Functional Interaction among KCa and TRP Channels for Cardiovascular Physiology: Modern Perspectives on Aging and Chronic Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
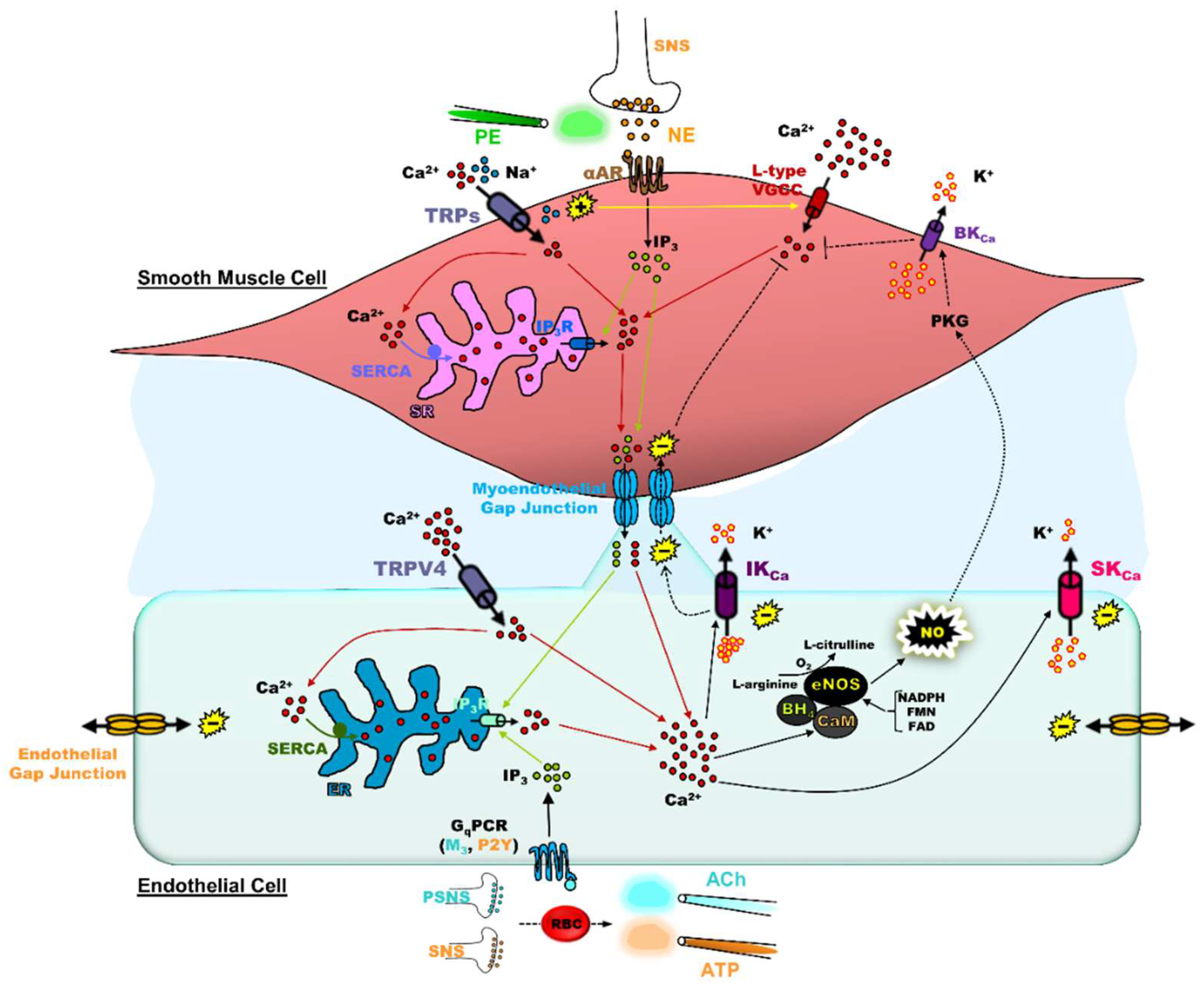
2. Significance of the Relationship of [Ca2+]i and Vm in the Vascular Wall of Blood Vessels
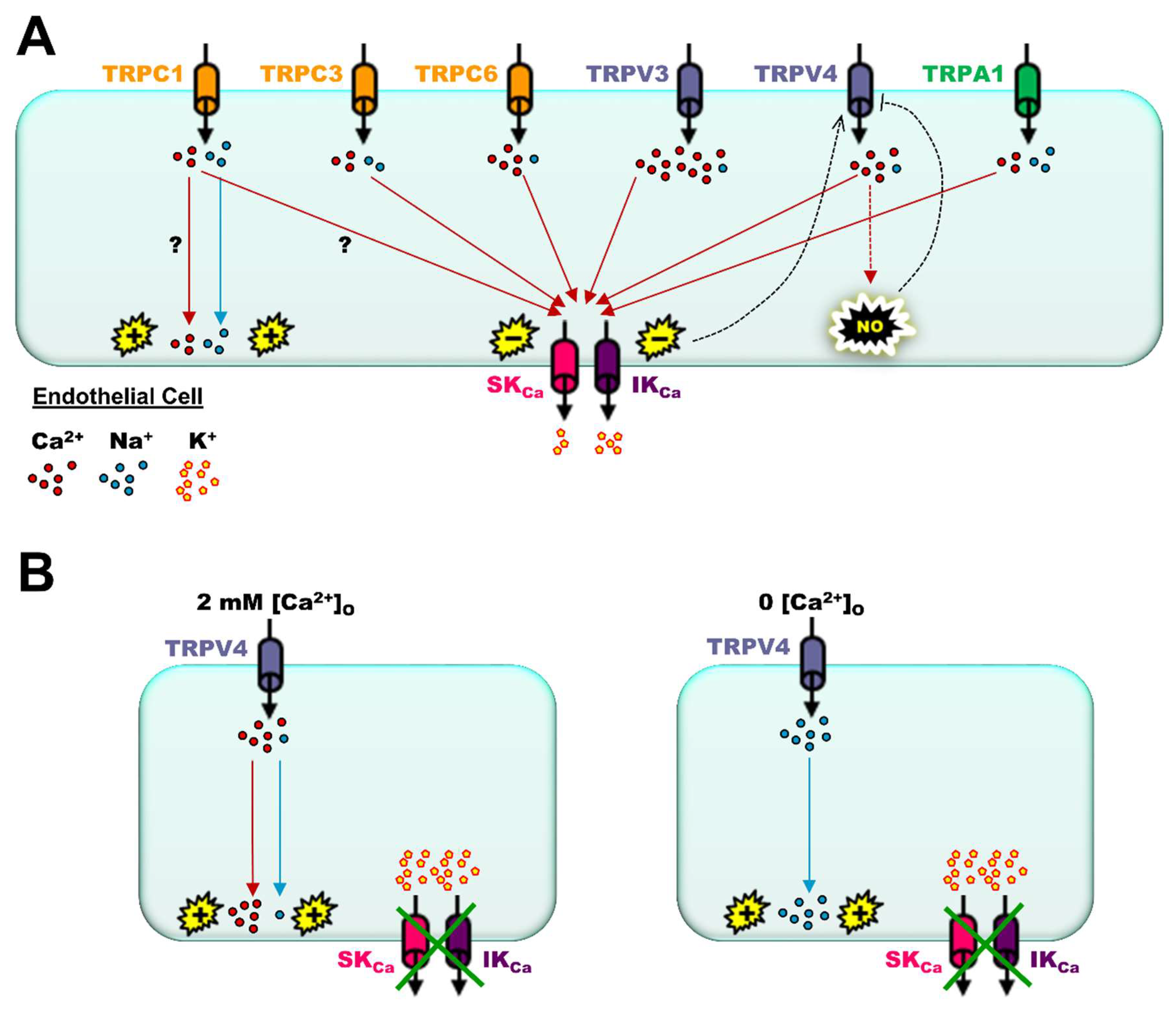
3. Significance of SKCa/IKCa and TRP Channels in the Blood Vasculature
4. Impact of Adrenergic Tone on SKCa/IKCa and TRP Channels & Significance of In Vivo vs. Ex Vivo Observations
5. Role of Familial Mutations in KCa and TRP Channels in the Emergence of Cardiovascular Disease
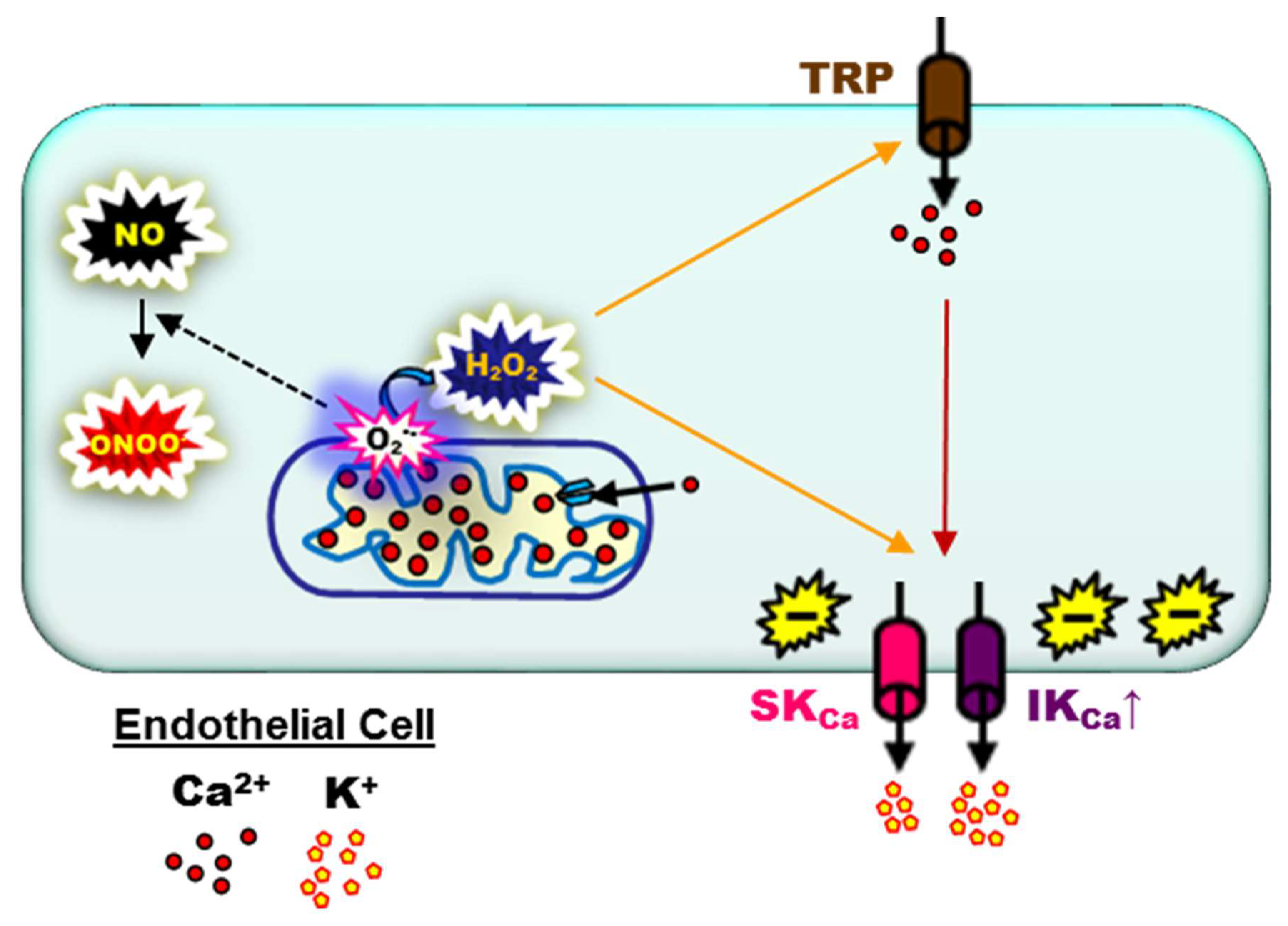
6. Endothelial SKCa and IKCa during Aging and Chronic Pathology
7. What May Be Next for Investigative Studies of Endothelial Function: Novel Physiological and Pharmacological Molecular Signaling Pathways
8. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Socha, M.J.; Segal, S.S. Microvascular mechanisms limiting skeletal muscle blood flow with advancing age. J. Appl. Physiol. (1985) 2018, 125, 1851–1859. [Google Scholar] [CrossRef] [PubMed]
- Himmel, H.M.; Whorton, A.R.; Strauss, H.C. Intracellular calcium, currents, and stimulus-response coupling in endothelial cells. Hypertension 1993, 21, 112–127. [Google Scholar] [PubMed]
- Behringer, E.J.; Segal, S.S. Membrane potential governs calcium influx into microvascular endothelium: Integral role for muscarinic receptor activation. J. Physiol. 2015, 593, 4531–4548. [Google Scholar] [CrossRef] [PubMed]
- Di Giuro, C.M.L.; Shrestha, N.; Malli, R.; Groschner, K.; van Breemen, C.; Fameli, N. Na+/Ca2+ exchangers and Orai channels jointly refill endoplasmic reticulum (ER) Ca2+ via ER nanojunctions in vascular endothelial cells. Pflug. Arch. 2017, 469, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Avila-Medina, J.; Mayoral-Gonzalez, I.; Dominguez-Rodriguez, A.; Gallardo-Castillo, I.; Ribas, J.; Ordonez, A.; Rosado, J.A.; Smani, T. The Complex Role of Store Operated Calcium Entry Pathways and Related Proteins in the Function of Cardiac, Skeletal and Vascular Smooth Muscle Cells. Front. Physiol. 2018, 9, 257. [Google Scholar]
- Trebak, M. STIM/Orai signalling complexes in vascular smooth muscle. J. Physiol. 2012, 590, 4201–4208. [Google Scholar] [CrossRef]
- Zhang, X.; Gueguinou, M.; Trebak, M. Store-Independent Orai Channels Regulated by STIM. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Jr., Eds.; CRC Press: Boca Raton, FL, USA, 2018; pp. 197–214. [Google Scholar]
- Behringer, E.J. Calcium and electrical signaling in arterial endothelial tubes: New insights into cellular Physiol.ogy and cardiovascular function. Microcirculation 2017, 24. [Google Scholar] [CrossRef]
- Ledoux, J.; Taylor, M.S.; Bonev, A.D.; Hannah, R.M.; Solodushko, V.; Shui, B.; Tallini, Y.; Kotlikoff, M.I.; Nelson, M.T. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc. Natl. Acad. Sci. USA 2008, 105, 9627–9632. [Google Scholar] [CrossRef]
- Bagher, P.; Beleznai, T.; Kansui, Y.; Mitchell, R.; Garland, C.J.; Dora, K.A. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc. Natl. Acad. Sci. USA 2012, 109, 18174–18179. [Google Scholar] [CrossRef]
- Sandow, S.L.; Neylon, C.B.; Chen, M.X.; Garland, C.J. Spatial separation of endothelial small- and intermediate-conductance calcium-activated potassium channels (KCa) and connexins: Possible relationship to vasodilator function? J. Anat. 2006, 209, 689–698. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Dalsgaard, T.; Bonev, A.D.; Hill-Eubanks, D.C.; Kotlikoff, M.I.; Scott, J.D.; Santana, L.F.; Nelson, M.T. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci. Signal. 2014, 7, ra66. [Google Scholar] [CrossRef]
- Emerson, G.G.; Segal, S.S. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: Role in vasomotor control. Circ. Res. 2000, 87, 474–479. [Google Scholar] [CrossRef]
- Garland, C.J.; Bagher, P.; Powell, C.; Ye, X.; Lemmey, H.A.L.; Borysova, L.; Dora, K.A. Voltage-dependent Ca2+ entry into smooth muscle during contraction promotes endothelium-mediated feedback vasodilation in arterioles. Sci. Signal. 2017, 10, eaal3806. [Google Scholar] [PubMed]
- Billaud, M.; Lohman, A.W.; Johnstone, S.R.; Biwer, L.A.; Mutchler, S.; Isakson, B.E. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharm. Rev. 2014, 66, 513–569. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Sessa, W.C. Endothelial NOS: Perspective and recent developments. Br. J. Pharm. 2019, 176, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [PubMed]
- Feil, R.; Lohmann, S.M.; de Jonge, H.; Walter, U.; Hofmann, F. Cyclic GMP-dependent protein kinases and the cardiovascular system: Insights from genetically modified mice. Circ. Res. 2003, 93, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Irvine, J.C.; Favaloro, J.L.; Kemp-Harper, B.K. NO- activates soluble guanylate cyclase and Kv channels to vasodilate resistance arteries. Hypertension 2003, 41, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Halling, D.B.; Hall, A.W.; Aldrich, R.W. EF hands at the N-lobe of calmodulin are required for both SK channel gating and stable SK-calmodulin interaction. J. Gen. Physiol. 2009, 134, 281–293. [Google Scholar] [CrossRef]
- Behringer, E.J.; Segal, S.S. Spreading the signal for vasodilatation: Implications for skeletal muscle blood flow control and the effects of ageing. J. Physiol. 2012, 590, 6277–6284. [Google Scholar] [CrossRef]
- Emerson, G.G.; Neild, T.O.; Segal, S.S. Conduction of hyperpolarization along hamster feed arteries: Augmentation by acetylcholine. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H102–H109. [Google Scholar] [CrossRef]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995, 268 Pt 1, C799–C822. [Google Scholar] [CrossRef]
- Dora, K.A.; Doyle, M.P.; Duling, B.R. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc. Natl. Acad. Sci. USA 1997, 94, 6529–6534. [Google Scholar] [CrossRef]
- Sandow, S.L.; Hill, C.E. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium-derived hyperpolarizing factor-mediated responses. Circ. Res. 2000, 86, 341–346. [Google Scholar] [CrossRef]
- Shimokawa, H.; Yasutake, H.; Fujii, K.; Owada, M.K.; Nakaike, R.; Fukumoto, Y.; Takayanagi, T.; Nagao, T.; Egashira, K.; Fujishima, M.; et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric Circulation. J. Cardiovasc. Pharm. 1996, 28, 703–711. [Google Scholar] [CrossRef]
- Haas, T.L.; Duling, B.R. Morphology favors an endothelial cell pathway for longitudinal conduction within arterioles. Microvasc. Res. 1997, 53, 113–120. [Google Scholar] [CrossRef]
- Hakim, C.H.; Jackson, W.F.; Segal, S.S. Connexin isoform expression in smooth muscle cells and endothelial cells of hamster cheek pouch arterioles and retractor feed arteries. Microcirculation 2008, 15, 503–514. [Google Scholar] [CrossRef]
- Sandow, S.L.; Tare, M.; Coleman, H.A.; Hill, C.E.; Parkington, H.C. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ. Res. 2002, 90, 1108–1113. [Google Scholar] [CrossRef]
- Nagaraja, S.; Kapela, A.; Tran, C.H.; Welsh, D.G.; Tsoukias, N.M. Role of microprojections in myoendothelial feedback—A theoretical study. J. Physiol. 2013, 591, 2795–2812. [Google Scholar] [CrossRef]
- Tran, C.H.; Taylor, M.S.; Plane, F.; Nagaraja, S.; Tsoukias, N.M.; Solodushko, V.; Vigmond, E.J.; Furstenhaupt, T.; Brigdan, M.; Welsh, D.G. Endothelial Ca2+ wavelets and the induction of myoendothelial feedback. Am. J. Physiol.-Cell Physiol. 2012, 302, C1226–C1242. [Google Scholar] [CrossRef]
- Cole, W.C.; Welsh, D.G. Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch. Biochem. Biophys. 2011, 510, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Uhrenholt, T.R.; Domeier, T.L.; Segal, S.S. Propagation of calcium waves along endothelium of hamster feed arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1634–H1640. [Google Scholar] [CrossRef]
- Socha, M.J.; Behringer, E.J.; Segal, S.S. Calcium and electrical signalling along endothelium of the resistance vasculature. Basic Clin. Pharm. Toxicol. 2012, 110, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Kerr, P.M.; Wei, R.; Tam, R.; Sandow, S.L.; Murphy, T.V.; Ondrusova, K.; Lunn, S.E.; Tran, C.H.; Welsh, D.G.; Plane, F. Activation of endothelial IKCa channels underlies NO-dependent myoendothelial feedback. Vasc. Pharm. 2015, 74, 130–138. [Google Scholar] [CrossRef]
- Stankevicius, E.; Dalsgaard, T.; Kroigaard, C.; Beck, L.; Boedtkjer, E.; Misfeldt, M.W.; Nielsen, G.; Schjorring, O.; Hughes, A.; Simonsen, U. Opening of small and intermediate calcium-activated potassium channels induces relaxation mainly mediated by nitric-oxide release in large arteries and endothelium-derived hyperpolarizing factor in small arteries from rat. J. Pharm. Exp. 2011, 339, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Segal, S.S. Tuning electrical conduction along endothelial tubes of resistance arteries through Ca2+-activated K+ channels. Circ. Res. 2012, 110, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Shaw, R.L.; Westcott, E.B.; Socha, M.J.; Segal, S.S. Aging impairs electrical conduction along endothelium of resistance arteries through enhanced Ca2+-activated K+ channel activation. Arter. Thromb. Vasc. Biol. 2013, 33, 1892–1901. [Google Scholar] [CrossRef] [PubMed]
- Emerson, G.G.; Segal, S.S. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ. Res. 2000, 86, 94–100. [Google Scholar] [CrossRef]
- Wolfle, S.E.; Chaston, D.J.; Goto, K.; Sandow, S.L.; Edwards, F.R.; Hill, C.E. Non-linear relationship between hyperpolarisation and relaxation enables long distance propagation of vasodilatation. J. Physiol. 2011, 589 Pt 10, 2607–2623. [Google Scholar] [CrossRef]
- Hakim, M.A.; Buchholz, J.N.; Behringer, E.J. Electrical dynamics of isolated cerebral and skeletal muscle endothelial tubes: Differential roles of G-protein-coupled receptors and K+ channels. Pharm. Res. Perspect. 2018, 6, e00391. [Google Scholar] [CrossRef]
- Stocker, M. Ca2+-activated K+ channels: Molecular determinants and function of the SK family. Nat. Rev. Neurosci. 2004, 5, 758–770. [Google Scholar] [CrossRef]
- Kohler, M.; Hirschberg, B.; Bond, C.T.; Kinzie, J.M.; Marrion, N.V.; Maylie, J.; Adelman, J.P. Small-conductance, calcium-activated potassium channels from mammalian brain. Science 1996, 273, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Bonev, A.D.; Gross, T.P.; Eckman, D.M.; Brayden, J.E.; Bond, C.T.; Adelman, J.P.; Nelson, M.T. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ. Res. 2003, 93, 124–131. [Google Scholar] [CrossRef]
- Bond, C.T.; Sprengel, R.; Bissonnette, J.M.; Kaufmann, W.A.; Pribnow, D.; Neelands, T.; Storck, T.; Baetscher, M.; Jerecic, J.; Maylie, J.; et al. Respiration and parturition affected by conditional overexpression of the Ca2+-activated K+ channel subunit, SK3. Science 2000, 289, 1942–1946. [Google Scholar] [CrossRef]
- Ishii, T.M.; Silvia, C.; Hirschberg, B.; Bond, C.T.; Adelman, J.P.; Maylie, J. A human intermediate conductance calcium-activated potassium channel. Proc. Natl. Acad. Sci. USA 1997, 94, 11651–11656. [Google Scholar] [CrossRef]
- Wei, A.D.; Gutman, G.A.; Aldrich, R.; Chandy, K.G.; Grissmer, S.; Wulff, H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharm. Rev. 2005, 57, 463–472. [Google Scholar] [CrossRef]
- Kohler, R.; Ruth, P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflug Arch. Eur. J. Phys. 2010, 459, 969–976. [Google Scholar] [CrossRef]
- Logsdon, N.J.; Kang, J.; Togo, J.A.; Christian, E.P.; Aiyar, J. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J. Biol. Chem. 1997, 272, 32723–32726. [Google Scholar] [CrossRef]
- Grgic, I.; Kaistha, B.P.; Hoyer, J.; Kohler, R. Endothelial Ca2+-activated K+ channels in normal and impaired EDHF-dilator responses--relevance to cardiovascular pathologies and drug discovery. Br. J. Pharm. 2009, 157, 509–526. [Google Scholar] [CrossRef]
- Si, H.; Heyken, W.T.; Wolfle, S.E.; Tysiac, M.; Schubert, R.; Grgic, I.; Vilianovich, L.; Giebing, G.; Maier, T.; Gross, V.; et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ. Res. 2006, 99, 537–544. [Google Scholar] [CrossRef]
- Brahler, S.; Kaistha, A.; Schmidt, V.J.; Wolfle, S.E.; Busch, C.; Kaistha, B.P.; Kacik, M.; Hasenau, A.L.; Grgic, I.; Si, H.; et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 2009, 119, 2323–2332. [Google Scholar] [CrossRef] [PubMed]
- Milkau, M.; Kohler, R.; de Wit, C. Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J. 2010, 24, 3572–3579. [Google Scholar] [CrossRef] [PubMed]
- Radtke, J.; Schmidt, K.; Wulff, H.; Kohler, R.; de Wit, C. Activation of KCa3.1 by SKA-31 induces arteriolar dilatation and lowers blood pressure in normo- and hypertensive connexin40-deficient mice. Br. J. Pharm. 2013, 170, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Yap, F.C.; Weber, D.S.; Taylor, M.S.; Townsley, M.I.; Comer, B.S.; Maylie, J.; Adelman, J.P.; Lin, M.T. Endothelial SK3 channel-associated Ca2+ microdomains modulate blood pressure. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1151–H1163. [Google Scholar] [PubMed]
- Doughty, J.M.; Plane, F.; Langton, P.D. Charybdotoxin and apamin block EDHF in rat mesenteric artery if selectively applied to the endothelium. Am. J. Physiol. 1999, 276 Pt 2, H1107–H1112. [Google Scholar] [CrossRef]
- Sandow, S.L.; Haddock, R.E.; Hill, C.E.; Chadha, P.S.; Kerr, P.M.; Welsh, D.G.; Plane, F. What’s where and why at a vascular myoendothelial microdomain signalling complex. Clin. Exp. Pharm. Physiol. 2009, 36, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Dora, K.A. EDH: Endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. (Oxf.) 2017, 219, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Otani, S.; Nagaoka, T.; Omae, T.; Tanano, I.; Kamiya, T.; Ono, S.; Hein, T.W.; Kuo, L.; Yoshida, A. Histamine-Induced Dilation of Isolated Porcine Retinal Arterioles: Role of Endothelium-Derived Hyperpolarizing Factor. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4791–4798. [Google Scholar] [CrossRef] [PubMed]
- Hannah, R.M.; Dunn, K.M.; Bonev, A.D.; Nelson, M.T. Endothelial SKCa and IKCa channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J. Cereb. Blood Flow Metab. 2011, 31, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Marrelli, S.P.; Eckmann, M.S.; Hunte, M.S. Role of endothelial intermediate conductance KCa channels in cerebral EDHF-mediated dilations. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1590–H1599. [Google Scholar] [PubMed]
- McNeish, A.J.; Sandow, S.L.; Neylon, C.B.; Chen, M.X.; Dora, K.A.; Garland, C.J. Evidence for involvement of both IKCa and SKCa channels in hyperpolarizing responses of the rat middle cerebral artery. Stroke 2006, 37, 1277–1282. [Google Scholar]
- Feher, A.; Broskova, Z.; Bagi, Z. Age-related impairment of conducted dilation in human coronary arterioles. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1595–H1601. [Google Scholar] [CrossRef]
- Liu, Y.; Terata, K.; Chai, Q.; Li, H.; Kleinman, L.H.; Gutterman, D.D. Peroxynitrite inhibits Ca2+-activated K+ channel activity in smooth muscle of human coronary arterioles. Circ. Res. 2002, 91, 1070–1076. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Jian, M.Y.; Taylor, M.S.; Cioffi, D.L.; Yap, F.C.; Liedtke, W.; Townsley, M.I. Functional coupling of TRPV4, IK, and SK channels contributes to Ca2+-dependent endothelial injury in rodent lung. Pulm. Circ. 2015, 5, 279–290. [Google Scholar] [CrossRef]
- Marziano, C.; Hong, K.; Cope, E.L.; Kotlikoff, M.I.; Isakson, B.E.; Sonkusare, S.K. Nitric Oxide-Dependent Feedback Loop Regulates Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Cooperativity and Endothelial Function in Small Pulmonary Arteries. J. Am. Heart Assoc. 2017, 6, e007157. [Google Scholar]
- Sinkler, S.Y.; Segal, S.S. Rapid versus slow ascending vasodilatation: Intercellular conduction versus flow-mediated signalling with tetanic versus rhythmic muscle contractions. J. Physiol. 2017, 595, 7149–7165. [Google Scholar] [CrossRef] [PubMed]
- Bussemaker, E.; Popp, R.; Binder, J.; Busse, R.; Fleming, I. Characterization of the endothelium-derived hyperpolarizing factor (EDHF) response in the human interlobar artery. Kidney Int. 2003, 63, 1749–1755. [Google Scholar] [CrossRef]
- Salomonsson, M.; Brasen, J.C.; Sorensen, C.M. Role of renal vascular potassium channels in Physiology and pathophysiology. Acta Physiol. (Oxf.) 2017, 221, 14–31. [Google Scholar] [CrossRef]
- Waeckel, L.; Bertin, F.; Clavreul, N.; Damery, T.; Kohler, R.; Paysant, J.; Sansilvestri-Morel, P.; Simonet, S.; Vayssettes-Courchay, C.; Wulff, H.; et al. Preserved regulation of renal perfusion pressure by small and intermediate conductance KCa channels in hypertensive mice with or without renal failure. Pflug. Arch. 2015, 467, 817–831. [Google Scholar]
- Kochukov, M.Y.; Balasubramanian, A.; Abramowitz, J.; Birnbaumer, L.; Marrelli, S.P. Activation of endothelial transient receptor potential C3 channel is required for small conductance calcium-activated potassium channel activation and sustained endothelial hyperpolarization and vasodilation of cerebral artery. J. Am. Heart Assoc. 2014, 3, e000913. [Google Scholar] [CrossRef]
- Chaston, D.J.; Baillie, B.K.; Grayson, T.H.; Courjaret, R.J.; Heisler, J.M.; Lau, K.A.; Machaca, K.; Nicholson, B.J.; Ashton, A.; Matthaei, K.I.; et al. Polymorphism in endothelial connexin40 enhances sensitivity to intraluminal pressure and increases arterial stiffness. Arter. Thromb. Vasc. Biol. 2013, 33, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, A.; Gudermann, T. TRP channels in the cardiopulmonary vasculature. Adv. Exp. Med. Biol. 2011, 704, 781–810. [Google Scholar] [PubMed]
- Yue, Z.; Xie, J.; Yu, A.S.; Stock, J.; Du, J.; Yue, L. Role of TRP channels in the cardiovascular system. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H157–H182. [Google Scholar] [CrossRef]
- Poteser, M.; Graziani, A.; Rosker, C.; Eder, P.; Derler, I.; Kahr, H.; Zhu, M.X.; Romanin, C.; Groschner, K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 2006, 281, 13588–13595. [Google Scholar] [CrossRef]
- Ma, X.; Cao, J.; Luo, J.; Nilius, B.; Huang, Y.; Ambudkar, I.S.; Yao, X. Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4-c1 heteromeric channels to the plasma membrane. Arter. Thromb. Vasc. Biol. 2010, 30, 2249–2255. [Google Scholar] [CrossRef]
- Zhang, P.; Mao, A.Q.; Sun, C.Y.; Zhang, X.D.; Pan, Q.X.; Yang, D.T.; Jin, J.; Tang, C.L.; Yang, Z.Y.; Yao, X.Q.; et al. Translocation of PKG1alpha acts on TRPV4-C1 heteromeric channels to inhibit endothelial Ca2+ entry. Acta Pharm. Sin. 2016, 37, 1199–1207. [Google Scholar] [CrossRef]
- Greenberg, H.Z.E.; Carlton-Carew, S.R.E.; Khan, D.M.; Zargaran, A.K.; Jahan, K.S.; Vanessa Ho, W.S.; Albert, A.P. Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vasc. Pharm. 2017, 96–98, 53–62. [Google Scholar] [CrossRef]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef]
- Thorneloe, K.S.; Sulpizio, A.C.; Lin, Z.; Figueroa, D.J.; Clouse, A.K.; McCafferty, G.P.; Chendrimada, T.P.; Lashinger, E.S.; Gordon, E.; Evans, L.; et al. N-((1S)-1-{[4-((2S)-2-{[(2,4-dichlorophenyl)sulfonyl]amino}-3-hydroxypropanoyl)-1 -piperazinyl]carbonyl}-3-methylbutyl)-1-benzothiophene-2-carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J. Pharm. Exp. 2008, 326, 432–442. [Google Scholar]
- Cheung, M.; Bao, W.; Behm, D.J.; Brooks, C.A.; Bury, M.J.; Dowdell, S.E.; Eidam, H.S.; Fox, R.M.; Goodman, K.B.; Holt, D.A.; et al. Discovery of GSK2193874: An Orally Active, Potent, and Selective Blocker of Transient Receptor Potential Vanilloid 4. ACS Med. Chem. Lett. 2017, 8, 549–554. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef]
- Bubolz, A.H.; Mendoza, S.A.; Zheng, X.; Zinkevich, N.S.; Li, R.; Gutterman, D.D.; Zhang, D.X. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: Role of Ca2+ entry and mitochondrial ROS signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H634–H642. [Google Scholar] [CrossRef]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef]
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263. [Google Scholar] [CrossRef]
- Strotmann, R.; Schultz, G.; Plant, T.D. Ca2+-dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. J. Biol. Chem. 2003, 278, 26541–26549. [Google Scholar] [CrossRef]
- Schmidt, K.; Dubrovska, G.; Nielsen, G.; Fesus, G.; Uhrenholt, T.R.; Hansen, P.B.; Gudermann, T.; Dietrich, A.; Gollasch, M.; de Wit, C.; et al. Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br. J. Pharm. 2010, 161, 1722–1733. [Google Scholar] [CrossRef]
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef]
- Earley, S.; Waldron, B.J.; Brayden, J.E. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ. Res. 2004, 95, 922–929. [Google Scholar] [CrossRef]
- Garland, C.J.; Smirnov, S.V.; Bagher, P.; Lim, C.S.; Huang, C.Y.; Mitchell, R.; Stanley, C.; Pinkney, A.; Dora, K.A. TRPM4 inhibitor 9-phenanthrol activates endothelial cell intermediate conductance calcium-activated potassium channels in rat isolated mesenteric artery. Br. J. Pharm. 2015, 172, 1114–1123. [Google Scholar] [CrossRef]
- Behringer, E.J.; Scallan, J.P.; Jafarnejad, M.; Castorena-Gonzalez, J.A.; Zawieja, S.D.; Moore, J.E., Jr.; Davis, M.J.; Segal, S.S. Calcium and electrical dynamics in lymphatic endothelium. J. Physiol. 2017, 595, 7347–7368. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.; Qian, X.; Charbel, C.; Ledoux, J.; Parker, J.C.; Taylor, M.S. Automated region of interest analysis of dynamic Ca2+ signals in image sequences. Am. J. Physiol. Cell Physiol. 2012, 303, C236–C243. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.; Waldrup, J.; Qian, X.; Taylor, M.S. Automated analysis of dynamic Ca2+ signals in image sequences. J. Vis. Exp. 2014. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Francis, M.; Kohler, R.; Solodushko, V.; Lin, M.; Taylor, M.S. Positive feedback regulation of agonist-stimulated endothelial Ca2+ dynamics by KCa3.1 channels in mouse mesenteric arteries. Arter. Thromb. Vasc. Biol. 2014, 34, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.; Waldrup, J.R.; Qian, X.; Solodushko, V.; Meriwether, J.; Taylor, M.S. Functional Tuning of Intrinsic Endothelial Ca2+ Dynamics in Swine Coronary Arteries. Circ. Res. 2016, 118, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Advancing Age Decreases Pressure-Sensitive Modulation of Calcium Signaling in the Endothelium of Intact and Pressurized Arteries. J. Vasc. Res. 2016, 53, 358–369. [Google Scholar] [CrossRef]
- Chung, M.K.; Lee, H.; Mizuno, A.; Suzuki, M.; Caterina, M.J. 2-aminoethoxydiphenyl borate activates and sensitizes the heat-gated ion channel TRPV3. J. Neurosci. 2004, 24, 5177–5182. [Google Scholar] [CrossRef] [PubMed]
- Earley, S.; Gonzales, A.L.; Garcia, Z.I. A dietary agonist of transient receptor potential cation channel V3 elicits endothelium-dependent vasodilation. Mol. Pharm. 2010, 77, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Sullivan, M.N.; Pritchard, H.A.; Robinson, J.J.; Earley, S. Unitary TRPV3 channel Ca2+ influx events elicit endothelium-dependent dilation of cerebral parenchymal arterioles. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H2031–H2041. [Google Scholar] [CrossRef] [PubMed]
- Story, G.M.; Peier, A.M.; Reeve, A.J.; Eid, S.R.; Mosbacher, J.; Hricik, T.R.; Earley, T.J.; Hergarden, A.C.; Andersson, D.A.; Hwang, S.W.; et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 2003, 112, 819–829. [Google Scholar] [CrossRef]
- Earley, S.; Gonzales, A.L.; Crnich, R. Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ. Res. 2009, 104, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Francis, M.; Solodushko, V.; Earley, S.; Taylor, M.S. Recruitment of dynamic endothelial Ca2+ signals by the TRPA1 channel activator AITC in rat cerebral arteries. Microcirculation 2013, 20, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 2015, 8, ra2. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, K.; Xu, X.; Mozhayeva, G.; Kuo, T.; Pessah, I.; Mignery, G.; Zhu, X.; Birnbaumer, L.; Muallem, S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature 1998, 396, 478. [Google Scholar] [PubMed]
- Senadheera, S.; Kim, Y.; Grayson, T.H.; Toemoe, S.; Kochukov, M.Y.; Abramowitz, J.; Housley, G.D.; Bertrand, R.L.; Chadha, P.S.; Bertrand, P.P.; et al. Transient receptor potential canonical type 3 channels facilitate endothelium-derived hyperpolarization-mediated resistance artery vasodilator activity. Cardiovasc. Res. 2012, 95, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.F. Endothelial cell ion channel expression and function in arterioles and resistance arteries. In Vascular Ion Channels in Physiology and Disease; Levitan, I., Dopico, A.M., Eds.; Springer: Cham, Switzerland, 2016; pp. 3–36. [Google Scholar]
- Hong, K.; Cope, E.L.; DeLalio, L.J.; Marziano, C.; Isakson, B.E.; Sonkusare, S.K. TRPV4 (Transient Receptor Potential Vanilloid 4) Channel-Dependent Negative Feedback Mechanism Regulates Gq Protein-Coupled Receptor-Induced Vasoconstriction. Arter. Thromb. Vasc. Biol. 2018, 38, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Nausch, L.W.; Bonev, A.D.; Heppner, T.J.; Tallini, Y.; Kotlikoff, M.I.; Nelson, M.T. Sympathetic nerve stimulation induces local endothelial Ca2+ signals to oppose vasoconstriction of mouse mesenteric arteries. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H594–H602. [Google Scholar] [CrossRef] [PubMed]
- Isakson, B.E.; Ramos, S.I.; Duling, B.R. Ca2+ and inositol 1,4,5-trisphosphate-mediated signaling across the myoendothelial junction. Circ. Res. 2007, 100, 246–254. [Google Scholar] [CrossRef]
- Wei, R.; Lunn, S.E.; Tam, R.; Gust, S.L.; Classen, B.; Kerr, P.M.; Plane, F. Vasoconstrictor stimulus determines the functional contribution of myoendothelial feedback to mesenteric arterial tone. J. Physiol. 2018, 596, 1181–1197. [Google Scholar]
- Aguirre, J.A.; Lucchinetti, E.; Clanachan, A.S.; Plane, F.; Zaugg, M. Unraveling Interactions between Anesthetics and the Endothelium: Update and Novel Insights. Anesth. Analg. 2016, 122, 330–348. [Google Scholar] [CrossRef]
- Tran, C.H.; Gordon, G.R. Astrocyte and microvascular imaging in awake animals using two-photon microscopy. Microcirculation 2015, 22, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Martinez, A.D.; Peirce, S.M.; Isakson, B.E.; Nice, M.; Wang, J.; Lounsbury, K.M.; Scallan, J.P.; Murfee, W.L. Induction of microvascular network growth in the mouse mesentery. Microcirculation 2018, 25, e12502. [Google Scholar] [PubMed]
- Biwer, L.A.; Lechauve, C.; Vanhoose, S.; Weiss, M.J.; Isakson, B.E. A Cell Culture Model of Resistance Arteries. J. Vis. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Dabertrand, F.; Earley, S. Isolation and Cannulation of Cerebral Parenchymal Arterioles. J. Vis. Exp. 2016. [Google Scholar] [CrossRef]
- Wilson, C.; Saunter, C.D.; Girkin, J.M.; McCarron, J.G. Pressure-dependent regulation of Ca2+ signalling in the vascular endothelium. J. Physiol. 2015, 593, 5231–5253. [Google Scholar] [PubMed]
- Bagher, P.; Segal, S.S. The mouse cremaster muscle preparation for intravital imaging of the microCirc.ulation. J. Vis. Exp. 2011. [Google Scholar] [CrossRef] [PubMed]
- Koot, B.G.; Alders, M.; Verheij, J.; Beuers, U.; Cobben, J.M. A de novo mutation in KCNN3 associated with autosomal dominant idiopathic non-cirrhotic portal hypertension. J. Hepatol. 2016, 64, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Ellinor, P.T.; Lunetta, K.L.; Glazer, N.L.; Pfeufer, A.; Alonso, A.; Chung, M.K.; Sinner, M.F.; de Bakker, P.I.; Mueller, M.; Lubitz, S.A.; et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genet. 2010, 42, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, Y.Q.; Gong, X.Q.; Wang, X.H.; Li, R.G.; Tan, H.W.; Liu, X.; Fang, W.Y.; Bai, D. Novel germline GJA5/connexin40 mutations associated with lone atrial fibrillation impair gap junctional intercellular communication. Hum. Mutat. 2013, 34, 603–609. [Google Scholar]
- Ghanshani, S.; Coleman, M.; Gustavsson, P.; Wu, A.C.; Gargus, J.J.; Gutman, G.A.; Dahl, N.; Mohrenweiser, H.; Chandy, K.G. Human calcium-activated potassium channel gene KCNN4 maps to chromosome 19q13.2 in the region deleted in diamond-blackfan anemia. Genomics 1998, 51, 160–161. [Google Scholar] [CrossRef]
- Rapetti-Mauss, R.; Lacoste, C.; Picard, V.; Guitton, C.; Lombard, E.; Loosveld, M.; Nivaggioni, V.; Dasilva, N.; Salgado, D.; Desvignes, J.P.; et al. A mutation in the Gardos channel is associated with hereditary xerocytosis. Blood 2015, 126, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Szallasi, A. Transient receptor potential channels as drug targets: From the Science of basic research to the art of medicine. Pharm. Rev. 2014, 66, 676–814. [Google Scholar] [CrossRef] [PubMed]
- Letavernier, E.; Rodenas, A.; Guerrot, D.; Haymann, J.P. Williams-BEur.en syndrome hypercalcemia: Is TRPC3 a novel mediator in calcium homeostasis? Pediatrics 2012, 129, e1626–e1630. [Google Scholar] [PubMed]
- Daumy, X.; Amarouch, M.Y.; Lindenbaum, P.; Bonnaud, S.; Charpentier, E.; Bianchi, B.; Nafzger, S.; Baron, E.; Fouchard, S.; Thollet, A.; et al. Targeted resequencing identifies TRPM4 as a major gene predisposing to progressive familial heart block type I. Int. J. Cardiol. 2016, 207, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.Y.; deBlois, D.; Laporte, S.A.; Gelinas, D.; Tardif, J.C.; Thorin, E.; Shi, Y.; Raignault, A.; Menard, A. Novel Pathogenesis of Hypertension and Diastolic Dysfunction Caused by M3R (Muscarinic Cholinergic 3 Receptor) Signaling. Hypertension 2018, 72, 755–764. [Google Scholar] [PubMed]
- Wang, L.; Guo, D.C.; Cao, J.; Gong, L.; Kamm, K.E.; Regalado, E.; Li, L.; Shete, S.; He, W.Q.; Zhu, M.S.; et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am. J. Hum. Genet. 2010, 87, 701–707. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [PubMed]
- Abete, P.; Della-Morte, D.; Gargiulo, G.; Basile, C.; Langellotto, A.; Galizia, G.; Testa, G.; Canonico, V.; Bonaduce, D.; Cacciatore, F. Cognitive impairment and cardiovascular diseases in the elderly. A heart-brain continuum hypothesis. Ageing Res. Rev. 2014, 18, 41–52. [Google Scholar] [CrossRef]
- Berridge, M.J. Vitamin D, reactive oxygen species and calcium signalling in ageing and disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150434. [Google Scholar] [CrossRef]
- Bugiardini, R.; Manfrini, O.; Pizzi, C.; Fontana, F.; Morgagni, G. Endothelial function predicts future development of coronary artery disease: A study of women with chest pain and normal coronary angiograms. Circulation 2004, 109, 2518–2523. [Google Scholar]
- Schachinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef]
- Segal, S.S. Integration and Modulation of Intercellular Signaling Underlying Blood Flow Control. J. Vasc. Res. 2015, 52, 136–157. [Google Scholar] [CrossRef]
- Climent, B.; Moreno, L.; Martinez, P.; Contreras, C.; Sanchez, A.; Perez-Vizcaino, F.; Garcia-Sacristan, A.; Rivera, L.; Prieto, D. Upregulation of SK3 and IK1 channels contributes to the enhanced endothelial calcium signaling and the preserved coronary relaxation in obese Zucker rats. PLoS ONE 2014, 9, e109432. [Google Scholar] [CrossRef]
- Chadha, P.S.; Haddock, R.E.; Howitt, L.; Morris, M.J.; Murphy, T.V.; Grayson, T.H.; Sandow, S.L. Obesity up-regulates intermediate conductance calcium-activated potassium channels and myoendothelial gap junctions to maintain endothelial vasodilator function. J. Pharm. Exp. 2010, 335, 284–293. [Google Scholar] [CrossRef]
- Giachini, F.R.; Carneiro, F.S.; Lima, V.V.; Carneiro, Z.N.; Dorrance, A.; Webb, R.C.; Tostes, R.C. Upregulation of intermediate calcium-activated potassium channels counterbalance the impaired endothelium-dependent vasodilation in stroke-prone spontaneously hypertensive rats. Transl. Res. 2009, 154, 183–193. [Google Scholar] [CrossRef]
- Schach, C.; Resch, M.; Schmid, P.M.; Riegger, G.A.; Endemann, D.H. Type 2 diabetes: Increased expression and contribution of IKCa channels to vasodilation in small mesenteric arteries of ZDF rats. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1093–H1102. [Google Scholar] [CrossRef]
- Yada, T.; Shimokawa, H.; Tachibana, H. Endothelium-dependent hyperpolarization-mediated vasodilatation compensates nitric oxide-mediated endothelial dysfunction during ischemia in diabetes-induced canine coronary collateral microcirculation in vivo. Microcirculation 2018, 25, e12456. [Google Scholar] [CrossRef]
- Mokhtar, S.S.; Vanhoutte, P.M.; Leung, S.W.; Yusof, M.I.; Wan Sulaiman, W.A.; Mat Saad, A.Z.; Suppian, R.; Rasool, A.H. Endothelium dependent hyperpolarization-type relaxation compensates for attenuated nitric oxide-mediated responses in subcutaneous arteries of diabetic patients. Nitric Oxide 2016, 53, 35–44. [Google Scholar] [CrossRef]
- Bondarenko, A.I.; Panasiuk, O.; Okhai, I.; Montecucco, F.; Brandt, K.J.; Mach, F. Ca2+-dependent potassium channels and cannabinoid signaling in the endothelium of apolipoprotein E knockout mice before plaque formation. J. Mol. Cell. Cardiol. 2018, 115, 54–63. [Google Scholar] [CrossRef]
- Stead, R.; Musa, M.G.; Bryant, C.L.; Lanham, S.A.; Johnston, D.A.; Reynolds, R.; Torrens, C.; Fraser, P.A.; Clough, G.F. Developmental conditioning of endothelium-derived hyperpolarizing factor-mediated vasorelaxation. J. Hypertens. 2016, 34, 452–463. [Google Scholar] [CrossRef]
- Choi, S.; Kim, J.A.; Li, H.Y.; Shin, K.O.; Oh, G.T.; Lee, Y.M.; Oh, S.; Pewzner-Jung, Y.; Futerman, A.H.; Suh, S.H. KCa3.1 upregulation preserves endothelium-dependent vasorelaxation during aging and oxidative stress. Aging Cell 2016, 15, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Behringer, E.J.; Segal, S.S. Impact of Aging on Calcium Signaling and Membrane Potential in Endothelium of Resistance Arteries: A Role for Mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.S.; Walker, B.R. Endothelial-dependent dilation following chronic hypoxia involves TRPV4-mediated activation of endothelial BK channels. Pflug. Arch. 2018, 470, 633–648. [Google Scholar] [CrossRef]
- Jobs, A.; Schmidt, K.; Schmidt, V.J.; Lubkemeier, I.; van Veen, T.A.; Kurtz, A.; Willecke, K.; de Wit, C. Defective Cx40 maintains Cx37 expression but intact Cx40 is crucial for conducted dilations irrespective of hypertension. Hypertension 2012, 60, 1422–1429. [Google Scholar] [CrossRef]
- Givvimani, S.; Narayanan, N.; Armaghan, F.; Pushpakumar, S.; Tyagi, S.C. Attenuation of conducted vasodilation in skeletal muscle arterioles during hyperhomocysteinemia. Pharmacology 2013, 91, 287–296. [Google Scholar] [CrossRef]
- Lemmey, H.A.L.; Ye, X.; Ding, H.C.; Triggle, C.R.; Garland, C.J.; Dora, K.A. Hyperglycaemia disrupts conducted vasodilation in the resistance vasculature of db/db mice. Vasc. Pharm. 2018, 103–105, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Bachschmid, M.M.; Schildknecht, S.; Matsui, R.; Zee, R.; Haeussler, D.; Cohen, R.A.; Pimental, D.; Loo, B. Vascular aging: Chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann. Med. 2013, 45, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Muller-Delp, J.M.; Gurovich, A.N.; Christou, D.D.; Leeuwenburgh, C. Redox balance in the aging microCirc.ulation: New friends, new foes, and new clinical directions. Microcirculation 2012, 19, 19–28. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Chidgey, J.; Fraser, P.A.; Aaronson, P.I. Reactive oxygen species facilitate the EDH response in arterioles by potentiating intracellular endothelial Ca2+ release. Free Radic. Biol. Med. 2016, 97, 274–284. [Google Scholar] [CrossRef]
- Feletou, M. Endothelium-Dependent Hyperpolarization and Endothelial Dysfunction. J. Cardiovasc. Pharm. 2016, 67, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Hart, J.L.; Woodman, O.L. Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br. J. Pharm. 2011, 162, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Long, D.A.; Newaz, M.A.; Prabhakar, S.S.; Price, K.L.; Truong, L.D.; Feng, L.; Mu, W.; Oyekan, A.O.; Johnson, R.J. Loss of nitric oxide and endothelial-derived hyperpolarizing factor-mediated responses in aging. Kidney Int. 2005, 68, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Chennupati, R.; Lamers, W.H.; Koehler, S.E.; De Mey, J.G. Endothelium-dependent hyperpolarization-related relaxations diminish with age in murine saphenous arteries of both sexes. Br. J. Pharm. 2013, 169, 1486–1499. [Google Scholar]
- Seki, T.; Goto, K.; Kiyohara, K.; Kansui, Y.; Murakami, N.; Haga, Y.; Ohtsubo, T.; Matsumura, K.; Kitazono, T. Downregulation of Endothelial Transient Receptor Potential Vanilloid Type 4 Channel and Small-Conductance of Ca2+-Activated K+ Channels Underpins Impaired Endothelium-Dependent Hyperpolarization in Hypertension. Hypertension 2017, 69, 143–153. [Google Scholar] [PubMed]
- Gradel, A.K.J.; Salomonsson, M.; Sorensen, C.M.; Holstein-Rathlou, N.H.; Jensen, L.J. Long-term diet-induced hypertension in rats is associated with reduced expression and function of small artery SKCa, IKCa, and Kir2.1 channels. Clin. Sci. (Lond.) 2018, 132, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Wandall-Frostholm, C.; Dalsgaard, T.; Bajoriunas, V.; Olivan-Viguera, A.; Sadda, V.; Beck, L.; Mogensen, S.; Stankevicius, E.; Simonsen, U.; Kohler, R. Genetic deficit of KCa 3.1 channels protects against pulmonary Circ.ulatory collapse induced by TRPV4 channel activation. Br. J. Pharmacol. 2015, 172, 4493–4505. [Google Scholar] [PubMed]
- Simonsen, U.; Wandall-Frostholm, C.; Olivan-Viguera, A.; Kohler, R. Emerging roles of calcium-activated K channels and TRPV4 channels in lung oedema and pulmonary Circ.ulatory collapse. Acta Physiol. (Oxf.) 2017, 219, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, T.; Sonkusare, S.K.; Teuscher, C.; Poynter, M.E.; Nelson, M.T. Pharmacological inhibitors of TRPV4 channels reduce cytokine production, restore endothelial function and increase survival in septic mice. Sci. Rep. 2016, 6, 33841. [Google Scholar] [PubMed]
- Jones, J.L.; Peana, D.; Veteto, A.B.; Lambert, M.D.; Nourian, Z.; Karasseva, N.G.; Hill, M.A.; Lindman, B.R.; Baines, C.P.; Krenz, M.; et al. TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc. Res. 2019, 115, 46–56. [Google Scholar]
- Nachman, R.L.; Jaffe, E.A. Endothelial cell culture: Beginnings of modern vascular biology. J. Clin. Investig. 2004, 114, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Lee, M.D.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Girkin, J.M.; Saunter, C.D.; McCarron, J.G. Mitochondrial ATP production provides long-range control of endothelial inositol trisphosphate-evoked calcium signaling. J. Biol. Chem. 2019, 294, 737–758. [Google Scholar] [CrossRef]
- Durand, M.J.; Ait-Aissa, K.; Levchenko, V.; Staruschenko, A.; Gutterman, D.D.; Beyer, A.M. Visualization and Quantification of Mitochondrial Structure in the Endothelium of Intact Arteries. Cardiovasc. Res. 2018. [Google Scholar] [CrossRef]
- Busija, D.W.; Rutkai, I.; Dutta, S.; Katakam, P.V. Role of Mitochondria in Cerebral Vascular Function: Energy Production, Cellular Protection, and Regulation of Vascular Tone. Compr. Physiol. 2016, 6, 1529–1548. [Google Scholar] [PubMed]
- Kaczara, P.; Motterlini, R.; Rosen, G.M.; Augustynek, B.; Bednarczyk, P.; Szewczyk, A.; Foresti, R.; Chlopicki, S. Carbon monoxide released by CORM-401 uncouples mitochondrial respiration and inhibits glycolysis in endothelial cells: A role for mitoBKCa channels. Biochim. Biophys. Acta 2015, 1847, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Kanagy, N.L.; Kevil, C.G. The pleiotropic effects of hydrogen sulfide. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1–H2. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.S.; Osmond, J.M.; Walker, B.R.; Kanagy, N.L. Hydrogen sulfide-induced vasodilation mediated by endothelial TRPV4 channels. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1437–H1444. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Yang, G.; Jiang, B.; Ju, Y.; Wu, L.; Wang, R. H2S is an endothelium-derived hyperpolarizing factor. Antioxid. Redox Signal. 2013, 19, 1634–1646. [Google Scholar] [CrossRef] [PubMed]
- Durante, W. Targeting heme oxygenase-1 in vascular disease. Curr. Drug Targets 2010, 11, 1504–1516. [Google Scholar] [CrossRef]
- McRae, K.E.; Pudwell, J.; Peterson, N.; Smith, G.N. Inhaled carbon monoxide increases vasodilation in the microvascular circulation. Microvasc. Res. 2019, 123, 92–98. [Google Scholar] [CrossRef]
- Rezkalla, S.; Kloner, R.A. Cardiovascular effects of marijuana. Trends Cardiovasc. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.S.; Zheng, X.; Zhang, D.X. Role of endothelial TRPV4 channels in vascular actions of the endocannabinoid, 2-arachidonoylglycerol. Br. J. Pharm. 2015, 172, 5251–5264. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.P.; O’Sullivan, S.E. Cyclooxygenase metabolism mediates vasorelaxation to 2-arachidonoylglycerol (2-AG) in human mesenteric arteries. Pharm. Res. 2014, 81, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bondarenko, A.I.; Panasiuk, O.; Drachuk, K.; Montecucco, F.; Brandt, K.J.; Mach, F. The quest for endothelial atypical cannabinoid receptor: BKCa channels act as cellular sensors for cannabinoids in in vitro and in situ endothelial cells. Vasc. Pharm. 2018, 102, 44–55. [Google Scholar] [CrossRef]
- Brandes, R.P.; Schmitz-Winnenthal, F.H.; Feletou, M.; Godecke, A.; Huang, P.L.; Vanhoutte, P.M.; Fleming, I.; Busse, R. An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice. Proc. Natl. Acad. Sci. USA 2000, 97, 9747–9752. [Google Scholar] [CrossRef] [PubMed]
- Limbu, R.; Cottrell, G.S.; McNeish, A.J. Characterisation of the vasodilation effects of DHA and EPA, n-3 PUFAs (fish oils), in rat aorta and mesenteric resistance arteries. PLoS ONE 2018, 13, e0192484. [Google Scholar] [CrossRef]
- Idris Khodja, N.; Chataigneau, T.; Auger, C.; Schini-Kerth, V.B. Grape-derived polyphenols improve aging-related endothelial dysfunction in rat mesenteric artery: Role of oxidative stress and the angiotensin system. PLoS ONE 2012, 7, e32039. [Google Scholar]
- Han, J.; Xu, H.H.; Chen, X.L.; Hu, H.R.; Hu, K.M.; Chen, Z.W.; He, G.W. Total Flavone of Rhododendron Improves Cerebral Ischemia Injury by Activating Vascular TRPV4 to Induce Endothelium-Derived Hyperpolarizing Factor-Mediated Responses. Evid.-Based Complement. Altern. Med. 2018, 2018, 8919867. [Google Scholar] [CrossRef]
- Ruamyod, K.; Watanapa, W.B.; Shayakul, C. Testosterone rapidly increases Ca2+-activated K+ currents causing hyperpolarization in human coronary artery endothelial cells. J. Steroid Biochem. Mol. Biol. 2017, 168, 118–126. [Google Scholar] [CrossRef]
- Mazzuca, M.Q.; Mata, K.M.; Li, W.; Rangan, S.S.; Khalil, R.A. Estrogen receptor subtypes mediate distinct microvascular dilation and reduction in [Ca2+]i in mesenteric microvessels of female rat. J. Pharm. Exp. 2015, 352, 291–304. [Google Scholar] [CrossRef]
- Aird, W.C. Spatial and temporal dynamics of the endothelium. J. Thromb. Haemost. 2005, 3, 1392–1406. [Google Scholar]
- Fishman, A.P. Endothelium: A distributed organ of diverse capabilities. Ann. N. Y. Acad. Sci. 1982, 401, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hielscher, D.; Kaebisch, C.; Braun, B.J.V.; Gray, K.; Tobiasch, E. Stem Cell Sources and Graft Material for Vascular Tissue Engineering. Stem Cell Rev. 2018, 14, 642–667. [Google Scholar] [CrossRef] [PubMed]
- Mauricio, M.D.; Guerra-Ojeda, S.; Marchio, P.; Valles, S.L.; Aldasoro, M.; Escribano-Lopez, I.; Herance, J.R.; Rocha, M.; Vila, J.M.; Victor, V.M. Nanoparticles in Medicine: A Focus on Vascular Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 6231482. [Google Scholar] [CrossRef]
- Phan, D.T.; Bender, R.H.F.; Andrejecsk, J.W.; Sobrino, A.; Hachey, S.J.; George, S.C.; Hughes, C.C. Blood-brain barrier-on-a-chip: MicroPhysiol.ogical systems that capture the complexity of the blood-central nervous system interface. Exp. Biol. Med. (Maywood) 2017, 242, 1669–1678. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behringer, E.J.; Hakim, M.A. Functional Interaction among KCa and TRP Channels for Cardiovascular Physiology: Modern Perspectives on Aging and Chronic Disease. Int. J. Mol. Sci. 2019, 20, 1380. https://doi.org/10.3390/ijms20061380
Behringer EJ, Hakim MA. Functional Interaction among KCa and TRP Channels for Cardiovascular Physiology: Modern Perspectives on Aging and Chronic Disease. International Journal of Molecular Sciences. 2019; 20(6):1380. https://doi.org/10.3390/ijms20061380
Chicago/Turabian StyleBehringer, Erik J., and Md A. Hakim. 2019. "Functional Interaction among KCa and TRP Channels for Cardiovascular Physiology: Modern Perspectives on Aging and Chronic Disease" International Journal of Molecular Sciences 20, no. 6: 1380. https://doi.org/10.3390/ijms20061380
APA StyleBehringer, E. J., & Hakim, M. A. (2019). Functional Interaction among KCa and TRP Channels for Cardiovascular Physiology: Modern Perspectives on Aging and Chronic Disease. International Journal of Molecular Sciences, 20(6), 1380. https://doi.org/10.3390/ijms20061380