang, V.C. Caffeic acid phenethyl ester inhibits proliferation and migration, and induces apoptosis in platelet-derived growth factor-BB-stimulated human coronary smooth muscle cells. J. Vasc. Res. 2012, 49, 24–32. [Google Scholar] [CrossRef]
- Paeng, S.H.; Jung, W.K.; Park, W.S.; Lee, D.S.; Kim, G.Y.; Choi, Y.H.; Seo, S.K.; Jang, W.H.; Choi, J.S.; Lee, Y.M.; et al. Caffeic acid phenethyl ester reduces the secretion of vascular endothelial growth factor through the inhibition of the ROS, PI3K and HIF-1alpha signaling pathways in human retinal pigment epithelial cells under hypoxic conditions. Int. J. Mol. Med. 2015, 35, 1419–1426. [Google Scholar] [CrossRef]
- Armutcu, F.; Akyol, S.; Ustunsoy, S.; Turan, F.F. Therapeutic potential of caffeic acid phenethyl ester and its anti-inflammatory and immunomodulatory effects (Review). Exp. Ther. Med. 2015, 9, 1582–1588. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Semenz1, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Colombo, R.; Siqueira, R.; Conzatti, A.; de Lima Seolin, B.G.; Fernandes, T.R.; Godoy, A.E.; Litvin, I.E.; Silva, J.M.; Tucci, P.J.; da Rosa Araujo, A.S.; et al. Exercise training contributes to H2O2/VEGF signaling in the lung of rats with monocrotaline-induced pulmonary hypertension. Vascul. Pharmacol. 2016, 87, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.X.; Xu, X.Y.; Zhao, Z.; Zhao, F.Y.; Gao, Y.M.; Yan, X.H.; Wan, Y. Hydrogen peroxide is a critical regulator of the hypoxia-induced alterations of store-operated Ca(2+) entry into rat pulmonary arterial smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L477–L487. [Google Scholar] [CrossRef]
- Liao, H.F.; Chen, Y.Y.; Liu, J.J.; Hsu, M.L.; Shieh, H.J.; Liao, H.J.; Shieh, C.J.; Shiao, M.S.; Chen, Y.J. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J. Agric. Food Chem. 2003, 51, 7907–7912. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, G.; Karim, S.; Akram, M.R.; Khan, S.A.; Azhar, S.; Mumtaz, A.; Bin Asa4, M.H. Caffeic acid phenethyl ester and therapeutic potentials. Biomed. Res. 2014, 2014, 145342. [Google Scholar] [CrossRef]
- Dai, Z.; Zhu, M.M.; Peng, Y.; Machireddy, N.; Evans, C.E.; Machado, R.; Zhang, X.; Zhao, Y.Y. Therapeutic Targeting of Vascular Remodeling and Right Heart Failure in PAH with HIF-2alpha Inhibitor. Am. J. Respir. Crit. Care Med. 2018, 198, 1423–1434. [Google Scholar] [CrossRef]
- Perros, F.; Montani, D.; Dorfmuller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O.; et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef]
- Sweatt, A.J.; Hedlin, H.K.; Balasubramanian, V.; Hsi, A.; Blum, L.K.; Robinson, W.H.; Hadda, F.; Hickey, P.M.; Condliffe, R.A.; Lawrie, A.; et al. Discovery of Distinct Immune Phenotypes Using Machine Learning in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 904–919. [Google Scholar] [CrossRef]
- Sanchez, O.; Marcos, E.; Perros, F.; Fadel, E.; Tu, L.; Humbert, M.; Dartevelle, P.; Simonneau, G.; Adnot, S.; Eddahibi, S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2007, 176, 1041–1047. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.L.; Law, T.C. Chronic hypoxia- and monocrotaline-induced elevation of hypoxia-inducible factor-1 alpha levels and pulmonary hypertension. J. Biomed. Sci. 2004, 11, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Ayla, S.; Tunali, G.; Bilgic, B.E.; Sofuoglu, K.; Ozdemir, A.A.; Tanriverdi, G.; Ozdemir, S.; Soner, B.C.; Ozturk, B.; Karahuseyinoglu, S.; et al. Antioxidant activity of CAPE (caffeic acid phenethyl ester) in vitro can protect human sperm deoxyribonucleic acid from oxidative damage. Acta Histochem. 2018, 120, 117–121. [Google Scholar] [CrossRef]
- Toyoda, T.; Tsukamoto, T.; Takasu, S.; Shi, L.; Hirano, N.; Ban, H.; Kumagai, T.; Tatematsu, M. Anti-inflammatory effects of caffeic acid phenethyl ester (CAPE), a nuclear factor-kappaB inhibitor, on Helicobacter pylori-induced gastritis in Mongolian gerbils. Int. J. Cancer 2009, 125, 1786–1795. [Google Scholar] [CrossRef]
- Song, J.J.; Lim, H.W.; Kim, K.; Kim, K.M.; Cho, S.; Chae, S.W. Effect of caffeic acid phenethyl ester (CAPE) on H(2)O(2) induced oxidative and inflammatory responses in human middle ear epithelial cells. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Jung, W.K.; Lee, C.M.; Yea, S.S.; Choi, Y.H.; Kim, G.Y.; Lee, D.S.; Na, G.; Park, S.G.; Seo, S.K.; et al. Caffeic acid phenethyl ester inhibits the inflammatory effects of interleukin-1beta in human corneal fibroblasts. Immunopharmacol. Immunotoxicol. 2014, 36, 371–377. [Google Scholar] [CrossRef]
- Choi, D.; Han, J.; Lee, Y.; Choi, J.; Han, S.; Hong, S.; Jeon, H.; Kim, Y.M.; Jung, Y. Caffeic acid phenethyl ester is a potent inhibitor of HIF prolyl hydroxylase: Structural analysis and pharmacological implication. J. Nutr. Biochem. 2010, 21, 809–817. [Google Scholar] [CrossRef]
- Kim, H.G.; Han, E.H.; Im, J.H.; Lee, E.J.; Jin, S.W.; Jeong, H.G. Caffeic acid phenethyl ester inhibits 3-MC-induced CYP1A1 expression through induction of hypoxia-inducible factor-1alpha. Biochem. Biophys. Res. Commun. 2015, 465, 562–568. [Google Scholar] [CrossRef]
- Mermis, J.; Gu, H.; Xue, B.; Li, F.; Tawfik, O.; Buch, S.; Bartolome, S.; O’Brien-Ladner, A.; Dhillon, N.K. Hypoxia-inducible factor-1 alpha/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respir. Res. 2011, 12, 103. [Google Scholar] [CrossRef]
- Kim, E.Y.; Ryu, J.H.; Kim, A.K. CAPE promotes TRAIL-induced apoptosis through the upregulation of TRAIL receptors via activation of p38 and suppression of JNK in SK-Hep1 hepatocellular carcinoma cells. Int. J. Oncol. 2013, 43, 1291–1300. [Google Scholar] [CrossRef]
- God, N.; Dozier, S.J.; Johnson, R.S. HIF-1 in cell cycle regulation, apoptosis, and tumor progression. Antioxid. Redox Signal. 2003, 5, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Minet, E.; Arnoul, T.; Michel, G.; Rolan, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett. 2000, 468, 53–58. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Roch, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Xu, R.; Hu, Z.; Tian, Y.; Zhu, Y.; Gu, L.; Zhou, L. PI3K and ERK-induced Rac1 activation mediates hypoxia-induced HIF-1alpha expression in MCF-7 breast cancer cells. PLoS ONE 2011, 6, e25213. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Li, H.Z.; Wang, Y.H.; Peng, X.; Shao, H.J.; Li, H.X.; Bai, S.Z.; Lu, X.X.; Wu, L.Y.; Wang, R.; et al. Exogenous spermine inhibits the proliferation of human pulmonary artery smooth muscle cells caused by chemically-induced hypoxia via the suppression of the ERK1/2- and PI3K/AKT-associated pathways. Int. J. Mol. Med. 2016, 37, 39–46. [Google Scholar] [CrossRef]
- Zhang, L.; Pu, Z.; Wang, J.; Zhang, Z.; Hu, D.; Wang, J. Baicalin inhibits hypoxia-induced pulmonary artery smooth muscle cell proliferation via the AKT/HIF-1alpha/p27-associated pathway. Int. J. Mol. Sci. 2014, 15, 8153–8168. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and Methodological Rigor in Pulmonary Arterial Hypertension Preclinical and Translational Research. Circ. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef]
- Raghavan, A.; Zhou, G.; Zhou, Q.; Ibe, J.C.; Ramchandran, R.; Yang, Q.; Racherl, H.; Raychaudhuri, P.; Raj, J.U. Hypoxia-induced pulmonary arterial smooth muscle cell proliferation is controlled by forkhead box M1. Am. J. Respir. Cell Mol. Biol. 2012, 46, 431–436. [Google Scholar] [CrossRef]
- Ciuclan, L.; Bonneau, O.; Hussey, M.; Duggan, N.; Holmes, A.M.; Goo4, R.; Stringer, R.; Jones, P.; Morrell, N.W.; Jarai, G.; et al. A novel murine model of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2011, 184, 1171–1182. [Google Scholar] [CrossRef]
- Thomas, M.; Docx, C.; Holmes, A.M.; Beach, S.; Duggand, N.; Englan, K.; Leblanc, C.; Lebret, C.; Schindler, F.; Raz, F.; et al. Activin-like kinase 5 (ALK5) mediates abnormal proliferation of vascular smooth muscle cells from patients with familial pulmonary arterial hypertension and is involved in the progression of experimental pulmonary arterial hypertension induced by monocrotaline. Am. J. Pathol. 2009, 174, 380–389. [Google Scholar]
- Sutendr, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.-C.; Chi, P.-L.; Shen, M.-C.; Shu, C.-W.; Wann, S.-R.; Liu, C.-P.; Tseng, C.-J.; Huang, W.-C. Caffeic Acid Phenethyl Ester Rescues Pulmonary Arterial Hypertension through the Inhibition of AKT/ERK-Dependent PDGF/HIF-1α In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 1468. https://doi.org/10.3390/ijms20061468
Cheng C-C, Chi P-L, Shen M-C, Shu C-W, Wann S-R, Liu C-P, Tseng C-J, Huang W-C. Caffeic Acid Phenethyl Ester Rescues Pulmonary Arterial Hypertension through the Inhibition of AKT/ERK-Dependent PDGF/HIF-1α In Vitro and In Vivo. International Journal of Molecular Sciences. 2019; 20(6):1468. https://doi.org/10.3390/ijms20061468
Chicago/Turabian StyleCheng, Chin-Chang, Pei-Ling Chi, Min-Ci Shen, Chih-Wen Shu, Shue-Ren Wann, Chun-Peng Liu, Ching-Jiunn Tseng, and Wei-Chun Huang. 2019. "Caffeic Acid Phenethyl Ester Rescues Pulmonary Arterial Hypertension through the Inhibition of AKT/ERK-Dependent PDGF/HIF-1α In Vitro and In Vivo" International Journal of Molecular Sciences 20, no. 6: 1468. https://doi.org/10.3390/ijms20061468
APA StyleCheng, C.-C., Chi, P.-L., Shen, M.-C., Shu, C.-W., Wann, S.-R., Liu, C.-P., Tseng, C.-J., & Huang, W.-C. (2019). Caffeic Acid Phenethyl Ester Rescues Pulmonary Arterial Hypertension through the Inhibition of AKT/ERK-Dependent PDGF/HIF-1α In Vitro and In Vivo. International Journal of Molecular Sciences, 20(6), 1468. https://doi.org/10.3390/ijms20061468